
Abstract
Objective
To explore the effects of wedelolactone (WEL) on sepsis-induced renal injury in the human renal proximal tubular epithelial cell line HK-2.
Methods
HK-2 cells were stimulated by 1 µg/ml lipopolysaccharide (LPS) to trigger renal injury in vitro. HK-2 cells were pretreated with or without WEL (0.1, 1 and 10 µM) before LPS stimulation. Protein and mRNA analyses were performed using enzyme-linked immunosorbent assays, Western blot analysis and quantitative reverse transcription–polymerase chain reaction. The MTT assay and flow cytometry were used to measure cell viability and the rate of cell apoptosis. Protein tyrosine phosphatase non-receptor type 2 (PTPN2) knockdown was induced by the transection of HK-2 cells with short hairpin RNA.
Results
Cell viability was significantly increased in a dose-dependent manner by WEL in LPS-induced HK-2 cells. WEL also decreased the levels of four inflammatory cytokines and cell apoptosis in LPS-induced HK-2 cells. The level of PTPN2 was increased after WEL treatment. PTPN2 knockdown partly abolished the inhibitory effects of WEL on cell apoptosis, the levels of inflammatory cytokines and on p38 mitogen-activated protein kinase/nuclear factor-kappaB signalling in LPS-induced HK-2 cells.
Conclusion
WEL improved renal injury by suppressing inflammation and cell apoptosis through upregulating PTPN2 in HK-2 cells. PTPN2 might be used as a potential therapeutic target for LPS-induced sepsis.
Keywords
Introduction
Sepsis is one of the leading causes of death in intensive care units worldwide due to its association with the dysfunction of multiple organs resulting from a dysregulated host response to infection induced by pathogens like lipopolysaccharide (LPS)-releasing gram-negative bacteria.1,2 It has been reported that approximately half of acute renal injury cases are associated with severe sepsis, which is induced by an overwhelming immune response and lots of inflammatory cytokines. 3 Renal parenchymal inflammation, intrarenal thrombosis and tubular injury are associated with inflammatory cytokine secretion. 4 Therefore, anti-inflammatory therapy is beneficial for the treatment of sepsis-induced renal injury. 5
Wedelolactone (WEL), of which the chemical structure is shown in Figure 1a, is a major component derived from the medicinal plant Eclipta prostrata. 6 It has been reported to hold immunomodulatory, antifibrotic, anti-inflammatory, antifree radical, antioxidative and antifatigue effects.7,8 Evidence shows that WEL plays a crucial role in regulating the inflammatory response in zymosan-induced mouse bone marrow-derived macrophages and inhibiting the levels of systemic inflammatory cytokines. 9 WEL can regulate the IkappaK/IkappaB/nuclear factor-kappaB (NF-κB) signalling pathway to alleviate oxidative stress and inhibit LPS-induced inflammation via the NF-κB pathway in RAW 264.7 cells, contributing to the attenuation of renal damage. 8 WEL exerts a protective effect on inflammation-related diseases and might be a potential treatment for renal injury.7–9 However, the previous study only investigated the role of WEL in podocytes. 8 The effect of WEL on LPS-induced HK-2 cells, a classical cell model of sepsis-induced renal injury in vitro, has not been reported.
