
Abstract
Objective
This study aimed to investigate regulation of the cyclic adenosine monophosphate (cAMP) signaling pathway on connective tissue growth factor (CTGF) during myocardial fibrosis (MF) in mice after myocardial infarction (MI).
Methods
An MI mouse model was established and cardiac function indices were detected by ultrasound. Quantitative reverse transcription polymerase chain reaction and western blotting were used to determine CTGF and transforming growth factor β1 (TGF-β1) cardiac expression. Mouse cardiac fibroblasts (MCFs) were used to study the mechanism of MF after MI.
Results
Cardiac function indices were lower after MI. Cardiac function indices were better in the MI + meglumine adenosine cyclophosphate (MAC) group than in the MI group, and CTGF expression in the MI + MAC group was downregulated. TGF-β1 expression was not different among the MI groups. Forskolin increased intracellular cAMP levels and inhibited CTGF expression in MCFs. Expression of p44/42 mitogen-activated protein kinase (MAPK) was significantly lower in the TGF-β1 + forskolin group than in the TGF-β1 group, while protein kinase A was significantly upregulated. CTGF expression was significantly lower in the TGF-β1 + forskolin + PD98509 group than in the TGF-β1 + forskolin group.
Conclusions
This study shows that cAMP upregulates protein kinase A expression through the p44/42MAPK signaling pathway and decreases p44/42MAPK phosphorylation levels, inhibiting CTGF expression.
Keywords
Introduction
Myocardial infarction (MI) is an ischemic cardiomyopathy caused by stenosis or obstruction of the coronary artery or its branches,1,2 and it seriously endangers human health.1,2 Myocardial fibrosis (MF) is a common pathological process in many cardiac and cardiovascular diseases. MF mainly occurs by secretion of collagen in the myocardial interstitium and an imbalance of the collagen I/collagen III ratio, which is important for left ventricular remodeling after MI.3,4 Cardiac repair of MI mainly depends on tissue fibrosis, and repair of fibrosis is conducive to maintaining integrity and function of the heart. 4 However, excessive MF leads to increased cardiac stiffness and decreased diastolic and systolic function, and a change in the normal electrophysiological structure of the heart, thus greatly increasing the incidence of heart failure.4,5 Therefore, MF after MI is an important issue for prognosis. However, the mechanism of MF after MI remains unclear.
The main factors involved in the process of MF are transforming growth factor β1 (TGF-β1), connective tissue growth factor (CTGF), and angiotensin II. 6 TGF-β1 and CTGF are important profibrotic factors.7,8 TGF-β1 and CTGF promote each other by cell phenotype transformation and induce increased synthesis of extracellular matrices.9,10 CTGF is a secretory protein with important biological functions, and its most prominent function is to promote collagen synthesis and fibrosis. 11 In animal models of MI, high CTGF expression is directly related to fibrosis, and this high expression can be detected in infarcted and non-infarcted areas of myocardial tissue.12,13 High CTGF expression is also found in many pathological processes, such as atherosclerosis, airway remodeling, liver fibrosis, and renal fibrosis, 14 and its expression levels gradually increase with time. 15
Cyclic adenosine monophosphate (cAMP) is an important second messenger signaling molecule in cells. 16 Elevation of intracellular cAMP levels selectively inhibit CTGF gene expression, as well as proliferation and collagen synthesis of lung and kidney fibroblasts.17,18 Levels of cAMP in atherosclerotic plaques of atherosclerotic vessels are significantly lower than those in normal vessels, and cAMP levels in fibroblasts and smooth muscle cells are also significantly reduced. 19 These studies suggest a relationship between intracellular cAMP and CTGF gene expression, and this regulates MF. Various regulatory factors regulate CTGF expression through specific signaling pathways. 20 In human lung fibroblasts, CTGF expression is mainly mediated by the c-Jun N-terminal kinase signaling pathway. The p44/42 mitogen-activated protein kinase (MAPK) signaling pathway plays an important role in fibrosis caused by CTGF overexpression induced by TGF-β1. 21 However, the mechanism by which cAMP regulates MF after MI remains unclear.
