
Abstract
We report a family with riboflavin-reactive multiple acyl-CoA dehydrogenase deficiency (RR-MADD) partially caused by a novel mutation in the electron transfer flavoprotein dehydrogenase gene (ETFDH). The RR-MADD family was identified by physical examination, electromyography, and muscle biopsy of the proband. Laboratory examination and electromyography suggested a muscle disease of the lipid storage myopathies. This was confirmed by a muscle biopsy that revealed lipid deposition in the muscle fibers. The proband’s sister previously had a similar disease, so the family underwent genetic testing. This revealed complex heterozygous ETFDH mutations c.389A > T (p. D130V) and c.1123C > A (p. P375T) in the proband and her sister, of which c.1123C > A (p. P375T) is a novel pathogenic mutation. The proband was treated with riboflavin and changes in physical symptoms and laboratory tests were evaluated before and after treatment. The discovery of a novel locus further expands the ETFDH mutation spectrum and suggests that genotyping is vital for early detection of RR-MADD as it can greatly improve the prognosis.
Keywords
Introduction
Lipid storage myopathies (LSMs) are genetic muscular disorders that result from defective lipid metabolic pathways, but exclude Fabry disease and some other mitochondrial diseases because of their unrelated mechanism for lipid deposition. Other metabolic disorders of skeletal muscle such as very long-chain acyl-CoA dehydrogenase deficiency and carnitine palmitoyltransferase 2 deficiency are also not usually considered LSMs because of the insignificant fat deposition that occurs in muscle fibers. Therefore, LSMs typically represent neutral lipid storage disease with ichthyosis (NLSDI), neutral lipid storage disease with myopathy (NLSDM), primary carnitine deficiency (PCD), and multiple acyl-CoA dehydrogenase deficiency (MADD).1–3
Among these, NLSDI and NLSDM are caused by mutations in CGI-58 and PNPLA2 genes, respectively, which encode adipose triglyceride lipase. The mutations lead to a dysfunctional lipase enzyme, causing triglyceride breakdown disorder and fat deposition. At present, no effective therapeutic methods are available to treat these diseases. PCD is a disorder of defective carnitine transport, caused by mutations in the SLC22A5 gene and resulting in dysfunction of sodium-dependent organic cationic carnitine transporter protein. This impairs carnitine uptake and reabsorption in renal tubules, resulting in intracellular carnitine deficiency. The symptoms of PCD can be greatly improved by the administration of L-carnitine.
MADD is also known as glutaric acidemia type II. It is caused by mutations in the electron transfer flavoprotein dehydrogenase gene (ETFDH) or electron transfer flavoprotein gene (ETFA/B), which lead to the dysfunction of a variety of mitochondrial respiratory chain dehydrogenases that then fail to transfer electrons. This causes lipid dysmetabolism along with other metabolic disorders. The analysis of organic acids in urine during the start of late-onset MADD revealed an increased concentration of glutaric acid and other organic acids, suggesting glutaric acidemia type II. 4
Late-onset MADD often shows a dramatic response to riboflavin, hence it is also known as RR-MADD. 5 Several findings have suggested that ETFDH deficiency may be a major cause of RR-MADD. 6 , 7 In this study, the family of an RR-MADD patient was analyzed to understand the clinical features of disease and response to the riboflavin treatment. We also report a novel causative mutation site that we believe will improve the clinicians’ understanding of the disease.
Case report
The proband was a 59-year-old woman from northern China who was admitted to the Department of Neurology of our hospital for 1 month because of general weakness accompanied by mastication weakness. Her symptoms commenced 1 month previously. Initial symptoms included weakness of upper and lower limbs, being prone to falling while walking, and difficulty squatting and holding heavy objects. These symptoms improved with rest and worsened in cold environments. Noticeably, during this period, she had no symptoms of hypoglycemia, acidosis, or high blood ammonia. However, she had a history of coronary heart disease for 3 years. Her 57-year-old sister had also been admitted to another hospital 1 year previously for the treatment of weakness in her limbs. The main manifestations were similar to those of the proband in that her upper limbs could not hold heavy objects and she could not walk for long periods. At that time, although muscle biopsy showed lipid deposition myopathy, no genetic tests were performed as no other family members had similar symptoms (Figure 1).