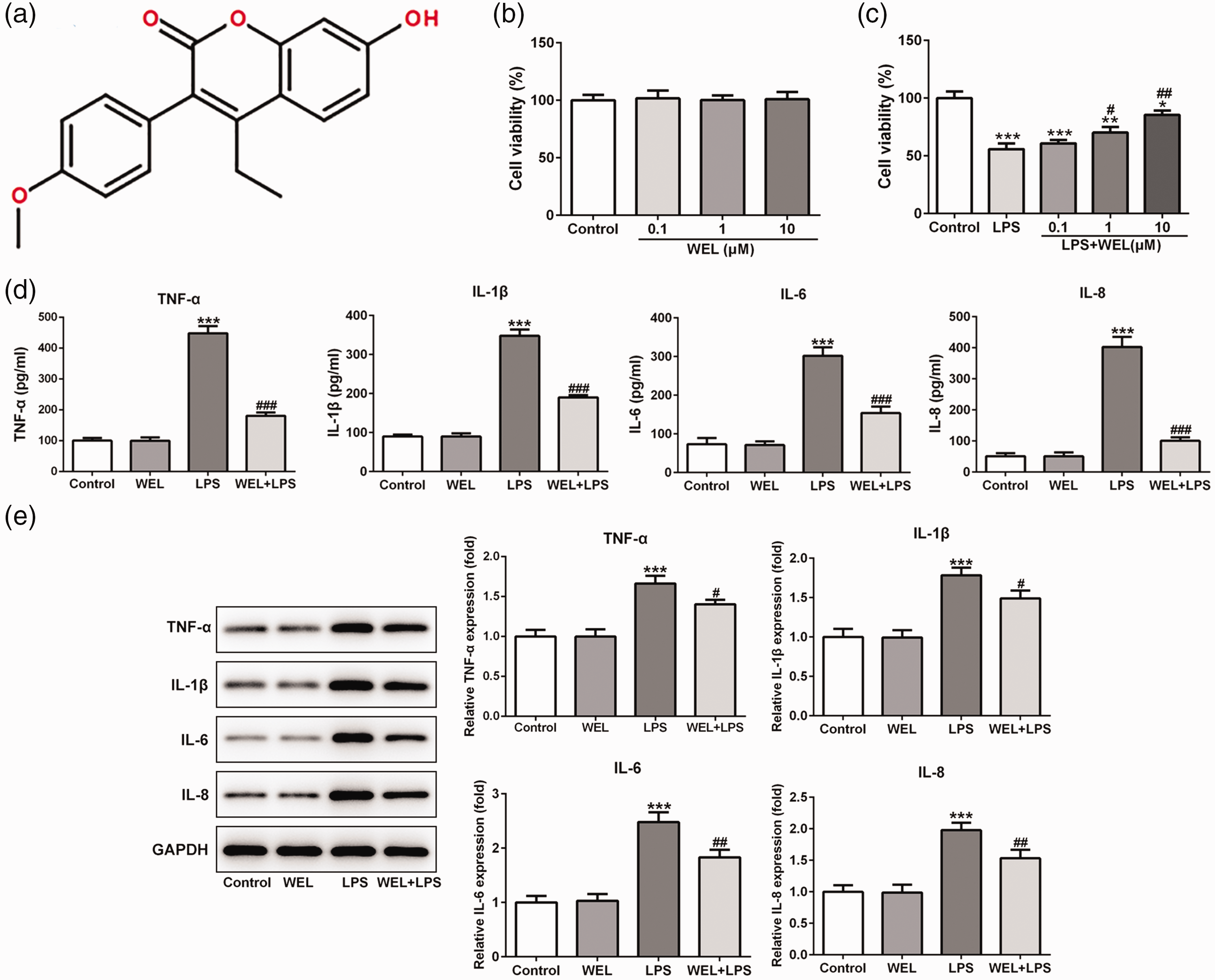
The structural formula of wedelolactone (WEL) (a). The viability of HK-2 cells treated with WEL (0.1, 1 and 10 μM) (b) and/or 1 μg/ml lipopolysaccharide (LPS) (c). The effects of 10 μM WEL in the presence or absence of 1 μg/ml LPS on the production of tumour necrosis factor (TNF)-α, interleukin (IL)-1β, IL-6 and IL-8 in HK-2 cells as examined by enzyme-linked immunosorbent assay (d) and Western blot analysis (e). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as the internal control. Data presented as mean ± SD; *P < 0.05, **P < 0.01 and ***P < 0.001 versus control group; #P < 0.05, ##P < 0.01 and ###P < 0.001 versus LPS group; one-way analysis of variance.
By using the STITCH website, 10 WEL was shown to combine with protein tyrosine phosphatase non-receptor type 2 (PTPN2) and regulate its expression. PTPN2, also known as T cell protein tyrosine phosphatase, is one of the 17 intracellular and non-receptor protein tyrosine phosphatases, which is ubiquitously expressed in intestinal and renal epithelium, fibroblasts and hepatocytes. 11 It has been reported that PTPN2 can negatively regulate proinflammatory signalling cascades, thus participating into various inflammatory disorders, including inflammatory bowel disease, rheumatoid arthritis and type 1 diabetes.12–14 The anti-inflammatory role of PTPN2 has been highlighted due to the fact that PTPN2-deficient mice died a few weeks after birth because of systemic inflammation characterized by chronic myocarditis, gastritis, nephritis and sialadenitis; as well having elevated serum interferon-γ levels. 15 In addition, PTPN2 improved renal injury and fibrosis by suppressing signal transducer and activator of transcription-induced inflammation in early diabetic nephropathy, suggesting that PTPN2 has anti-inflammatory effects. 16 PTPN2 could be induced by an inflammatory response and oxidative stress; and its deficiency decreased glioma cell growth through inhibiting cell colony formation and inducing cell apoptosis. 17 Although these results suggest the important role of PTPN2 in metabolic diseases and inflammation, it remains unknown whether PTPN2 can contribute to the progression of kidney injury due to sepsis.
As a consequence of its important anti-inflammatory and antiapoptotic roles, PTPN2 might represent an interesting target of WEL therapy in sepsis or other inflammatory disorders. This current study aimed to investigate the therapeutic effects of WEL and the underlying function of PTPN2 in LPS-induced renal injury in HK-2 cells.