Therefore, this study aimed to investigate regulation of the cAMP signaling pathway on CTGF during MF in mice after MI.
Materials and methods
Mice
Specific pathogen-free grade C57BL/6J adult male mice (body weight, 24–26 g) and C57BL/6J neonatal mice within 1 week of birth were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd. (Beijing, China). All procedures performed in the current study were approved by the Ethics Committee of Hebei Medical University (20160802-03). Animal experiments in the present study strictly abided by ethical guidelines of the Hospital Ethics Committee for Experimental Animals and the principles of “Guidelines for the Care of Laboratory Animals” published by the NIH of the United States and met the requirements of ethical standards for animal experiments.
Adult male C57BL/6J mice were accurately weighed, injected with 3.3% chloral hydrate intraperitoneally (0.014 mL/g) for anesthesia, and fixed on the operating table for skin preparation. Intraoperative electrocardiographic monitoring was performed using the BL-420F biological function signal acquisition system (Techman, Chengdu, China). The chest of mice in the experimental group was sterilized with cotton soaked in alcohol. An oblique incision was made on the left side of the sternum along the rib margin to expose the fourth and fifth intercostals and the heart. The pericardium was removed and the left anterior descending coronary artery was located. The anterior descending artery was ligated 2 to 3 mm from the root of the left coronary artery. The thoracic incision was then closed, thoracic air was extruded, and the wound was sutured. The MI model was judged as successful when the myocardium below the ligation point changed from red to white, apical beating weakened, and an intraoperative electrocardiogram indicated that the ST segment arch rose upward. Mice in the sham group were treated in the same way as the experimental group, except that the left anterior descending coronary artery was not ligated. Mice in the MI + MAC group were treated with meglumine adenosine cyclophosphate (MAC) via intraperitoneal injection (Sigma-Aldrich, St. Louis, MO, USA) 1 week after successful construction of the MI model. MAC is a new synthetic derivative of cAMP that has increased fat solubility and can be well absorbed by the body. After the operation, the mice were subjected to routine feeding and intramuscular injection of antibiotics (penicillin sodium, 2 × 104 U/day) was performed during the first 3 days to prevent postoperative infection.
Ultrasonography
At the end of the fifth week after the operation, adult male C57BL/6J mice were injected with 3.3% chloral hydrate intraperitoneally (0.014 mL/g) for anesthesia, and fixed on the operating table for skin preparation. A small amount of ultrasound coupling agent was applied. A special high-frequency probe for small animals was used to determine cardiac parameters in mice, including the left ventricular diameter, left ventricular anterior wall thickness, and left ventricular posterior wall thickness during systolic/diastolic phases. The ejection fraction (EF%) and short axis fractional shortening (FS%) of the left ventricle were evaluated by data analysis with software of the ultrasound system (Shenzhen Mindray Bio-Medical Electronics Co., Ltd., Shenzhen, China).
Cells
To isolate primary mouse cardiac fibroblasts (MCFs), C57BL/6J neonatal mice were sacrificed within 1 week of birth to remove their hearts. After removing atria and cardiac connective tissues, the left ventricle was preserved. Left ventricular tissue was cut and mixed with 5 mL of digestive juice (equal volumes of 0.125% trypsin and 0.1% collagenase II) before being bathed in water at 37°C for 10 minutes. The supernatant was removed after remaining still for 10 minutes. This digestion was repeated several times until the myocardial tissue mass completely disappeared. The supernatant was filtered by a 200-mesh cell sieve and centrifuged at 4°C and 1500 × g for 10 minutes. After discarding the supernatant, 10% fetal bovine serum (Thermo Fisher Scientific, Waltham, MA, USA) was added to resuspend the cells, which were then cultured under 37°C and 5% CO2 for 90 minutes. Cellular adherence was observed under an inverted phase contrast microscope (Thermo Fisher Scientific) and non-adherent cells were removed. The third generation of cells was used in subsequent experiments. To stimulate CTGF expression in MCFs, recombinant human TGF-β1 (Thermo Fisher Scientific) was added and incubated for 24 hours. To increase intracellular levels of cAMP, forskolin, which is an adenylate cyclase agonist, was added to MCFs. To test whether the p44/42MAPK signaling pathway is involved in regulation of CTGF by cAMP, PD98509 (Merck KGaA, Darmstadt, Germany), which is an inhibitor of mitogen-activated protein kinase kinase (MAPKK), was used to treat MCFs that were stimulated by recombinant human TGF-β1 and forskolin.