Family tree.
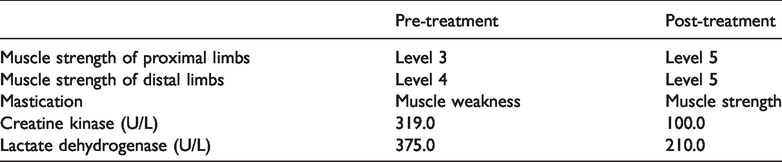
Physical examination of the proband showed no abnormalities in the heart, lung, or abdomen. There was also no obvious fasciculation, muscle atrophy, muscle tenderness, or pain. The patient’s upper limbs were naturally sagging, her elbow joint was slightly bent, and her forearm was pronated. We held her shoulder blade with one hand, and applied pressure to the elbow joint with the other hand so that she could complete a shoulder joint forward flexion and lifting movement of 90° but she was unable to do so. In a sitting position her lower leg sagged naturally. We held her pelvis with one hand and asked her to flex her hip joint while we applied resistance to the proximal end of her knee joint, but she was unable to complete this action. Similarly, her resistance was incomplete when we asked her to stretch and flex her wrist and carry out plantar flexion and dorsiflexion. Her muscle strength was 3/5 in proximal limbs and 4/5 in distal limbs (Medical Research Council Scale). Limb tendon reflexes were weakened but there were no obvious positive signs of the nervous system.
Venous blood was collected from the proband and creatine kinase and lactate dehydrogenase levels were measured by a creatine kinase assay kit and visible spectrophotometry. The concentrations of creatinine kinase and lactate dehydrogenase were 319.0 U/L (normal, 18–198 U/L) and 375.0 U/L (normal, 109–245 U/L), respectively. Urinary organic analysis showed that 2-hydroxy glutaric acid and 3-hydroxy glutaric acid levels were raised. Serum acylcarnitine analysis showed an increased level of C8–C16 fatty acylcarnitine. Myogenic lesions were evident in the electromyography of the left tibialis anterior, right biceps, and right deltoid muscles. The clinical phenotypes of the proband were mainly difficult to detect, involving fluctuating muscle weakness and exercise intolerance. The involvement of proximal extremities and masticatory muscles was evident.
Muscle biopsy
An open muscle biopsy of the left biceps of the proband was performed under local anesthesia after receiving her written informed consent. Muscle tissue specimens of 0.5 × 1 × 0.5 cm were placed vertically on a small piece of cork within a beaker containing isopentane, which was positioned in a liquid nitrogen container. When the temperature of the isopentane had dropped to −160°C, the muscle specimens were clamped with long tweezers and immersed in isopentane for rapid freezing. They were then sliced into 8-µm sections within a frozen slicer at −22°C for about 30 minutes. Various staining procedures were performed on the frozen sections and the morphological changes of muscle fibers were observed under a light microscope.
Hematoxylin and eosin (HE) and modified Gomori trichrome staining revealed small round scattered vacuoles in most muscle fibers, and showed that some were fused into cracks. Oil red O (ORO) staining revealed the accumulation of lipid droplets in muscle tissue. Nicotinamide adenine dinucleotide-tetrazolium reductase, succinate dehydrogenase, and non-specific esterase staining showed hyperchromasia of individual atrophic muscle fibers. Periodic acid-Schiff staining revealed no obvious abnormalities. ATPase (pH 4.5, pH 10.2) staining showed the major involvement of type I muscle fibers but that two types of vacuole-like muscle fibers were distributed in a checkerboard pattern (Figure 2).