Materials and methods
Cell culture and WEL or LPS treatment
The human renal proximal tubular epithelial cell line, HK-2, was obtained from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). The cells were then cultured in the F12/Dulbecco’s modified Eagle’s medium (Gibco BRL, Life Technologies Inc., Gaithersburg, MD, USA) supplemented with 10% fetal bovine serum (Sigma-Aldrich, St Louis, MO, USA), 100 U/ml penicillin and 0.1 mg/ml streptomycin (both from Invitrogen, Carlsbad, CA, USA) at 37°C with 5% CO2 and saturation humidity.
For WEL treatment (purity ≥ 95%; Jilin Xidian Pharmaceutical Co. Ltd., Panshi, China), HK-2 cells (1 × 105 cells/ml) were seeded into a 6-well plate and incubated for 48 h. Cells were exposed to cell culture medium alone (blank control), low dose of WEL (0.1 µM WEL), medium dose of WEL (1 µM WEL) and high dose of WEL (10 µM WEL) for 12 h at 37°C with 5% CO2 and saturation humidity.
For LPS induction, HK-2 cells (1 × 105 cells/ml) were plated into 6-well plates and cultured for 48 h at 37°C with 5% CO2 and saturation humidity. A solution of 1 µg/ml LPS (Sigma-Aldrich) or saline solution was added and the cells were incubated for 12 h at 37°C with 5% CO2 and saturation humidity to induce inflammatory injury. After that, HK-2 cells were harvested and prepared for quantitative reverse transcription–polymerase chain reaction (qRT–PCR) or Western blot analysis.
MTT assay
A total of 1 × 104 HK-2 cells were cultured in each well of a 96-well plate for 24 h at 37°C with 5% CO2 and saturation humidity. Cells were exposed to LPS or/and different concentrations of WEL (0.1, 1 and 10 µM) and then placed in an incubator for 12 h at 37°C with 5% CO2 and saturation humidity. After treatment, 10 μl of 5 mg/ml MTT (Sigma-Aldrich) was added to the wells and then incubated for 4 h at 37°C with 5% CO2 and saturation humidity. Next, 100 μl dimethyl sulfoxide (Sigma-Aldrich) was added to each well and maintained at room temperature to solubilize the formazan crystals. A microplate reader (SpectraMax® i3x; Molecular Devices, San Jose, CA, USA) was used to measure the absorbance at 570 nm.
Enzyme-linked immunosorbent assay
HK-2 cells were incubated as described above. Tumour necrosis factor (TNF)-α, interleukin (IL)-1β, IL-6 and IL-8 were detected in cell culture supernatants using enzyme-linked immunosorbent assay (ELISA) kits (Shanghai Enzyme-linked Biotechnology Co. Ltd., Shanghai, China) according to the manufacturer's instructions. A microplate reader (SpectraMax® i3x; Molecular Devices) was used to read the absorbance at 450 nm and the data were analysed. All experiments were repeated three times. The minimum detectable concentrations were 31.25 pg/ml for TNF-α, 7.8125 pg/ml for IL-1β, 18.75 pg/ml for IL-6 and 62.5 pg/ml for IL-8. Intra- and interassay coefficients of variation for all ELISAs were <10% and <10%, respectively.
Flow cytometry analysis
Flow cytometry assay was used to determine the levels of cell apoptosis using an annexin V-fluorescein isothiocyanate (FITC)/ propidium iodide (PI) apoptosis detection kit (Bio Vision Inc., Milpitas, CA, USA). After HK-2 cells were treated as described above, they were harvested and washed with ice-cold 0.01 M phosphate-buffered saline (pH 7.2). Cells were suspended in binding buffer and then incubated with annexin V-FITC and PI at room temperature for 15 min in the dark. The apoptotic rate was measured using a flow cytometer (Attune NxT Flow Cytometer; Thermo Fisher Scientific Inc., Rockford, IL, USA) and analysed with BD CellQuest™ Pro version S7 (BD Biosciences, San Jose, CA, USA).
