
Abstract
We herein report two cases of primary adrenal insufficiency (AI) associated with antiphospholipid syndrome (APS). In both patients, the main finding that led to the diagnosis was hyponatraemia. The major difference between the two cases was the time at which AI evolved during the course of APS. In the first patient, AI developed acutely along with other presenting features of APS. In the second patient, the AI was unmasked during a stressful situation induced by severe inflammation that occurred 7 years after the first APS manifestation and had probably evolved slowly during the previous few years. These cases emphasise the importance of considering AI in patients with either suspected or newly diagnosed APS as well as in patients who have long been known to have APS. The symptoms and signs alerting the clinician to possible AI are general abdominal complaints, fever, hypotension, and hyponatraemia. Conversely, patients with primary AI should be questioned about the signs and symptoms of APS.
Keywords
Abbreviations
AI = adrenal insufficiency; APS = antiphospholipid syndrome; ACTH = adrenocorticotropic hormone; SLE = systemic lupus erythematosus; PE = pulmonary embolism; CT = computed tomography.
Introduction
Antiphospholipid syndrome (APS) is defined by the presence of antiphospholipid antibodies in patients with thrombotic and/or obstetrical events. 1 Laboratory tests are positive for several antiphospholipid antibodies (namely anticardiolipin, anti-beta-2 glycoprotein I, and/or lupus anticoagulant) on two or more occasions at least 12 weeks apart. 1 APS can be primary or associated with a systemic autoimmune disease, especially systemic lupus erythematosus (SLE).
Many clinical manifestations are possible in patients with APS because this disorder can affect any organ system in the body. Deep vein thrombosis, pulmonary embolism (PE), stroke, and transient ischaemic attacks are the most common manifestations. 2 Endocrine complications of APS are generally considered to be very rare. Among them, adrenal insufficiency (AI) is probably the most common, although hypopituitarism 3 , 4 and isolated adrenocorticotropic hormone (ACTH) deficiency 5 have also been described. The prevalence of AI in patients with APS is currently unknown. 6 In previous studies, adrenal failure was reportedly present in 10% to 26% of patients with catastrophic APS, a peracute form of the disease affecting at least three organ systems. 7 , 8 In a study by Cervera et al., 2 only 4 of 1000 patients with APS developed AI in the course of 5 years. Primary adrenal failure caused by bilateral venous thrombosis and/or adrenal haemorrhage is the most common mechanism, while another possible mechanism is autoimmune adrenal failure and microthrombi. The unique vascular structure of the adrenal gland, with three arteries and only one vein, may result in limited drainage of the organ’s blood supply, predisposing the patient to thrombosis and haemorrhage. 9 Additional risk factors for adrenal haemorrhage are inadequate anticoagulant therapy, recent surgery, and infection. 10 , 11 Clinically, AI commonly presents as abdominal pain (55%), hypotension (54%), and fever (40%). Weight loss and hyperpigmentation are seen in 13% and 10% of patients, respectively. 12 In patients with APS, spontaneous bilateral adrenal haemorrhage is an uncommon condition that may lead to acute AI and, if not recognised, to death. This was almost exclusively a postmortem diagnosis before computed tomography (CT) became widely available. 13 The estimated mortality rate of patients who have AI associated with APS or SLE is 3.81%. 14 The long-term outcome of patients who survive the acute phase is most likely favourable. 15
Case 1
A 27-year-old man presented with pain in the left hemithorax, fever (38.5°C), and dyspnoea. His medical history was positive for Raynaud’s phenomenon. Otherwise, he was a healthy athlete. His family history was positive for rheumatoid arthritis.
A chest radiograph was obtained to investigate the patient’s clinical condition, and it showed bilateral pleural effusion. The levels of inflammatory markers were high (Table 1). He was admitted to the hospital and treated for pneumonia. After treatment with empirical antibiotic therapy, the patient’s inflammatory markers remained elevated and he was still febrile. Serology for atypical pneumonia, human immunodeficiency virus, and hepatitis A, B, and C was negative, as were blood and pleural fluid cultures. Biochemically, the pleural fluid was an exudate.
Relevant laboratory test results in the first patient.

Abnormal results are indicated by boldface type.
