
Abstract
Paroxysmal sympathetic hyperactivity (PSH) is a clinical syndrome of episodic sympathetic hyperactivities following severe acquired brain injury. It is characterized by paroxysmal hyperthermia, tachycardia, hypertension, tachypnea, excessive diaphoresis, and specific posturing. Although the persistence of PSH increases the risk of several adverse events and worsens the prognosis, pharmacological treatments for PSH have not yet been clearly established. We report the valuable case of a 60-year-old man who developed PSH following hypoxic encephalopathy, which was effectively treated with a combination therapy of gabapentin and guanfacine. The present case suggests that combination therapy with gabapentin and guanfacine may be a therapeutic option for PSH.
Keywords
Introduction
Paroxysmal sympathetic hyperactivity (PSH) is a distinct syndrome of episodic sympathetic hyperactivities following severe acquired brain injury. It is characterized by paroxysmal hyperthermia, tachycardia, hypertension, tachypnea, excessive diaphoresis, and specific posturing (including dystonia, stiffness, or spasticity).1–4
Although there is a large potential for a worsened prognosis as a result of these symptoms, a consensus on the definition of this syndrome was clarified only recently, 4 and its pathophysiological bases and treatments are not yet clearly established.1–3 Nevertheless, several studies5,6 have reported the great efficacy of several drugs for PSH in clinical practice, including opioids, nonselective β-blockers, α2 agonists, bromocriptine, baclofen, gabapentin (GBP), and long-acting benzodiazepines.1–3 Both our experience with PSH and the relevant literature indicate that multidrug therapies are generally required and recommended. We consider that a combination therapy using GBP and guanfacine (GXR) is effective and safe from the points of view of the action mechanisms and tolerability of these drugs.7–13
We present a case of PSH that developed following cerebral hypoxia; this combination therapy was effective as a treatment.
Case report
A 60-year-old man, who was an ambulatory patient of a cardiologist in our hospital with a medical history of coronary artery disease and dyslipidemia, suddenly went into cardiac arrest in his house, witnessed by his family. He was taken in an ambulance to a hospital and given cardiopulmonary resuscitation. His initial electrocardiogram waveform indicated ventricular fibrillation, so defibrillation was performed and 1 mg of adrenaline was administered intravenously. His heartbeat recovered, and in the presence of an ST-segment elevation, immediate coronary angiography and percutaneous coronary intervention were performed for severe stenosis of the left main coronary trunk. His heartbeat recovered but his consciousness did not, and he had loss of circulation for approximately 26 minutes. He was attached to an artificial respirator and underwent therapeutic hypothermia under sedation with midazolam on days 1 and 2 of hospitalization. Sedation was stopped on day 5, but his depressed level of consciousness continued. On day 10, a neurologist diagnosed him with cerebral hypoxia based on his clinical course, physical and neurological examinations, brain magnetic resonance imaging, and an electroencephalogram (EEG). On day 20, he was subjected to a tracheostomy and continuous positive airway pressure. He was fed by a nasogastric tube. From this point onward, his general condition remained almost unchanged.
On day 37 of hospitalization, the patient was transferred to our hospital. His consciousness was assessed using the Glasgow Coma Scale as follows: eye response (E): 4, verbal response (V): T (tube), motor response (M): 1. He had mechanical ventilation and a nasogastric tube. His eyes were open, but he was unable to make eye contact, track, or respond to our requests. We observed that he had aimless movements and limb hypertonus. His vital signs were within the normal limits and there were no particular abnormalities in his laboratory data or a head computed tomography (CT) scan. However, an EEG showed abnormal findings, with poor organization of background activity, sporadic theta waves, and intermittent delta waves over the bilateral frontal regions, but no paroxysmal discharges. Following his transfer to our hospital, the patient’s consciousness and motor dysfunction gradually improved through nursing care and rehabilitation.