Transfection experiments
For silencing of PTPN2, two different commercially available plasmids constructed with the short hairpin RNA (shRNA) of the PTPN2 sequence (shRNA-PTPN2-1 and shRNA-PTPN2-2; 500 ng/μl; Shanghai GenePharma, Shanghai, China) were transfected into HK-2 cells using Lipofectamine® 3000 (Invitrogen) according to the manufacturer’s instructions. Scrambled shRNA (shRNA-NC; 500 ng/μl) was used as a negative transfection control. The transfection efficiency was confirmed by qRT–PCR as described below using the following PTPN2 shRNA primer sequences: sense 5′-
Western blot analysis
HK-2 cells (1 × 105 cells/ml) were plated into 6-well plates, and after treatment as described above, total protein from all of the HK-2 cells was harvested and lysed in RIPA lysis buffer (Beyotime, Jiangsu, China). The concentration of protein was detected using a BCA Protein Quantification kit (Beyotime). Proteins (30 µg/lane) were separated using 10% sodium dodecyl sulphate–polyacrylamide gel electrophoresis and transferred onto polyvinylidene fluoride membranes (Millipore, Billerica, MA, USA). After blocking with 5% skimmed milk at room temperature for 2 h, membranes were incubated with primary antibodies at 4°C overnight. Primary antibodies to the following proteins were used: PTPN2 (1:1000 dilution; Abcam, Cambridge, MA, UK); TNF-α (1:1000 dilution; Abcam); IL-1β (1:1000 dilution; Abcam), IL-6 (1:1000 dilution; Abcam), IL-8 (1:1000 dilution; Abcam), Bcl-2 (1:1000 dilution; Abcam), Bax (1:1000 dilution; Abcam), cleaved-caspase3 (1:500 dilution; Abcam)/9 (1:500 dilution; Abcam), caspase3 (1: 5000 dilution; Abcam)/9 (1:1000 dilution; Abcam) and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (1:1000 dilution; Cell Signaling Technology®, Danvers, MA, USA). The membranes were then washed with Tris-buffered saline-Tween 20 (TBST) three times and incubated with horseradish peroxidase-conjugated goat antirabbit secondary antibody (1:4000; Cell Signaling Technology®) for 1 h at room temperature. The membranes were washed with TBST three times, and the immunoreactive signals were detected using an enhanced chemiluminescence kit (Thermo Fisher Scientific Inc.). Quantitative densitometric analyses of the immunoblotted images were performed using Image J software (version 5.0; National Institutes of Health, USA). The experiment was repeated three times.
Quantitative reverse transcription–polymerase chain reaction
HK-2 cells (1 × 105 cells/ml) were plated into 6-well plates, and after treatment as described above, total RNA was extracted from all of the HK-2 cells using TRIzol® reagent (Invitrogen) according to the manufacturer’s instructions. NanoDrop™ 2000 (Thermo Fisher Scientific Inc.) was used to detect the quality and quantity of the extracted RNA. Complementary DNA (cDNA) was reverse-transcribed from quantified RNA using a PrimeScript TM RT reagent kit (Takara, Dalian, China) before qRT–PCR was used for gene amplification using the SYBR Premix Ex TaqTM GC (Takara) protocol according to their manufacturer’s instructions on a 7900 real-time PCR system (Applied Biosystems, Foster City, CA, USA). The cycling programme involved preliminary denaturation at 95°C for 10 min, followed by 40 cycles of denaturation at 94°C for 2 min, annealing at 60°C for 50 s, and elongation at 60°C for 60 s, followed by a final elongation step at 60°C for 5 min. Relative gene expression was measured using the 2−ΔΔCt method. The primers used are listed in Table 1. The ratio for the mRNA of interest was normalized to GAPDH.
Primers used for quantitative reverse transcription–polymerase chain reaction.

GAPDH, glyceraldehyde 3-phosphate dehydrogenase; PTPN2, protein tyrosine phosphatase non-receptor type 2.
Statistical analyses
All statistical analyses were performed using GraphPad Prism version 6.0 (GraphPad Software, San Diego, CA, USA). Data are presented as mean ± SD. One-way analysis of variance was used to evaluate significant differences between groups. A P-value < 0.05 was considered statistically significant.
Results
HK-2 cells were treated with different concentrations of WEL (0.1, 1 and 10 µM) in the absence or presence of 1 µg/ml LPS for 12 h. Then cell viability was assessed using the MTT assay to verify the role of WEL in the cell viability of HK-2 cells. WEL treatment at all three doses alone caused no change in the cell viability compared with the control (Figure 1b). Treatment with LPS significantly reduced cell viability compared with the control (P < 0.001) and this negative effect on cell viability was abrogated when LPS was used in combination with WEL in a dose-dependent manner (Figure 1c).
Following the experiments on the effect of WEL and LPS treatment on cell viability, the concentrations of four inflammatory cytokines in HK-2 cells, TNF-α, IL-1β, IL-6 and IL-8, were measured using ELISAs and Western blot assays. As presented in Figure 1d, 10 µM WEL treatment alone had no effect on the levels of these four inflammatory cytokines, whereas 1 µg/ml LPS treatment significantly elevated their levels in HK-2 cells compared with the control (P < 0.001 for all comparisons). The addition of 10 µM WEL to 1 µg/ml LPS significantly reduced the LPS-induced upregulation of TNF-α, IL-1β, IL-6 and IL-8 concentrations in the supernatant of HK-2 cells compared with LPS alone (P < 0.001 for all comparisons) (Figure 1d). Similar results were observed for the Western blot analysis in terms of the levels of the four inflammatory cytokines in HK-2 cells following treatment with 10 µM WEL and/or LPS (Figure 1e).