Quantitative reverse transcription polymerase chain reaction (RT-qPCR)
Total RNA from cells and heart tissue was extracted by an RNA extraction kit (Merck KGaA) and reverse-transcribed into cDNA. Target genes were amplified by RT-qPCR. Relative expression of target genes was calculated by the ΔΔCT method. 22
Western blotting
Cells or heart tissues were first submerged in liquid nitrogen and then lysed by radio immuno-precipitation assay (RIPA) buffer for protein extraction. Lysed protein samples were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (8% gel), transferred to a polyvinylidene fluoride membrane, and blocked with skimmed milk. The proteins were then reacted with a primary CTGF mouse monoclonal antibody (1: 1000; Abcam, Cambridge, UK) and then a secondary antibody (1: 1000; Abcam) before developing and imaging.
Enzyme-linked immunosorbent assay
MCFs in the third passage were trypsinized and centrifuged at 1500 × g for 10 minutes to collect the cells, which were then lysed, and the supernatant was collected for enzyme-linked immunosorbent assay. A mouse cAMP enzyme-linked immunosorbent assay kit (Merck KGaA) was used to determine the content of cAMP in cell supernatants. Concentrations of cAMP were determined from standard curves.
Statistical analysis
Statistical analysis was performed using the IBM Statistical Package for Social Science (version 22.0; IBM Corp., Armonk, NY, USA.). Measurement data are expressed as mean ± standard deviation. The independent sample t-test was used for comparison between two groups. Multigroup differences were compared by one-way analysis of variance. P < 0.05 indicated a significant difference.
Results
Indices of cardiac function in the MI group are worse than those in the sham group, but these are alleviated with treatment by MAC
Ultrasonography showed that the ventricular diameter in the MI and MI + MAC groups in left ventricular systole was significantly higher than that in the sham group (both P < 0.01) (Figure 1a). Similarly, the ventricular diameter in the MI and MI + MAC groups in left ventricular diastole was significantly higher than that in the sham group (both P < 0.01) (Figure 1b). The EF% and left ventricular FS% in the MI + MAC group were