CRP, C-reactive protein; ESR, erythrocyte sedimentation rate; WBC, white blood cell; AST, aspartate aminotransferase; ALT, alanine aminotransferase; GGT, gamma-glutamyltransferase; ALP, alkaline phosphatase; LDH, lactate dehydrogenase.
Because of his elevated liver enzymes, the patient underwent an abdominal ultrasound and CT scan; however, neither showed evidence of pathology. CT angiography of the pulmonary arteries was performed because of persistent dyspnoea. PE and multiple larger and smaller pulmonary infarcts in both lower lobes were diagnosed. Doppler ultrasound excluded deep vein thrombosis of the lower limbs. An echocardiogram showed no right ventricular strain, no valvular pathology, and no pericardial effusion. Therapy with low-molecular-weight heparin was introduced and was later gradually changed to a coumarin anticoagulant.
The patient became nauseous during hospitalisation, and 2 weeks after admission, hyponatraemia was first noted (serum sodium concentration of 129 mmol/L); the serum potassium concentration was within the reference range. AI was suspected and confirmed by a short ACTH stimulation test: basal cortisol concentration, 93 nmol/L (reference range, 138–690 nmol/L); cortisol at 30 minutes, 88.3 nmol/L (reference value, >500 nmol/L). Therefore, parenteral hydrocortisone replacement therapy was introduced by continuous infusion of 300 mg/day and then gradually decreased to 50 mg/day. The patient was subsequently switched to oral replacement therapy with hydrocortisone at 20 mg/day in divided doses. Further investigation showed a very high ACTH concentration of 106 pmol/L (reference value, <10.2 pmol/L), confirming primary AI. The plasma renin activity was within the reference range while he was receiving high doses of hydrocortisone; at the 1-month follow-up, however, it was elevated at 11.3 µg/L/h (reference value, <6.0 µg/L/h), and his potassium concentration showed a tendency to rise. Therefore, therapy with fludrocortisone at 0.05 mg/day was introduced.
To clarify the aetiology of the PE and AI, the patient underwent examination for systemic rheumatic disease and immunological tests (Table 2). He had microcytic anaemia (haemoglobin concentration, 124 g/L [reference range, 140–160 g/L] and mean corpuscular volume, 79.2 fl [reference range, 81–94 fl]) with a normal lactate dehydrogenase level (1.85 µkat/L [reference value, <4.13 µkat/L) and normal reticulocyte count 40 × 109/L (reference range, 34.0–120.6 × 109/L). The patient had no signs suggesting microangiopathic haemolytic anaemia because the haptoglobin concentration was normal at 1.32 g/L (reference range, 0.3–2.0 g/L) and no schistocytes were present. A direct Coombs test was positive for anti-C3d and negative for anti-IgG, anti-IgA, anti-IgM, and anti-C3c.
Rheumatologic test results in the first patient.
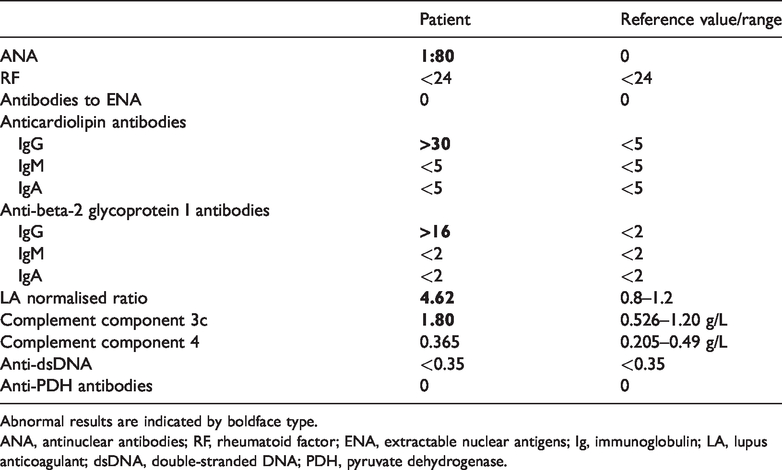
Abnormal results are indicated by boldface type.
ANA, antinuclear antibodies; RF, rheumatoid factor; ENA, extractable nuclear antigens; Ig, immunoglobulin; LA, lupus anticoagulant; dsDNA, double-stranded DNA; PDH, pyruvate dehydrogenase.