Cell apoptosis in HK-2 cells was detected using flow cytometry following 10 µM WEL treatment in the absence or presence of 1 µg/ml LPS (Figure 2a). WEL (10 µM) administration alone had no effect on cell apoptosis in HK-2 cells, but treatment with 1 µg/ml LPS resulted in a significant increase in the rate of cell apoptosis compared with the control group (P < 0.001). WEL (10 µM) significantly suppressed the LPS-induced cell apoptosis compared with LPS alone (P < 0.001). The levels of apoptosis-related factors in HK-2 cells were detected by Western blot analysis (Figure 2b). Treatment with 1 µg/ml LPS resulted in increases in Bax, cleaved-caspase3 and cleaved-caspase9 levels, but a decrease in the Bcl-2 level. The addition of 10 µM WEL to 1 µg/ml LPS abrogated the LPS-induced abnormal levels of these apoptosis-related factors in HK-2 cells. There were no significant changes in the levels of caspase3 and caspase9 in the four groups.

The effect of wedelolactone (WEL) on HK‑2 cell apoptosis. A flow cytometric assay was performed to assess the effect of 10 μM WEL and/or 1 μg/ml lipopolysaccharide (LPS) on apoptosis in HK‑2 cells and the cell apoptosis rate was quantified (A). Western blot analysis of the levels of Bcl-2, Bax and cleaved-caspase3/9 and caspase3/9 in HK-2 cells treated with 10 μM WEL and/or 1 μg/ml LPS. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as the internal control. Data are presented as mean ± SD; ***P < 0.001 versus control group; #P < 0.05, ##P < 0.01 and ###P < 0.001 versus LPS group; one-way analysis of variance.
Following treatment with different concentrations of WEL, the levels of PTPN2 were increased in a dose-dependent manner in HK-2 cells (Figure 3a). Treatment with 1 µg/ml LPS significantly reduced PTPN2 levels in HK-2 cells compared with the control group (P < 0.01). The addition of 10 µM WEL to 1 µg/ml LPS significantly increased the levels of PTPN2 in HK-2 cells compared with LPS treatment alone (P < 0.001) (Figure 3b).

The levels of protein tyrosine phosphatase non-receptor type 2 (PTPN2) were determined by Western blot analysis in HK-2 cells treated with wedelolactone (WEL) at different concentrations (0.1, 1, 10 μM) (a). The HK-2 cells were then stimulated with 1 μg/ml lipopolysaccharide (LPS) in addition to 10 μM WEL and the levels of PTPN2 were determined by Western blot analysis (b). Quantitative reverse transcription–polymerase chain reaction was used to measure the levels of PTPN2 mRNA in HK-2 cells after transfection with short hairpin RNA (shRNA)-PTPN2-1, shRNA-PTPN2-2 or the scrambled shRNA-NC negative control (c). HK-2 cells were transfected with shRNA-NC or shRNA-PTPN2-2 and then treated with 1 μg/ml LPS and 10 μM WEL. The production of tumour necrosis factor (TNF)-α, interleukin (IL)-1β, IL-6 and IL-8 in HK-2 cells were examined by enzyme-linked immunosorbent assays (d). Western blot analysis for TNF-α, IL-1β, IL-6 and IL-8 levels were also undertaken (e). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as the internal control. Data are presented as mean ± SD; **P < 0.01; ***P < 0.001 versus control group; #P < 0.05, ##P < 0.01 and ###P < 0.001 versus LPS group; &P < 0.05, &&P < 0.01 and &&&P < 0.001 versus shRNA-NC; one-way analysis of variance.
After the transfection of HK-2 cells with shRNA-PTPN2-1 and shRNA-PTPN2-2 plasmids, the vector encoding shRNA-PTPN2-2 was more efficient in knocking down PTPN2 expression compared with shRNA-PTPN2-1 according to qRT–PCR analysis (Figure 3c). HK-2 cells were transfected with shRNA-PTPN2-2 and the production of TNF-α, IL-1β, IL-6 and IL-8 were measured by ELISAs and Western blot analysis. The levels of these four inflammatory cytokines that were significantly suppressed when 10 µM WEL was added to 1 µg/ml LPS (P < 0.001 for all comparisons with LPS alone) were subsequently significantly elevated following transfection with shRNA-PTPN2 when compared with the sh-NC control group (P < 0.05 for all comparisons) (Figure 3d). Similar results were observed for the Western blot analysis in terms of the levels of the four inflammatory cytokines in HK-2 cells transfected with shRNA-PTPN2-2 following treatment with 10 µM WEL and 1 µg/ml LPS (Figure 3e).