On approximately day 50, the patient developed a sudden high fever that was accompanied by tachycardia, hypertension, tachypnea, excessive sweating, and specific posturing, such as hypertonia and left inferior limb torsion to the right side. These episodes occurred three to four times per day. Based on his clinical course, physical and neurological examinations, laboratory data, and a chest X-ray, we excluded any presumed causes of these features, such as infections, malignant hyperthermia, malignant neuroleptic syndrome, serotonin syndrome, withdrawal syndrome, and endocrine disorders. However, a brain CT and an EEG were not able to be performed because of his restlessness. We then suspected PSH and evaluated him using the PSH-Assessment Measure (PSH-AM), 4 which resulted in an assessment of severe PSH from his Clinical Feature Scale (CFS) score of 14. He was also diagnosed with probable PSH from his PSH-AM score of 25 (the result of a CFS score of 14 and a Diagnostic Likelihood Tool [DLT] score of 11).
On day 58, we diagnosed the patient with PSH and obtained informed consent from him and his family to treat him with a combination therapy of GBP and GXR. After beginning the therapy, the drugs were gradually increased to GBP 2.4 mg/day and GXR 5 mg/day. Under this treatment regimen, his severe symptoms of PSH were gradually improved and were ameliorated by around day 90. During this drug therapy, the patient continued with his rehabilitation, and he recovered his articulation, walking ability, and oral ingestion, although not entirely. There were no adverse events caused by these drugs. However, even though the patient’s specific posturing was gradually reduced from the viewpoint of frequency and severity, mild paroxysmal posturing has persisted up to the current time.
Discussion
PSH has been described as occurring after severe brain injury, and is associated with paroxysmal hyperthermia, tachycardia, hypertension, tachypnea, excessive diaphoresis, and specific posturing (including dystonia, hypertonia, or spasticity) during episodes.1–4 A number of conditions predispose an individual to developing PSH, the most common of which are traumatic brain injury (80%), hypoxic encephalopathy developed after cardiac arrest (10%), and cerebrovascular accident (5.5%). 14 The persistence of PSH increases the risk of adverse effects, such as cardiac overload, arrhythmias, dehydration, muscle loss, and contractures, which further contribute to patient morbidity and worsen the prognosis.1,14 Thus, it is very important to diagnose and appropriately treat PSH.
There are no specific diagnosis criteria for PSH. In 2014, the Consensus Working Group3,4 proposed a conceptual definition, nomenclature, and diagnostic criteria for PSH. They recommended the use of the PSH-AM, which consists of the CFS, the DLT, and the Interpretation of Scores (Table 1). As stated in the DLT, other causes can have similar clinical features. The exclusion of other diagnoses, such as infections, malignant hyperthermia, malignant neuroleptic syndrome, serotonin syndrome, withdrawal syndrome, and endocrine disorders such as thyroid storm, is therefore of primary importance.
The PSH-AM. Adapted from Baguley et al., 4 by permission of Mary Ann Liebert, Inc.

CFS, Clinical Feature Scale; DLT, Diagnostic Likelihood Tool; PSH-AM, Paroxysmal Sympathetic Hyperactivity-Assessment Measure.
In the present case, we excluded a range of presumed causes and used the PSH-AM, which resulted in a CFS score of 14, DLT score of 11, and PSH-AM score of 25. The PSH severity was severe, and the patient was diagnosed with probable PSH.
Several theories exist regarding the pathophysiology of PSH. Of these, the disconnection theory and the excitatory/inhibitory ratio (EIR) model are the most accepted.2,3 The disconnection theory proposes that sympathetic tone is normally modulated by the connections between cortical inhibitory centers (dorsolateral prefrontal cortex [dlPFC], amygdala, and basal ganglia) and sympathetic control centers (brainstem, hypothalamus, and diencephalon), which are responsible for supraspinal and spinal excitement. After severe brain injury, a disconnection occurs between the cortical inhibitory centers and the sympathetic centers. Therefore, the cortical inhibitory centers do not work well, and the sympathetic control centers become dysfunctional or overactive without the top-down regulation. Finally, sympathetic tone becomes excessive (Figure 1). In contrast, the EIR model proposes a two-stage process, with the loss of descending inhibition allowing the development of excitatory spinal circuits (Figure 2). At the level of the brain, the brainstem centers modulate inhibitory drive to the spinal reflex area. At the spinal level, the spinal centers provide upward feedback about sensory and perception stimuli, and about motor and sympathetic efferent output. The balance between inhibitory and excitatory interneuron activity is thereby maintained. In the EIR model of PSH, impaired descending inhibition leads to spinal circuit excitation, and non-noxious stimuli increase motor and sympathetic output (spinally) and potentially become perceived as noxious (centrally).