significantly higher than those in the MI group (both P < 0.05) (Figure 1c and d). Stroke volume in the MI + MAC group was significantly larger than that in the MI group (P < 0.05) (Figure 1e). These results suggest that the indices of cardiac function in mice in the MI group are worse than those in sham group, but treatment with MAC alleviates these indices.
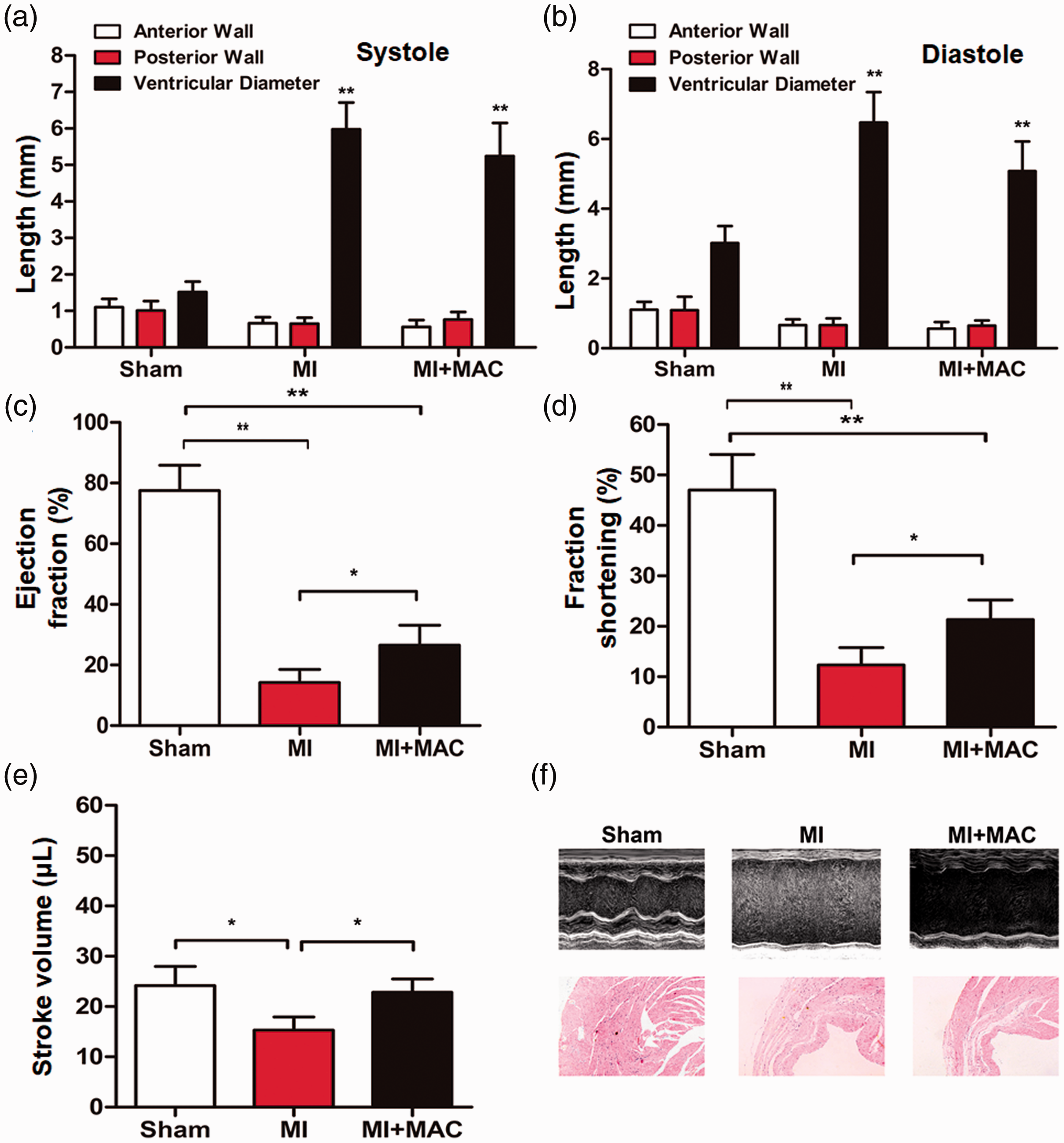
Detection of cardiac function indicators in mice determined by ultrasound. (a, b) Anterior wall thickness, posterior wall thickness, and ventricular diameter of the left ventricle in (a) systole and (b) diastole. **P < 0.01 compared with the sham group. (c) Left ventricular ejection fraction. (d) Left ventricular short axis fractional shortening rate. (e) Left ventricular stroke volume. *P < 0.05.
cAMP decreases CTGF expression in heart tissue of mice with MI
CTGF mRNA and protein expression in the infarct area (Figure 2a and c) and non-infarct area (Figure 2b and d) in the MI and MI + MAC groups was significantly higher than that in the sham group (all P < 0.05), but that in the MI + MAC group was significantly lower than that in the MI group (all P < 0.05) (Figure 2a–d). Moreover, TGF-β1 mRNA and protein expression in the infarct area (Figure 2e and g) and non-infarct area (Figure 2f and h) in the MI and MI + MAC groups was significantly higher than that in the sham group (all P < 0.05), but that in the MI + MAC group was not different from that in the MI group (Figure 2e–h). These results indicate that cAMP decreases CTGF levels in heart tissue of mice with MI.

Effect of cyclic adenosine monophosphate on CTGF and TGF-β1 expression in heart tissue from mice. (a, b) CTGF mRNA expression in (a) infarct and (b) non-infarct areas in the heart of mice in the sham, MI, and MI + MAC groups. (c, d) CTGF protein expression in (c) infarct and (d) non-infarct areas in the heart of mice in the sham, MI, and MI + MAC groups. (e, f) TGF-β1 mRNA expression in (e) infarct and (f) non-infarct areas in the heart of mice in the sham, MI, and MI + MAC groups. (g, h) TGF-β1 protein expression in (g) infarct and (h) non-infarct areas in the heart of mice in the sham, MI, and MI + MAC groups. *P < 0.05; **P < 0.01; n.s., P > 0.05.