After HK-2 cells were transfected with shRNA-PTPN2, HK-2 cell apoptosis was measured using flow cytometry and Western blotting following 1 µg/ml LPS stimulation and 10 µM WEL treatment. After LPS stimulation of HK-2 cells for 48 h, flow cytometric analysis showed that the rate of cell apoptosis after transfection with shRNA-PTPN2 was significantly increased compared with the sh-NC group after 10 µM WEL treatment (P < 0.001) (Figure 4a). The result suggested that downregulation of PTPN2 levels could promote the cell apoptosis that was suppressed by WEL. Subsequently, the data showed that knockdown of PTPN2 could result in the downregulation of Bcl-2 levels (Figure 4b). Meanwhile, the protein levels of Bax, cleaved-caspase3 and cleaved-caspase9 showed the opposite effect (Figure 4b). These results suggest that cell apoptosis was increased in HK-2 cells when the levels of PTPN2 were decreased.
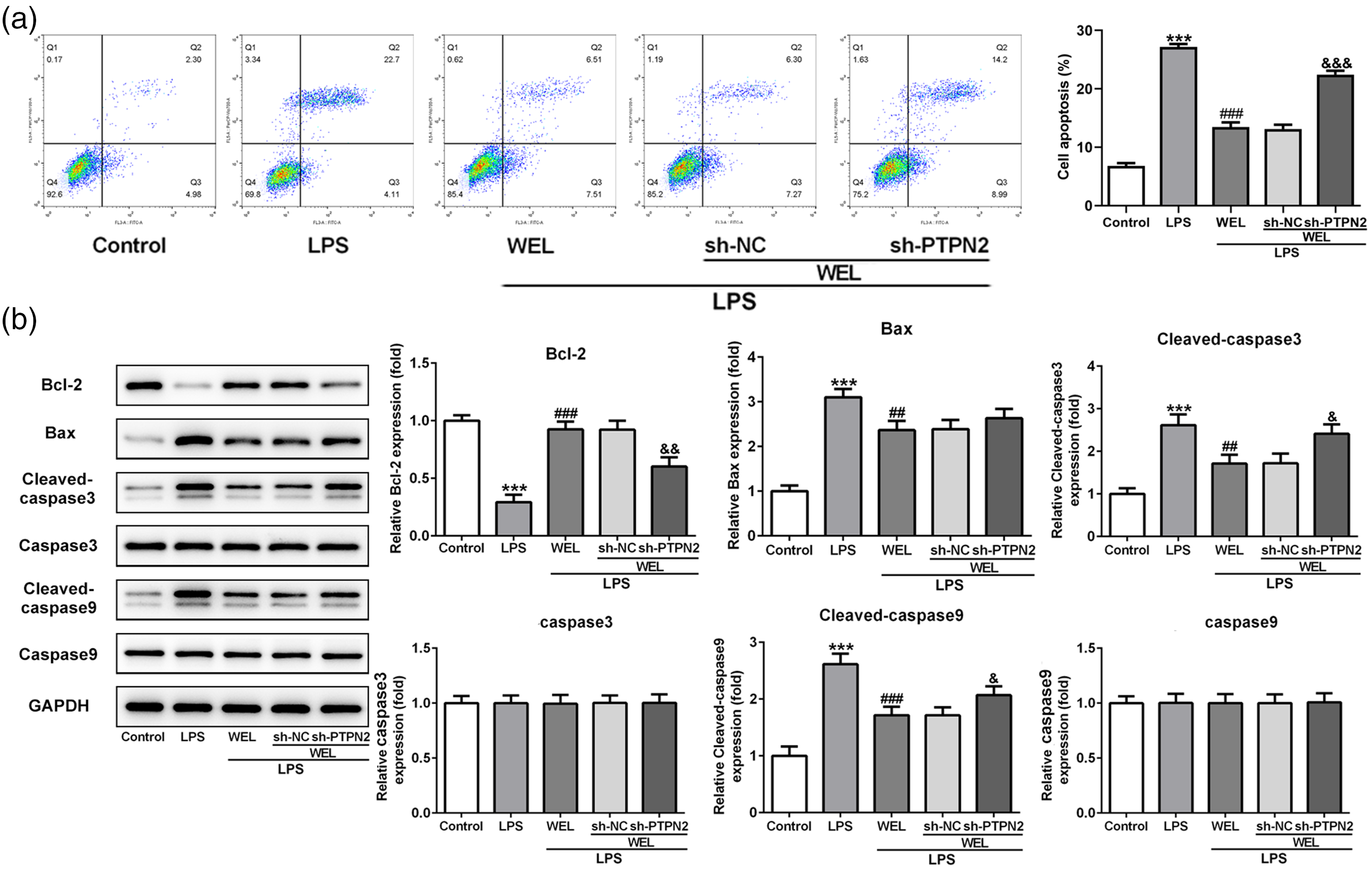
The effects of wedelolactone (WEL) inhibited lipopolysaccharide (LPS)-induced HK-2 cell apoptosis by regulating protein tyrosine phosphatase non-receptor type 2 (PTPN2) levels. HK-2 cells were treated with 10 μM WEL in the absence or presence of 1 μg/ml LPS after transfection with short hairpin RNA (shRNA)-PTPN2-2 or the scrambled shRNA-NC negative control. The rate of apoptosis was determined using flow cytometry (A) and the protein levels of Bax, Bcl-2, p53, cleaved-caspase3/9 and caspase3/9 were measured using Western blot analysis (B). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as the internal control. Data are presented as mean ± SD; ***P < 0.001 versus control group; ##P < 0.01 and ###P < 0.001 versus LPS group; &P < 0.05, &&P < 0.01 and &&&P < 0.001 versus shRNA-NC; one-way analysis of variance.