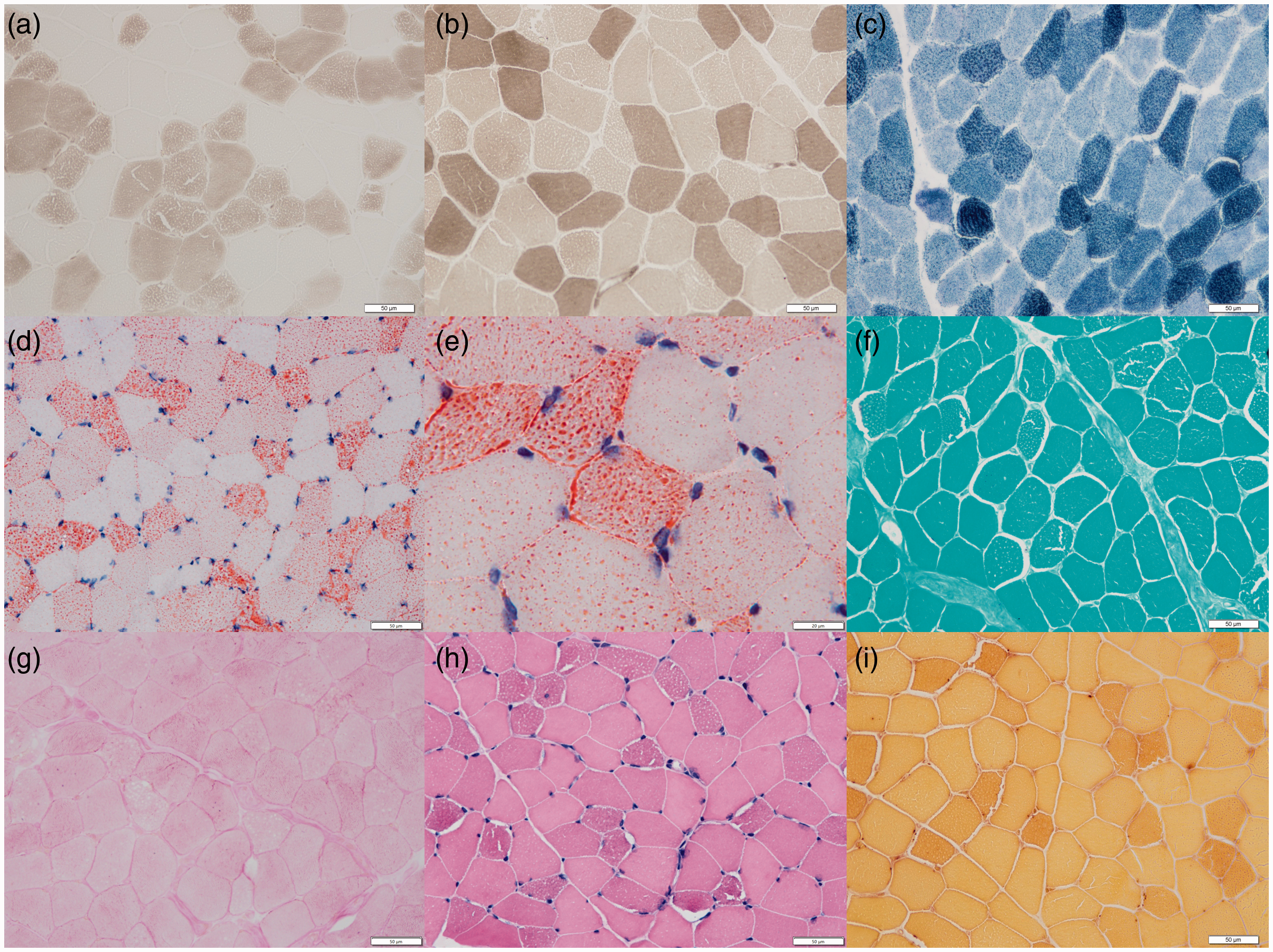
(a, b) ATPase staining (magnification × 400) showing the major involvement of type I muscle fibers. (c) NADH-TR staining (magnification ×400) showing hyperchromasia of individual atrophic muscle fibers. (d) ORO staining (magnification ×400) showing the accumulation of lipid droplets in muscle tissue. (e) ORO staining (magnification ×1000) showing red vacuoles in the cytoplasm. (f) MGT staining (magnification ×400) showing small vacuoles in some of the cytoplasm. (g) PAS staining (magnification ×400) showing no obvious abnormalities. (h) HE staining (magnification ×400) showing muscle fiber atrophy, and some cavitation. (i) NSE staining (magnification ×400) showing hyperchromasia of individual atrophic muscle fibers.
