
Abstract
Objectives
To investigate levels of cell-division cycle 42 (Cdc42) protein, and their relationship with Golgi apparatus function in peripheral lymphocytes, in patients following ischaemic stroke.
Methods
Patients with acute cerebral ischaemic stroke (within 24–72 h of the onset of focal neurological symptoms) and healthy control subjects were enrolled in this prospective case–control study. The cellular location of Cdc42 in peripheral lymphocytes was demonstrated using immunofluorescence. Protein levels of Cdc42 and trans-golgi network protein 2 (TGN46) in peripheral lymphocytes were determined by immunocytochemical staining and Western blotting.
Results
A total of 38 patients with stroke and 38 control subjects were studied. The mean ± SD percentage of Cdc42-positive lymphocytes from patients with stroke was significantly lower than that in control subjects (39.53 ± 13.55% versus 66.61 ± 23.30%, respectively). Similar findings were demonstrated for TGN46. Cdc42 levels were positively correlated with TGN46 levels (r = 0.92).
Conclusions
Acute ischaemic stroke was associated with reduced levels of Cdc42 protein. These findings might lead to the development of drugs that could have therapeutic benefits in patients with acute ischaemic stroke.
Keywords
Introduction
A growing body of evidence shows that initiation of the inflammatory cascade participates in the development of acute ischaemic stroke.1,2 Post-ischaemic inflammatory responses, which are dependent on the involvement of the inflammatory cells and their related cytokines, occur in the infarcted brain and the peripheral circulation, and contribute to the ischaemic damage that occurs subsequent to the stroke.3,4 As important inflammatory cells, the lymphocytes (both T and B cells) are essential during ischaemic injury. Studies have shown that lymphocytes promote the ischaemic injury response after cerebral ischaemia by releasing proinflammatory factors, such as tumour necrosis factor (TNF)-α and interleukin (IL)-1.5,6 Other research indicates that lymphocytes play a protective role in ischaemic tissue, by secreting anti-inflammatory cytokines and related antibodies.7,8 These findings suggest that the different roles played by lymphocytes during the ischaemic process can be attributed to the involvement of different lymphocyte subtypes. Despite their different roles, the first step after cerebral ischaemia is the migration and infiltration of lymphocytes from the circulation to the infarcted site.5–8
Experiments in animal models of stroke, and in patients with ischaemic stroke, show that lymphocytes in the peripheral circulation infiltrate into infarcted sites mainly within 24–72 h after stroke onset.9,10 Treatment strategies that interfere with lymphocyte infiltration might be able to prevent or limit the development of cerebral ischaemic injury.3,11,12 Cell infiltration and migration are multistep processes (at a subcellular level) that result from the rearrangement of the cytoskeleton and adhesion complexes. 13 Cell-division cycle 42 (Cdc42) protein, which is a member of the Ras homolog (Rho) GTPase (enzymes that hydrolyse guanosine triphosphate [GTP]) family, is a crucial factor in mediating cell infiltration and migration.13,14 Activation of Cdc42 is known to be modulated by IL-1, TNF-α and granulocyte colony-stimulating factor.15,16 Cdc42 protein levels decrease during the infiltration and migration of lymphocytes due to their integrin-related adhesion. 17 In addition, Cdc42 has been implicated in the regulation of diverse cellular functions including oxidant generation, membrane trafficking, apoptosis and cell-cycle control, by interacting with specific target proteins and activating their downstream effectors. 14 Cdc42 also affects the production of antibodies produced by B lymphocytes, which was demonstrated by the decreased production of immunoglobulin (Ig) G and IgM by Cdc42-deficient B lymphocytes.18,19 In general, Cdc42 is involved in the migration and infiltration of peripheral lymphocytes and in the production of certain proteins, but whether Cdc42 protein levels in peripheral lymphocytes are changed during the acute phase of cerebral ischaemia remains unknown.