To further clarify how WEL or PTPN2 exerted their effects, the proteins associated with the p38 mitogen-activated protein kinase (MAPK)/NF-κB signalling pathway were measured using Western blot analysis. As shown in Figure 5, 1 µg/ml LPS stimulation significantly increased the levels of p-NF-κB p65 and p-p38 MAPK in HK-2 cells compared with the control group (P < 0.001 for both comparisons). Treatment with 10 µM WEL significantly reduced the levels of p-NF-κB p65 and p-p38 MAPK compared with LPS alone (P < 0.001 for both comparisons). The inhibitory effect of WEL was partly abolished by PTPN2 knockdown with shRNA-PTPN2. The protein levels of NF-κB p65 and p38 MAPK were not changed by LPS or WEL treatment. These results suggest that p38 MAPK/NF-κB signalling was activated in LPS-induced HK-2 cells; and WEL and PTPN2 might exert effects by regulating the p38 MAPK/NF-κB signalling pathway.

HK-2 cells were treated with 10 μM wedelolactone (WEL) in the absence or presence of 1 μg/ml lipopolysaccharide (LPS) after transfection with short hairpin RNA (shRNA)-PTPN2-2 or the scrambled shRNA-NC negative control. Western blot analysis was undertaken to detect the protein levels of p-nuclear factor-kappaB (NF-κB) p65, NF-κB p65, p-p38 mitogen-activated protein kinase (MAPK) and p38 MAPK. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as the internal control. Data are presented as mean ± SD; ***P < 0.001 versus control group; ###P < 0.001 versus LPS group. &P < 0.05 versus shRNA-NC; one-way analysis of variance.
Discussion
Sepsis is a complex disease associated with high levels of morbidity and mortality worldwide. 18 The current treatment of renal injury caused by sepsis can lead to severe consequences. 18 Therefore, there is an urgent need to develop new drug interventions to treat or prevent renal injury. The present study demonstrated that pretreatment of LPS-stimulated HK-2 cells with WEL reduced the production of inflammatory cytokines and the rate of HK-2 cell apoptosis.
The pathogenesis of LPS-induced renal injury is closely associated with an abnormal inflammatory response and LPS induces renal injury via the induction of tubular epithelial cell apoptosis.19,20 Therefore, it is important to explore therapeutic strategies to inhibit LPS-induced inflammation and apoptosis during treatment for renal injury. In the present study, a model of renal injury was established using LPS-stimulated HK-2 cells. Treatment with LPS increased the production of inflammatory cytokines by HK-2 cells and resulted in HK-2 cell damage, loss of viability and increased apoptosis. Data from the current study demonstrated that WEL reduced the loss of viability caused by LPS; and reduced the LPS-induced production of inflammatory cytokines and the rate of apoptosis. These findings were in part consistent with previous findings that demonstrated that WEL negatively modulated the activation of p38 MAPK and ERK by LPS-induced inflammation in RAW264.7 cells. 21 WEL was found to prevent osteolytic bone metastasis in patients with bone metastasis of breast cancer origin;22,23 and to induce caspase-dependent apoptosis in prostate cancer cells. 24
The most recognized beneficial effects of WEL include anti-inflammatory, anti-hepatotoxic and trypsin inhibitory effects. 9 WEL inhibits the IκB kinase complex, resulting in suppression of LPS-induced caspase-11 expression. 25 WEL alleviates the inflammation and oxidative stress of doxorubicin-induced MPC-5 cells. 8 WEL induces inhibition of H3K27 methylation via EZH2 modulation and decreases cell proliferation in mantle cell lymphoma. 26 Similar to these previous studies, the current study found anti-apoptotic and anti-inflammatory effects of WEL in LPS-stimulated HK-2 cells. Previous research has shown that anti-inflammatory and anti-apoptotic therapies are beneficial for the treatment of sepsis-induced renal injury. For example, a previous study reported that aquaporin 1 exerted a protective role in sepsis-induced acute kidney injury because it conferred a survival advantage on LPS-induced HK-2 cells by inhibiting inflammation and apoptosis. 27 Another study demonstrated that flavonoid fisetin alleviated kidney inflammation and apoptosis to protect against LPS-induced septic AKI mice. 28 In the current study, the viability of LPS-stimulated HK-2 cells was increased by treatment with WEL, which was accompanied by reduced apoptosis and decreased production of inflammatory cytokines (TNF-α, IL-1β, IL-6 and IL-8). Furthermore, an obvious increase in the PTPN2 levels of HK-2 cells was observed after WEL treatment. PTPN2 levels were reduced following LPS stimulation, but WEL pretreatment restored the PTPN2 deficiency.