Mutation detection
A total of 3 mL venous blood was collected from the proband, her sister, and the proband’s children into 2% ethylenediaminetetraacetic acid anticoagulant tubes for next-generation sequencing using a metabolic myopathy gene panel after receiving written informed consent. Genomic DNA was extracted with a DNA extraction kit. Two primer pairs were designed to amplify exon 3 and exon 10 of ETFDH (NM-004453) as follows: exon 3-F: 5′-TTTTTATTTCTCCCAGGAGTGAAC-3′ and exon 3-R: 5′-TGAAAAGGGTTTCCTATATTCCA-3′; and exon 10-F: 5′-TTCAGCCTTTCCCTACAGCTC-3′ and exon 10-R: 5′-AAAGTGACAGATGCAATACAAATCTT-3′. PCR amplification was performed using the following conditions: denaturation at 95°C for 5 minutes, then 32 cycles of denaturation at 95°C for 30 s, annealing at 59°C (for exon 3) or 60°C (for exon 10) for 30 s, and extension at 72°C for 30 s, with a final extension at 72°C for 7 minutes. PCR products of 340 bp and 371 bp, respectively, were then purified and sequenced by Sanger sequencing.
Next-generation sequencing revealed that the proband and her sister both carried compound heterozygous mutations of ETFDH, c.389A > T (p.D130V) and c.1123C > A (p.P375T) (Figure 3), which were causative of their LSM phenotypes. The proband's eldest son, third son, and daughter were all shown to carry ETFDH c.1123C > A (p.P375T), and the second son carried ETFDH c.389A > T (p.D130V). None of the children had an LSM phenotype.

ETFDH sequencing in the proband. Red arrow indicates the mutation.
ETFDH c.389A > T (exon 3, NM_004453), resulting in p.D130V, is a missense mutation. According to American College of Medical Genetics and Genomics (ACMG) guidelines, it is likely to be pathogenic (PM3_Strong +PM1+PM2+PP3). ETFDH c.1123C > A (exon 10, NM_004453), resulting in p.P375T, is also a missense mutation. ACMG guidelines state that this mutation is of uncertain clinical significance (PM2 + PP3). REVEL software (MyGenostics, Beijing, China), used to predict the pathogenicity of missense variants, predicted that ETFDH c.389A > T and c.1123C > A were harmful. In silico tools SIFT, PolyPhen, and MutationTaster (MyGenostics, Beijing, China) also indicated these mutations to be detrimental. SWISS-MODEL showed that the predicted c.389A > T mutant protein has one less hydrogen bond than the wild-type, which may affect the tension between amino acids and the spatial structure of the protein. In c.1123C > A, no major structural changes were observed between the mutant and the wild-type.
Therapeutic effect and prognosis
The proband was instructed to consume a high-carbohydrate and low-fat diet. She was also treated with riboflavin (60 mg/d), coenzyme Q10 (150 mg/d), and carnitine. After 7 days, her clinical symptoms reduced, and after 1 month her ability to take part in physical labor and exercise had returned to normal (Table 1). At the 1-year follow-up, she requested to keep taking riboflavin but had ceased coenzyme Q10 and carnitine treatment and had fully returned to regular work and life.