Forskolin elevates intracellular cAMP levels and inhibits CTGF expression in MCFs
To test the effect of cAMP on CTGF expression in MCFs, recombinant human TGF-β1 was used to stimulate overexpression of CTGF in MCFs and forskolin was added to elevate cAMP levels in MCFs. CTGF mRNA and protein levels in MCFs treated with TGF-β1 were significantly higher than those in untreated MCFs in the control group (both P < 0.05) (Figure 3a and b). Levels of cAMP in the TGF-β1 group were not significantly different from those in the control group. Levels of cAMP in the TGF-β1 + forskolin group were significantly elevated compared with those in the TGF-β1 group (P < 0.05) (Figure 3c). Additionally, CTGF mRNA and protein levels in the TGF-β1 + forskolin group were significantly lower than those in the TGF-β1 group (both P < 0.05) (Figure 3d and e). The results suggest that forskolin elevates intracellular cAMP levels and inhibits CTGF expression in MCFs.
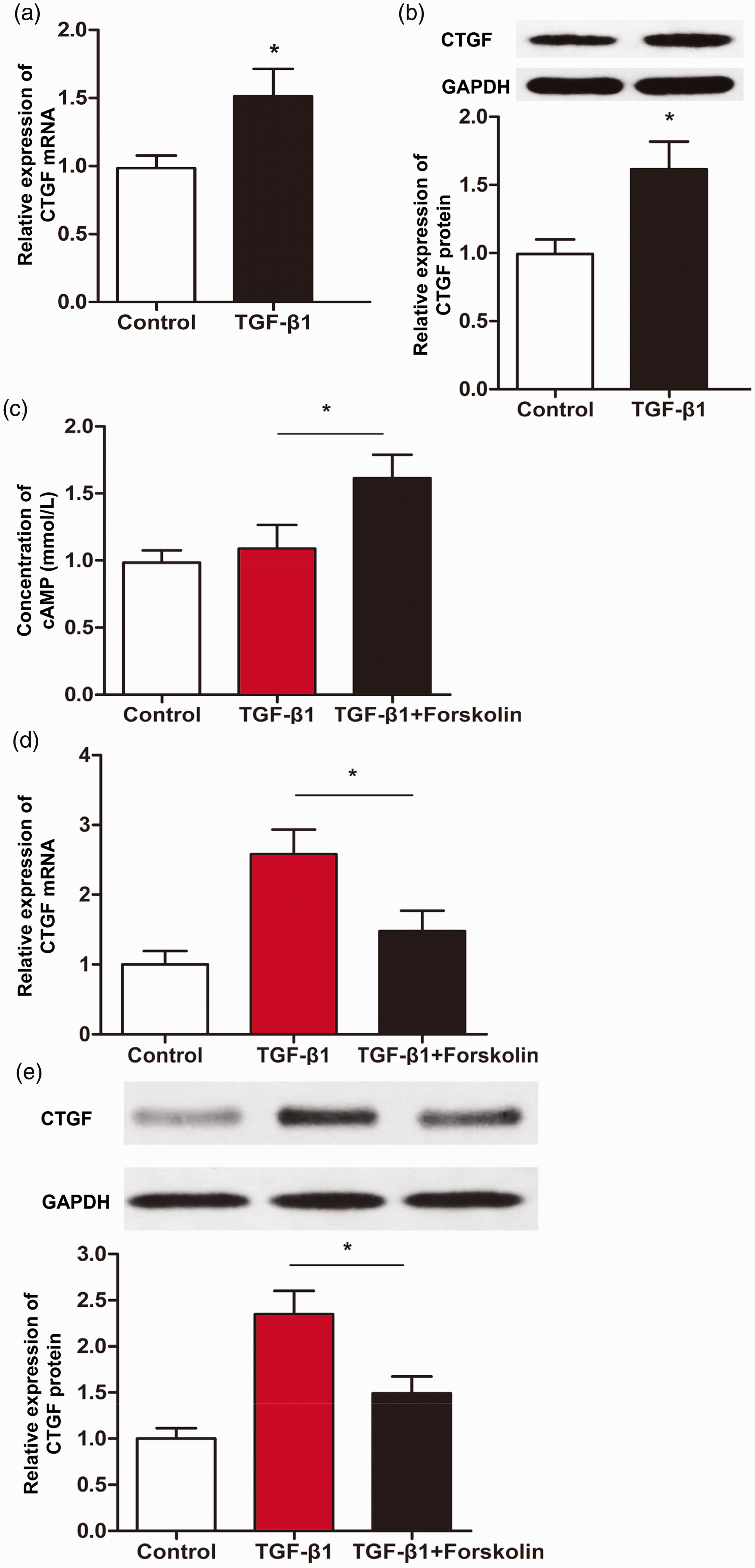
Effect of forskolin on CTGF expression in mouse cardiac fibroblasts. (a, b) CTGF (a) mRNA and (b) protein expression in untreated mouse cardiac fibroblasts (control group) or mouse cardiac fibroblasts treated with TGF-β1 for 24 hours. *P < 0.05 compared with the control group. (c) Concentrations of cAMP in untreated mouse cardiac fibroblasts (control group), mouse cardiac fibroblasts treated with TGF-β1 for 24 hours, and mouse cardiac fibroblasts treated with TGF-β1 and forskolin for 24 hours. *P < 0.05. (d, e) CTGF (d) mRNA and (e) protein expression in untreated mouse cardiac fibroblasts (control group), mouse cardiac fibroblasts treated with TGF-β1 for 24 hours, and mouse cardiac fibroblasts treated with TGF-β1 and forskolin for 24 hours. *P < 0.05.
cAMP inhibits CTGF expression in MCFs via the p44/42MAPK signaling pathway