During follow-up, abdominal CT showed atrophy of the adrenal glands. At the time of this writing, the patient was doing well and had normal serum electrolyte levels. He was continuing to regularly visit the rheumatology outpatient clinic, and apart from Raynaud’s phenomenon, he had developed no clinical signs or symptoms of possible underlying other autoimmune disease. Additionally, his autoimmune screening laboratory test results remained negative.
Case 2
A 54-year-old man had a 6-year history of APS and an undifferentiated connective tissue disease suggestive of SLE. We did not classify the disease as SLE, however, because not all criteria could be counted; the APS was thought to be the most likely cause of the patient’s several manifestations. 16
The patient had suffered from arthritis of the small joints for several years, and he developed two deep vein thromboses 6 years before presentation. At that time, he had high titres of anticardiolipin and anti-beta-2 glycoprotein I, and his antinuclear antibodies remained high and anti-DNA antibodies remained negative at several consequent assessments. A low C4 complement level, thrombocytopaenia, and positive Coombs test were also confirmed, and he began regularly taking a coumarin anticoagulant. He was later admitted to the hospital because of pharyngitis, conjunctivitis, and neck lymph node enlargement. He had a fever (39°C), severe pain in the throat, and difficulty swallowing. Physical examination showed aphthous mouth ulcers, conjunctival injection and periorbital oedema, enlarged lymph nodes on the left side of the neck, and general hyperpigmentation of the skin. Laboratory results indicated inflammation with a C-reactive protein concentration of 336 mg/L (reference value, <5 mg/L), erythrocyte sedimentation rate of 79 mm/h (reference value, <20 mm/h), and procalcitonin concentration of 0.85 µg/L (reference value, <0.5 µg/L). Chest radiograph and abdominal ultrasound findings were normal. Nasopharyngeal swabs were negative for pathogenic bacteria, as were blood and urine cultures. Polymerase chain reactions were negative for Epstein–Barr virus, Cytomegalovirus, human immunodeficiency virus, varicella zoster virus, and herpes simplex 1, 2, and 6. The patient began treatment with an empirical antibiotic. At the very beginning of hospitalisation, hyponatraemia (serum sodium concentration of 126 mmol/L) was noted. After a careful review of his previous laboratory results from the outpatient clinic, we realised that mild hyponatraemia had been present for several years. A short ACTH stimulation test confirmed AI: the basal cortisol concentration was 320 nmol/L, and 30 minutes after ACTH administration, it was even lower at 287 nmol/L. A CT scan of the abdomen showed bilateral adrenal atrophy, which was consistent with chronic AI. As expected, the plasma ACTH concentration was high at 133 pmol/L (reference value, <10.2 pmol/L). Repeatedly normal serum potassium (4.4 mmol/L) and plasma renin activity (2.54 µg/L/h) indicated that no mineralocorticoid deficiency was present. Oral hydrocortisone replacement therapy at 50 mg/day in divided doses was introduced and then gradually reduced to 20 mg/day in divided doses. The serum sodium concentration normalised (136 nmol/L) and the inflammatory markers finally started to decrease.
At the time of this writing, the patient’s main complaint was painful arthritis of the small hand joints, for which he was being treated with chloroquine. He was also regularly taking a coumarin and hydrocortisone therapy; fludrocortisone was not required.
Discussion
APS predisposes individuals to venous, arterial, or small vessel thrombosis (microthromboses); women are additionally predisposed to morbidity in pregnancy. 1 APS most commonly manifests as deep vein thrombosis, PE, stroke, or transient ischaemic attack. 2 Acute or chronic microangiopathy characterised by dominating endothelial cell injury, often associated with thrombotic necrotising lesions, has also been recognised as a manifestation of APS. 17
The main antibodies found in APS are anticardiolipin, anti-beta-2 glycoprotein I, and lupus anticoagulants. To fulfil the revised Sapporo criteria for APS, at least one type of antibody should be present on two separate occasions at least 12 weeks apart. 1
Since APS was first recognised in 1986, it has been reported as a cause of AI. 7 , 8 In 2003, Espinosa et al. 12 reviewed the clinical characteristics of 86 patients with adrenal involvement secondary to APS, and they suggested that the close relationship between APS and hypoadrenalism was more than coincidental. AI is now generally accepted to be the most common endocrine manifestation of APS, and routine testing of adrenal function has been suggested in patients with APS. 6 A study was undertaken to determine the significance of periodic evaluation of the hypothalamic-pituitary-adrenal axis in patients with APS, and no AI was found among 24 patients with a mean APS duration of 5.8 years. 18 In a cohort of 1000 patients with APS, only 4 patients developed AI in the course of 5 years. 2 However, further longitudinal studies are needed to estimate the need for routine evaluation of adrenal function in patients with APS.