Disconnection theory. Adapted from Zheng et al., 2 by permission of Frontiers Media S.A.
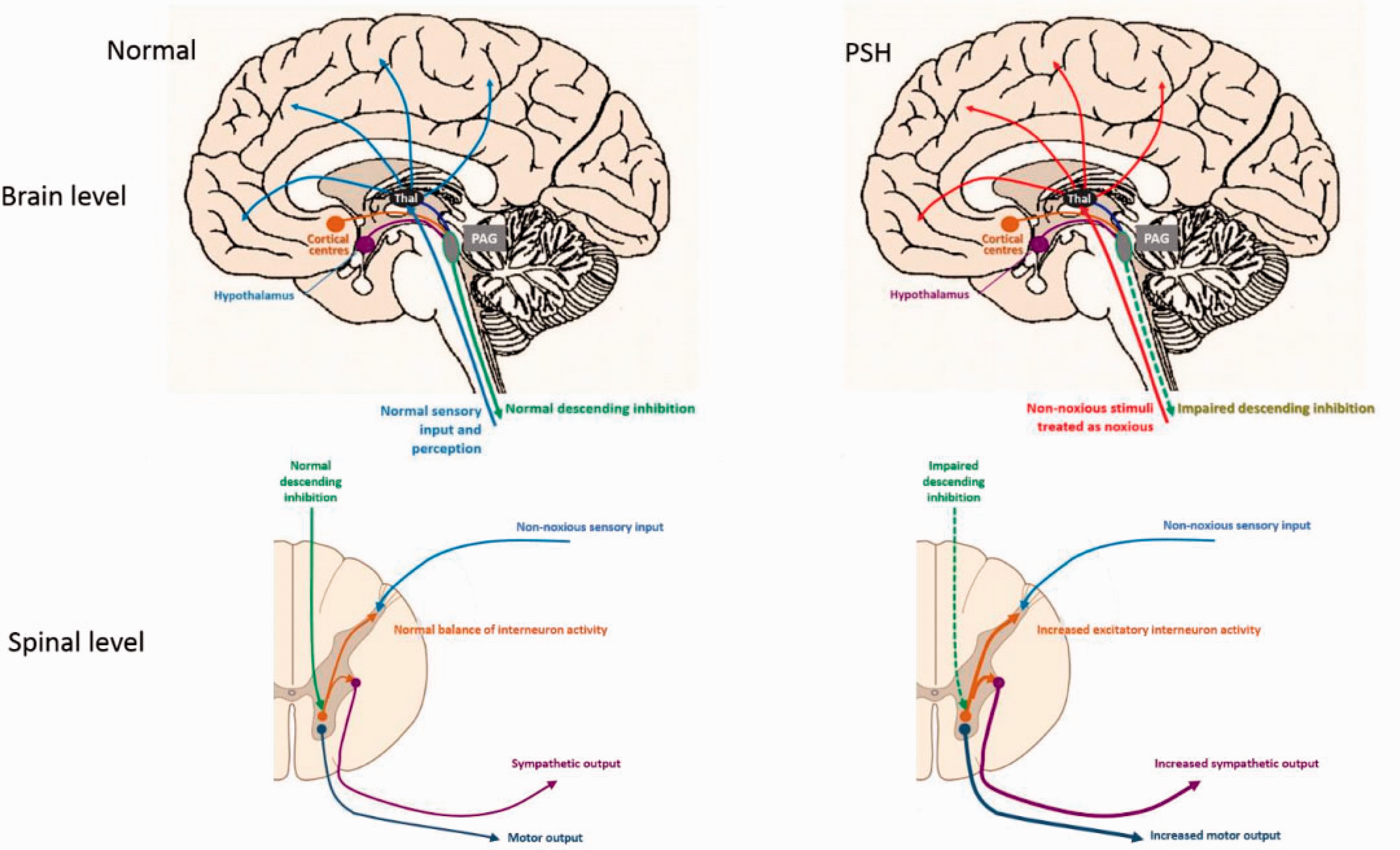
Excitatory–inhibitory ratio model for PSH pathogenesis. Adapted from Meyfroidt et al., 3 by permission of Elsevier Inc.