The metabolism of the Rho GTPase family in cells is related to the function of the Golgi apparatus. 20 Cdc42 is the first member of the Rho GTPase family to be found in the Golgi apparatus, and its synthesis and activity could be regulated by certain proteins within this apparatus.20–22 The Golgi apparatus in neurons might be impaired after an ischaemic attack by an extracellular environment full of free radicals, proteolytic enzymes, inflammatory cytokines and other cytotoxic substances. 23 Whether the Golgi apparatus in peripheral lymphocytes is affected by the peripheral environment during the acute phase of an ischaemic event, and what (if any) are its effects on Cdc42 expression in circulating lymphocytes, remain unknown.
The present study investigated the intracellular localization and levels of Cdc42 protein in peripheral lymphocytes from stroke patients after the onset of cerebral ischaemia. In addition, by using a recognized marker of Golgi apparatus function (trans-golgi network protein 2 [TGN46]), this study analysed the relationship between levels of Cdc42 protein and Golgi apparatus function in peripheral lymphocytes, following the onset of cerebral ischaemia.
Patients and methods
Patients
This prospective case–control study was undertaken in the Department of Neurology, Second Xiangya Hospital, Central South University, Changsha, Hunan Province, China. Consecutive patients with cerebral ischaemia were enrolled within 24–72 h of the onset of focal neurological symptoms, and the study took place between February 2010 and December 2011. Cerebral ischaemia was diagnosed on the basis of clinical history, neurological examination and a computed tomography (CT) or magnetic resonance imaging (MRI) scan of the brain. All patients with ischaemic stroke met World Health Organization (or similar Chinese National) criteria for stroke, and received standard stroke treatment according to the institutional guidelines of the Second Xiangya Hospital. 24 Healthy subjects (who underwent a health examination in the Medical Examination Centre of Second Xiangya Hospital between February 2010 and December 2010) served as the control group.
Exclusion criteria for patients with stroke were: patient was in a coma; neurological deficit was improving raplidly; head CT or MRI scan demonstrated intracranial haemorrhage or a diagnosis other than ischaemic stroke to explain the neurological deficit. Patients with stroke and control subjects were also excluded if they had any of the following active medical conditions: inflammatory disease; infection; cancer; bleeding; autoimmune disorder; myocardial infarction; liver or kidney disorder; endocrine disease; thromboembolic disease; chemotherapy or radiotherapy ≤1 month prior to study inclusion; history of ischaemic stroke ≤3 months prior to study inclusion. All patients with stroke had been excluded from acute thrombolytic therapies before study enrolment. This study was approved by the Ethics Committee of the Second Xiangya Hospital and all participants provided written informed consent.
Blood samples and isolation of lymphocytes
Blood samples were routinely taken from the cubital vein of patients with stroke and control subjects. Venous blood was treated with 1.5 mg/ml ethylenediaminetetra-acetic acid (EDTA) to prevent coagulation. Levels of blood leucocytes and lymphocytes were routinely quantified using an automated haematology analyser (ADVIA 2120; Bayer, Leverkusen, Germany) at the Central Laboratory of the Second Xiangya Hospital.
In addition to measuring routine haematological parameters, 10 ml of venous blood was drawn from each study participant and anticoagulated with 1.5 mg/ml EDTA, in preparation for analysis of peripheral blood mononuclear cells (PBMCs). PBMCs were isolated immediately by Percoll (GE Healthcare Biosciences, Piscataway, NJ, USA) density gradient centrifugation, according to the manufacturer’s instructions. PBMCs were collected from the interphase, washed twice with 0.01 M phosphate-buffered saline (PBS; pH 7.4) and counted using a haematocytometer. To separate cell subpopulations, PBMCs were consecutively incubated with Dynabeads® Magnetic Beads (Invitrogen, San Diego, CA, USA) directed against cluster of differentiation (CD) 68 (to label macrophages), CD14 (to label monocytes), CD3 (to label T lymphocytes) or CD19 (to label B lymphocytes), under rotation for 20 min at 4℃, and rinsed three times with 0.01 M PBS (pH 7.4) after separation. T and B lymphocytes were collected and a cell smear centrifugal machine (TXD3; Xiangyi, Changsha, China) was used to produce cytospin smears. Subsequently, cytospin smears of a lymphocyte suspension with 1 × 106 cells were dried in an oven at 37℃ for 10–15 min.