Emerging research suggests that the loss of PTPN2 enhances interferon (IFN)-γ, IL-6 and IL-1β secretion.13,14 PTPN2 regulates inflammasome activation and controls the onset of intestinal inflammation and colon cancer. 12 Dysfunction of PTPN2 in T cells drives the secretion of IFN-γ and IL-17, which promotes IL-18 and IL-1β secretion, respectively. 12 PTPN2 suppressed renal injury and fibrosis via downregulation of proinflammatory cytokines. 16 Decreased PTPN2 expression exacerbated IL-1β + IFN-γ-induced beta-cell apoptosis and turned IFN-γ alone into a proapoptotic signal. 29 WEL targeted EZH2-mediated histone H3K27 methylation in mantle cell lymphoma. 26 In the present study, loss of PTPN2 aggravated HK-2 cell damage via elevating the production of inflammatory cytokines and promoting apoptosis in the presence of LPS and WEL. Hence, elevating the levels of PTPN2 may be an effective therapeutic method for the treatment of LPS-induced renal injury. Nevertheless, to further confirm the results in this study, more experimental research is still needed.
The transcription factor NF-κB, widely expressed in cells and tissues, responds quickly to various inflammatory stimuli. 30 In the current study, NF-κB was activated in HK-2 cells in response to an inflammatory microenvironment. WEL has been reported to block NF-κB activity to exert a protective role in various diseases, such as osteoporosis and liver injury.31,32 Moreover, WEL demonstrated a protective effect in renal injury by alleviating inflammation and oxidative stress in podocytes via downregulating the NF-κB pathway. 8 This current study also found that the phosphorylation of NF-κB stimulated by LPS was inhibited by WEL treatment, further demonstrating the inhibitory effect of WEL on NF-κB signalling. In addition to NF-κB signalling, LPS-induced p38 MAPK signalling was also involved in the inflammatory microenvironment of kidney injury. 33 The activation of the MAPK and NF-κB signalling pathways induced by LPS was reduced following PTPN2 knockdown and PTPN2 acted as a positive regulator of LPS-induced inflammation. 34 In contrast, loss of PTPN2 was reported to promote the phosphorylation of p38 MAPK and contribute to NF-κB overexpression.14,35 The regulatory mechanism of PTPN2 in inflammatory events are controversial. In this current study, PTPN2 knockdown increased the levels of p-NF-κB and p-p38 MAPK, indicating that PTPN2 knockdown promoted the activity of the p38 MAPK/NF-κB signalling pathway. Considering that WEL could work in conjunction with PTPN2, the protective role of WEL in LPS-induced HK-2 cells might partly depend on PTPN2 expression via the p38 MAPK/NF-κB signalling pathway.
This current study had several limitations. First, the role and regulatory mechanism of PTPN2 remain controversial in different diseases. More in-depth investigations to determine the details of the mechanism of action underlying the role of PTPN2 in renal injury are required. Secondly, in vivo experiments are required in order to verify the present in vitro findings.
In conclusion, this current study demonstrated that WEL attenuated LPS-induced inflammatory cytokine secretion and cell apoptosis in HK-2 cells possibly via elevating PTPN2 levels. PTPN2 ameliorated renal cell damage and became nephroprotective in sepsis-induced renal injury by regulating p38 MAPK/NF-κB signalling. This study provides information about the therapeutic potential of WEL in renal injury and indicated PTPN2 may be a potential target for preventing the inflammatory response and improving the treatment outcomes of sepsis.