Although pharmacological treatments for PSH are not yet clearly established, several studies5,6 have reported the great efficacy of a range of drugs for PSH in clinical practice, including opioids, nonselective β-blockers (propranolol), α2 agonists (clonidine and dexmedetomidine), bromocriptine, baclofen, GBP, and long-acting benzodiazepines.1–3 In most of these studies, multidrug therapies are generally recommended. There are two main therapeutic strategies for PSH: the selection of drugs from the perspective of the aforementioned pathophysiology, or the selection of drugs according to symptoms. We considered that a combination therapy using GBP and GXR would be effective from the points of view of both the pathophysiology of PSH and their action mechanisms. Furthermore, we hypothesized that the therapy would be safe because of the tolerability of these two drugs.7–13
GBP is a γ-aminobutyric acid (GABA) agonist that acts on α2δ presynaptic voltage-gated Ca 2 + channels in the brain and spinal cord and recovers the inhibitory control of descending and afferent feedback at both the brain and spinal cord levels. As for GXR, the dlPFC plays an essential role in the interpretation of stimuli and behavioral responses,7–10 and GXR is a selective α2A agonist that enhances its function of top-down regulation of the sympathetic nervous system. It also reduces excessive locus coeruleus neuronal activity, resulting in a slower heart rate, decreased blood pressure, and the regulation of arousal. Although clonidine has been used a general drug for PSH, we propose that GXR is likely to be a better treatment for PSH than clonidine in terms of its action mechanisms and tolerability.5,7,8,11–13 For example, GXR is a selective α2A agonist, while clonidine is a general α2 agonist. To the best of our knowledge, the present report is the first to demonstrate the efficacy of combination therapy using GBP and GXR to treat PSH following cerebral hypoxia. It is especially novel because we demonstrated that GXR works well against PSH. GXR enhances noradrenergic activity in the dlPFC, resulting in augmented working memory, attention, and impulse control.7,8,15 The dlPFC then provides top-down regulation of the sympathetic nervous system through its extensive connections with the amygdala, basal ganglia, and brainstem, including the catecholamine neurons. Moderate levels of catecholamine release engage high-affinity α2A receptors, which strengthen dlPFC function but weaken that of the amygdala and basal ganglia. In contrast, under conditions of uncontrollable stress, there are high levels of catecholamine release that engage lower affinity α1 and β receptors, which enhance the amygdala and basal ganglia function but weaken that of the dlPFC9,10 (Figure 3).

Function of the prefrontal cortex, amygdala, basal ganglia, and brainstem under non-stressed and uncontrollable stress conditions. Adapted from Arnsten et al., 9 by permission of Elsevier Inc.
In the present case, we hypothesize that GBP recovered the inhibitory control, while GXR enhanced the dlPFC function and reduced excessive locus coeruleus neuronal activity, thus resulting in the amelioration of the severe symptoms of PSH.
Footnotes
Ethics statement
This study complied with the CARE guidelines for case reports, and was approved by the Ethics Committee of Itami City Hospital (reference number 1101). Written informed consent was received from the patient and his family.
Acknowledgements
We would like to thank all of the staff at the 2-East ward and the members of the psychiatric liaison team in our hospital. We also give special thanks to the patient and his family.
Declaration of conflicting interest
The authors declare that there is no conflict of interest.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.