We found that p44/42MAPK mRNA and protein levels in the TGF-β1 + forskolin group were significantly lower than those in the TGF-β1 group (both P < 0.05) (Figure 4a and b). By contrast, PKA mRNA and protein levels in the TGF-β1 + forskolin group were significantly higher than those in the TGF-β1 group (both P < 0.01) (Figure 4c and d). Moreover, treatment with PD98509 significantly reduced CTGF mRNA and protein levels in the TGF-β1 + forskolin group compared with no treatment of PD98509 in the TGF-β1 + forskolin group (both P < 0.05) (Figure 4e and f). These results indicate that cAMP upregulates PKA expression through the p44/42MAPK signaling pathway and reduces the phosphorylation level of downstream p44/42MAPK, thus inhibiting CTGF expression in MCFs.

Cyclic adenosine monophosphate regulates CTGF expression in MCFs via the p44/42MAPK signaling pathway. (a, b) Expression of p44/42MAPK (a) mRNA and (b) protein in untreated mouse cardiac fibroblasts (control group), mouse cardiac fibroblasts treated with TGF-β1 for 24 hours, and mouse cardiac fibroblasts treated with TGF-β1 and forskolin for 24 hours. *P < 0.05. (c, d) PKA (c) mRNA and (d) protein expression in untreated mouse cardiac fibroblasts (control group), mouse cardiac fibroblasts treated with TGF-β1 for 24 hours, and mouse cardiac fibroblasts treated with TGF-β1 and forskolin for 24 hours. **P < 0.01. (e, f) CTGF (e) mRNA and (f) protein expression in untreated mouse cardiac fibroblasts (control group), mouse cardiac fibroblasts treated with TGF-β1 and forskolin for 24 hours, and mouse cardiac fibroblasts treated with TGF-β1, forskolin, and PD98509 for 24 hours. *P < 0.05.
Discussion
Unfavorable factors for ventricular remodeling in the process of cardiac repair after MI are hotspots of current research. After repair of MF in the infarcted area after MI, MF does not disappear, but continues in infarcted areas or even in non-infarcted areas.23,24 Unbalanced fibrosis may occur in infarcted and non-infarcted areas. Secondary excessive interstitial fibrosis in non-infarcted areas may lead to decreased cardiac compliance, which greatly increases the incidence of heart failure. 4 Therefore, repair of fibrotic scarring after MI is important for recovery of cardiac function and structural integrity.