Before the availability of CT, bilateral adrenal haemorrhage was usually diagnosed by postmortem examination because signs and symptoms of AI are nonspecific and easily confused with those of the underlying condition. 19 The most consistent clinical feature is abdominal pain, which is usually accompanied by nausea, vomiting, fever, and hypotension. The main laboratory findings are hyponatraemia and hyperkalaemia. 12 Of the 28 reported cases of AI associated with APS, 17 had CT or magnetic resonance imaging findings revealing bilateral adrenal haemorrhage and adrenal enlargement or atrophy. 20 Histopathologic studies showed that haemorrhagic infarction with venous thrombosis (55%) and adrenal haemorrhage (27%) are the main causes of AI in patients with APS. 9 Additionally, intraparenchymal microhaemorrhages and microthromboses have been proposed as a possible mechanism of adrenal damage.17,21–23
In our patients, AI was confirmed before the CT scan was performed. The main finding that led to the diagnosis in both patients was hyponatraemia. The CT scan of the adrenal glands was performed to identify the aetiology of Addison’s disease. The adrenal imaging findings allowed us to exclude infiltration, haemorrhage, haematoma, malignancy, and adrenal infection. In the first patient, AI developed almost simultaneously with other manifestations of APS, suggesting an acute microthrombotic mechanism or, even more likely, an acute microangiopathic mechanism of adrenal damage. The patient’s elevated liver enzymes were also most likely the consequence of hepatic microthrombosis, as described in the case by Inam et al. 23 and in the review by Praprotnik et al. 17 The severe glucocorticoid and mineralocorticoid impairment of adrenal function suggested impairment of all layers of the adrenal cortex.
In the second patient, however, gradual and selective impairment of glucocorticoid function raised suspicion for chronic microangiopathy associated with antiphospholipid antibodies, as previously described in other organs. 17 In our first patient, the CT scan performed in the very early phase of the disease showed no pathologic abnormalities of the adrenal glands, and atrophy later developed. Additionally, adrenal atrophy was evident on abdominal CT in the second patient. These morphological findings are consistent with proposed microangiopathic adrenal damage, which was acute in one patient and chronic in the other. However, we cannot completely eliminate the possibility of autoimmune destruction of the adrenal glands because the ability to measure 21-hydroxylase antibodies was not available at our centre.
The average time at which AI is revealed in the course of APS has not yet been studied. In many cases, AI is the first manifestation of APS.24–26 Both of our patients presented with venous thromboembolic disease, namely PE in the first patient and deep vein thrombosis in the second. In the first patient, signs and symptoms of Addison’s disease occurred within the first week after PE. In the second patient, AI was revealed during management of severe infection, which occurred about 6 years after the first presentation of APS. However, AI had probably developed gradually during the last few years because mild hyponatraemia had been recorded at previous outpatient visits. Our cases and others published in recent years 27 , 28 indicate that AI must be considered in all patients with APS—both those with well-known APS and those with newly diagnosed APS.
Conclusion
AI has been reported as a rare but potentially fatal complication of APS. When we encounter patients with AI, we should inquire about thrombotic episodes and, in women, a history of spontaneous abortions or premature births. The diagnosis of AI should be suspected in any patient with a history of thrombophilia presenting with general abdominal complaints and hyponatraemia. We should consider the possibility of AI in all patients with diagnosed or suspected APS because AI can develop either rapidly in the course of APS or gradually, becoming apparent many years after the first presentation of APS, usually during a stressful event.
Footnotes
Declaration of conflicting interest
The authors declare that there is no conflict of interest.
Ethics statements
Both patients provided consent to submit these cases for publication. Ethics approval from an ethics review committee or institutional review board was not required for presentation of these cases.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.