Immunofluorescence confocal microscopy
Immunofluorescence staining was performed on the cell smears. These smears were fixed with 3.7% formaldehyde (pH 7.4) for 30 min at room temperature, dried in air for 5 min and washed with 0.01 M PBS (pH 7.4). Smears were incubated in 1% Triton™ X-100 (Sigma-Aldrich, St Louis, MO, USA) for 20 min, rinsed in 0.01 M PBS (pH 7.4), incubated in 3% H2O2 for 15 min and then rinsed in 0.01 M PBS (pH 7.4). Normal goat serum (5% in 0.01 M PBS [pH 7.4]; Santa Cruz Biotechnology, Santa Cruz, CA, USA) was used as an antigen blocking agent for 15 min at 37℃. Then, smears were incubated with primary antibody (mouse antiCdc42 antibody; 1 : 100 dilution in 0.01 M PBS; Millipore, Billerica, MA, USA) overnight at 4℃. After incubation, smears were thawed for 20 min, then rinsed three times with 0.01 M PBS (pH 7.4). The following steps were undertaken in the dark. Smears were incubated with donkey antimouse IgG conjugated to fluorescein isothiocyanate for 1 h (Santa Cruz Biotechnology; 1 : 100 dilution in 0.01 M PBS) at room temperature. After being rinsed three times with 0.01 M PBS (pH 7.4), smears were incubated with 4 µg/ml 2-(4-amidinophenyl)-6-indolecarbamidine dihydrochloride (DAPI) (1 : 5000; Roche, Basel, Switzerland) for nucleus staining. Smears were rinsed three times with 0.01 M PBS (pH 7.4), washed once with double-distilled water and cover slips were applied. Immunofluorescence images were obtained under a confocal fluorescence microscope (LSM 5; Zeiss, Gottingen, Germany) with lasers at both 359 and 488 nm. The cellular location of positively-stained proteins in the lymphocytes was observed.
Immunocytochemical methods
Immunocytochemical methods were also performed on the cell smears. These smears were fixed, dried in air and prepared as described above, with the exception that they were not incubated in Triton™ X-100 solution. After endogenous peroxidase activity was blocked with 3% H2O2 for 15 min at room temperature, smears were treated with 0.01 m/l citrate (pH 6.0) in a 500-W microwave oven for 15 min to facilitate antigen retrieval. Smears were naturally cooled and rinsed three times in 0.01 M PBS (pH 7.4), then preblocked with normal goat serum (5% in 0.01 M PBS [pH 7.4]) for 15 min at 37℃. Then, smears were incubated with two primary antibodies overnight at 4℃ in a humidified immunostaining chamber: mouse antiCdc42 antibody (1 : 100 dilution in 0.01 M PBS; Millipore) and rabbit antiTGN46 antibody (1 : 200 dilution in 0.01 M PBS; Abcam®, Cambridge, MA, USA).