Fibrotic scar tissue after MI is mainly composed of type I collagen fibers that are hard. Excessive fibrosis leads to decreased cardiac compliance and affects contractile function of the ventricular wall. 19 TGF-β1 is a major profibrotic factor, which is widely involved in the fibrosis process in cells. The profibrotic effect of TGF-β1 mainly depends on activating the downstream signal molecule CTGF, which is directly involved in fibrosis as an important signal molecule. 25 After MI, TGF-β1 only plays an important role in the initial stage of fibrosis, and its expression decreases gradually with time. 13 Sustained fibrosis is closely related to sustained CTGF expression, and its expression continues to rise until the 16th week after MI. 13 In a study of MI in rats, CTGF mRNA expression was continuously upregulated during the first and second weeks. 15 Once CTGF was activated by TGF-β1, CTGF continued to be expressed, even after withdrawing continuous stimulation by TGF-β1, and the fibrotic process continued. 15 These studies suggest that CTGF is more suitable than TGF-β1 as a target for anti-fibrosis therapy. Expression of CTGF can be selectively inhibited to suppress MF after MI.
cAMP is an important second messenger in cells that widely participates in cellular proliferation and differentiation.18,26 A significant decrease in cAMP levels and high CTGF expression occur in atherosclerotic lesions.23,24 CTGF is an important secretory protein and a member of the immediate early gene CTGF/cyr-61/nov family (CCN family).27,28 CTGF has a wide range of functions, including participating in normal embryonic development and differentiation, promoting angiogenesis, and extracellular matrix synthesis. However, the most important function of CTGF is to promote collagen synthesis and fibrosis.27,28 In our study, we found that CTGF expression in infarcted and non-infarcted areas was downregulated after intraperitoneal injection of MAC in mice with MI for 1 week. CTGF in different cells participates in different cellular events involving different signaling pathways. CTGF in smooth muscle cells mainly mediates fibrosis through the p38 signaling pathway. 29 In lung fibroblasts, CTGF mediates fibrosis mainly through the c-Jun N-terminal kinase signaling pathway. 20 The p44/42MAPK signaling pathway is involved in fibrosis caused by upregulation of CTGF, which is induced by TGF-β1.21,30 The present study showed that phosphorylated p44/42MAPK expression in MCFs after forskolin intervention was lower than that in the control group. This finding suggests that phosphorylation of p44/42MAPK plays an important role in the process of forskolin increasing intracellular cAMP levels and inhibiting CTGF expression in MCFs. We also found that forskolin inhibited CTGF mRNA. Additionally, PD98509 inhibited MAPKK. After combining forskolin and PD98509, CTGF expression was significantly decreased, which indicated that these two factors blocked CTGF expression with different signaling pathways.
TGF-β1 and CTGF are widely involved in the process of myocardial fibrosis after acute MI, and CTGF expression increases with time, reaching a peak at approximately 180 days. 13 This suggests that CTGF is not only involved in the initial stage of fibrosis, but is also closely related to the continuous progress of fibrosis after acute MI. 13 Therefore, CTGF is more targeted and specific than TGF-β1 in treatment of myocardial fibrosis after acute MI. 12 Our findings provide new evidence for clinical research and development of CTGF-targeted drugs.
In conclusion, the present study shows that increased intracellular cAMP levels in MCFs inhibit CTGF expression in these cells. High cAMP levels stimulate activation of PKA and inhibit phosphorylation of downstream p44/42MAPK and CTGF expression, thus suppressing fibrosis.
Footnotes
Acknowledgments
The authors wish to thank their department and research team for their help and dedication.
Author contributions
HZ and LD contributed to the design of the study. HZ performed the experiments and analyzed the data. HZ and LD interpreted the results and prepared the manuscript. The final version of the manuscript has been read and approved by both authors.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Declaration of conflicting interest
The authors declare that there is no conflict of interest.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.