Laboratory examination indexes, clinical signs and symptoms of the proband.
Discussion
LSMs are a group of fat metabolism myopathy disorders characterized by fat deposition in muscle fibers. RR-MADD is a special phenotype of LSMs that shows a dramatic response to riboflavin therapy. It is usually caused by mutations in the flavin adenine dinucleotide (FAD) structural domain of ETFDH. Clinical manifestations include progressive limb weakness, movement intolerance, mastication weakness, neck flexion weakness and dysphagia, and possible symptoms of the heart, kidney, and liver. 8 , 9 Electromyography of LSMs typically reveals myogenic impairment; however neurogenic or mixed impairment may occur in some patients.
Physical examination of our proband showed progressive limb weakness, movement intolerance, and mastication weakness. Biochemical tests revealed raised levels of 2-hydroxy glutaric acid and 3-hydroxy glutaric acid in the urine, and increased serum levels of C8-C16 fatty acylcarnitine, suggestive of a metabolic muscular disorder. Electromyography findings also suggested a muscle disease. To further confirm the diagnosis, a muscle biopsy was performed. HE, ORO, and ATPase staining revealed typical pathological changes of LSMs. Because the proband’s sister had previously been diagnosed with lipid deposition myopathy, we suspected that the proband had a hereditary disease.
Riboflavin has been shown to dramatically improve the symptoms of RR-MADD, even decreasing or removing lipid droplets in the muscle fibers of some patients, 10 so genetic testing is necessary for diagnosis and clinical treatment. 8 Previously reported ETFDH mutations include nonsense, shift, and missense mutations. Common mutation sites include c.1534G > A, c.1552C > G, c.176-1G > A, c.250G > A, c.770A > G, c.1227A > C, c.1285 + 2T > C, exon1-5 del, c.1351G > A, and c.511A > G. However, the ETFDH mutation spectrum differs between southern and northern China. For instance, c.250G > A is more common in southern China, whereas c. 389A > T is common in northern China. 11 Whether c.1123C > A (p.P375T) is also a common type in northern China remains to be determined. c.770A > G and c.1227A > C mutations are widely distributed and their prevalence rates are similar in northern and southern China. 11 , 12
Sequencing of the family members revealed that the proband and her sister carried compound heterozygous ETFDH mutations c.389A > T (p.D130V) and c.1123C > A (p.P375T). c.389A > T is a pathogenic mutation, and c.1123C > A is newly identified in this study. A change of cytosine to adenine at site 1123 changes the corresponding proline residue to threonine in the protein and renders the enzymes useless. None of the children carrying a single heterozygous mutation of ETFDH presented with clinical symptoms of LSMs. This indicates that the clinical manifestations of this family were caused by carrying both missense mutations.
Studies have shown that FAD binding plays an important role in the catalytic activity, folding, assembly, and stability of flavoprotein. Moreover, supplementation of the FAD precursor riboflavin can enhance the conformational stability of the mutated electron transfer flavoprotein ubiquinone oxidoreductase. 3 , 5 After 1 month of riboflavin treatment, our patient’s ability to perform physical labor or exercise had returned to normal, which was in line with the clinical transition characteristics of RR-MADD.
Conclusion
Although LSMs are genetic diseases, drug therapy greatly improves the clinical symptoms and quality of life of patients with a diagnosis of RR-MADD. This phenomenon differs from the idea that hereditary diseases are incurable. Because genotyping is crucial to achieve an accurate diagnosis, especially for RR-MADD patients, the identification of ETFDH c.1123C > A (p.P375T) as a novel pathogenic mutation in this study will help to further expand the ETFDH mutation database.
Footnotes
Acknowledgements
The authors thank the patient’s family for participating in this study.
Declaration of conflicting interest
The authors declare that there is no conflict of interest.
Ethics approval and consent to participate
This study was approved by the Institutional Review Board of The Second Hospital of Hebei Medical University. Informed consent was obtained from the patient and her family.