The next day, the smears were thawed, rinsed in 0.01 M PBS (pH 7.4), incubated with a biotinylated conjugated antimouse antibody (1 : 100 dilution in 0.01 M PBS; KPL, Gaithersburg, MA, USA) for 30 min at room temperature and washed again in 0.01 M PBS (pH 7.4). Then, smears were incubated in a commercially available avidin-conjugated horseradish peroxidase solution (1 µg/ml; Vector Laboratories, Burlingame, CA, USA) for 20 min at 37℃ and washed three times in 0.01 M PBS (pH 7.4). Finally, the smears were visualized using 0.05% diaminobenzidine and 0.02% H2O2 until the colour developed. The slides were then washed in tap water three times, counterstained with haematoxylin, cover slips were applied and cells were examined under a light microscope (DM-111; Motic®, Richmond, BC, Canada). Two independent investigators evaluated the number of positively-stained cells by counting five randomly selected high-powered fields. The proportion of positive lymphocytes was determined by dividing the number of positive lymphocytes by the total number of lymphocytes. Detailed observations of the immunostained product in the cytoplasm of lymphocytes were made using Motic® Images Advanced 3.2 digital microscopy analysis software (Motic®, Xiamen, China) on a Dell Vostro 430 personal computer (Dell, Xiamen, China).
Western blot analysis
For this analysis, 1 × 106 lymphocytes were lysed in 200 µl cell lysis buffer (10 mM Na2HPO4, pH 7.0, 300 mM NaCl, 1% v/v NP40, 1% w/v Na-deoxycholate, 2 mM EDTA, 1 mM dithiothreitol [DTT]), containing the complete Mini EDTA-free protease inhibitor cocktail solution (one tablet in 10ml cell lysis buffer, Roche Diagnostics, Mannheim, Germany) for 30 min on ice. Proteins were denatured by boiling in sample-loading buffer (10 mM Tris-HCl, pH 8.0, 1 mM EDTA, 1% w/v DTT, 2% w/v sodium dodecyl sulphate [SDS], 0.01% w/v bromophenol blue) for 5 min. Protein concentrations in solutions were measured using the Pierce® BCA Protein Assay Kit (Thermo Scientific, Houston, TX, USA), according to the manufacturer's instructions. Protein samples (10–20 µg) were electrophoretically separated using 10% SDS–polyacrylamide gel (SDS–PAGE), transferred to polyvinylidene fluoride membranes (Millipore) and blocked with 20 mM Tris-HCl, 5% fat-free milk solution for 1 h at room temperature. Then. the blots were incubated respectively with primary mouse antiCdc42 antibody (1 : 400 dilution in 5% fat-free milk; Millipore) and rabbit antiTGN46 antibody (1 : 500 dilution in 5% fat-free milk; Abcam®) overnight at 4℃ on a rotary platform with gentle agitation. Membranes were washed with 0.01 M Tris-buffered saline Tween® 20 (TBST; pH 7.4) three times at 15-min intervals, and subsequently probed with secondary horseradish peroxidase-conjugated antimouse or antirabbit IgG antibodies (1 : 4000 dilution in 5% fat-free milk; Sigma-Aldrich) at room temperature for 1 h. Next, the membranes were washed with 0.01 M TBST (pH 7.4) three times at 10-min intervals. Equal loading was confirmed by resolving 20 mg of total protein by SDS–PAGE and probing with antiβ-actin antibody (1 : 2000 dilution in 5% fat-free milk; Sigma-Aldrich). Detection was performed using an enhanced chemiluminescence kit (Millipore). To provide semiquantitative analysis of band intensity, band densitometry was determined from scanned images of unsaturated immunoblot films, using Scion Image software, version Beta 4.0.2 (Scion Corporation, Frederick, MD, USA).
Statistical analyses
Data were expressed as mean ± SD. All statistical analyses were performed using the SPSS® statistical software package, version 16.0 (SPSS Inc., Chicago, IL, USA) for Windows®. The percentages of Cdc42-positive and TGN46-positive cells out of all lymphocytes, and the level of Cdc42 and TGN46 proteins determined by Western blotting, were compared between patients with stroke and control subjects, using two-tailed Student's t-test or Mann–Whitney U-test. The correlation between the levels of Cdc42 and TGN46 proteins (as determined by Western blot analysis) was assessed by linear regression. A P-value < 0.05 was considered statistically significant.
Results
Demographic and haematological characteristics of patients with stoke (n = 38) and healthy control subjects (n = 38) who participated in a study investigating levels of cell-division cycle 42 protein and its relationship with Golgi apparatus function in peripheral lymphocytes.

Data presented as mean ± SD.
The two groups were compared with two-tailed Student's t-test if data were normally distributed or Mann–Whitney U-test if data were not normally distributed.
NS, not statistically significant (P ≥ 0.05).
Under fluorescent microscopy, lymphocytes exhibited round- or oval-shaped nuclei that were identified by blue fluorescence using DAPI. Cdc42 proteins in lymphocytes were identified by green fluorescence and were located in the cytoplasm. The overlaid immunofluorescence images showed that Cdc42 was mainly distributed around the nuclei (Figure 1).
Representative immunofluorescence images of cell-division cycle 42 (Cdc42) protein in lymphocytes, collected from patients within 24–72 h of the onset of focal neurological symptoms indicative of stroke. Under a fluorescence microscope equipped with a digital camera, nuclei (A; blue fluorescence) and the cytoplasmic location of Cdc42 protein in lymphocytes (B; green fluorescence) were observed. Fluorescence microscopy software presented an overlaid image of the stained nuclei and Cdc42 proteins (C). Cdc42 was unevenly distributed around the nuclei. Not all of the lymphocytes expressed Cdc42 in the cytoplasm.
Immunocytochemical images of Cdc42-positive lymphocytes showed blue–violet-stained round- or oval-shaped nuclei and brown-stained cytoplasm (Figure 2). The mean ± SD percentage of Cdc42-positive lymphocytes from patients with stroke was 39.53 ± 13.55%, which was significantly lower than that for control subjects (66.61 ± 23.30%) (P < 0.05). Brown-stained cytoplasm and blue-stained round nuclei were also found in TGN46-positive lymphocytes. Patients with stroke had a significantly lower mean ± SD percentage of TGN46-positive lymphocytes than control subjects (32.78 ± 10.98% versus 79.52 ± 13.23%, respectively; P < 0.001). Western blot analysis also confirmed that levels of Cdc42 and TGN46 proteins were decreased in the peripheral lymphocytes from patients with stroke compared with control subjects (Figure 3). Simple linear regression was used to test the relationship between Cdc42 and TGN46 protein levels in all patients with stroke and control subjects, as measured by Western blot analysis. Levels of Cdc42 protein were positively correlated with levels of TGN46 protein (r = 0.92, P < 0.05) (Figure 4).
Representative light photomicrographs of the immunocytochemical staining of cell-division cycle 42 (Cdc42) and trans-golgi network protein 2 (TGN46) proteins in peripheral lymphocytes, collected from patients within 24–72 h of the onset of focal neurological symptoms indicative of stroke (A and C, respectively) and peripheral lymphocytes from healthy control subjects (B and D, respectively). Cell smears were counterstained with haematoxylin. Quantification of the proportion of Cdc42-positive and TGN46-positive lymphocytes from each study group (E). *P < 0.05 and **P < 0.001 compared with control group; two-tailed Student's t-test if data were normally distributed; Mann–Whitney U-test if data were not normally distributed. Western blot analysis of levels of cell-division cycle 42 (Cdc42) and trans-golgi network protein 2 (TGN46) proteins in peripheral lymphocytes, collected from patients within 24–72 h of the onset of focal neurological symptoms indicative of stroke (Stroke) and peripheral lymphocytes from healthy control subjects (Control). β-actin used as loading control. Correlation between levels of cell-division cycle 42 (Cdc42) and trans-golgi network protein 2 (TGN46) proteins in peripheral lymphocytes, collected from patients within 24–72 h of the onset of focal neurological symptoms indicative of stroke, and in peripheral lymphocytes from healthy control subjects, as determined by Western blot analysis. Cdc42 protein levels were positively correlated with TGN46 protein levels (r = 0.92, P < 0.05).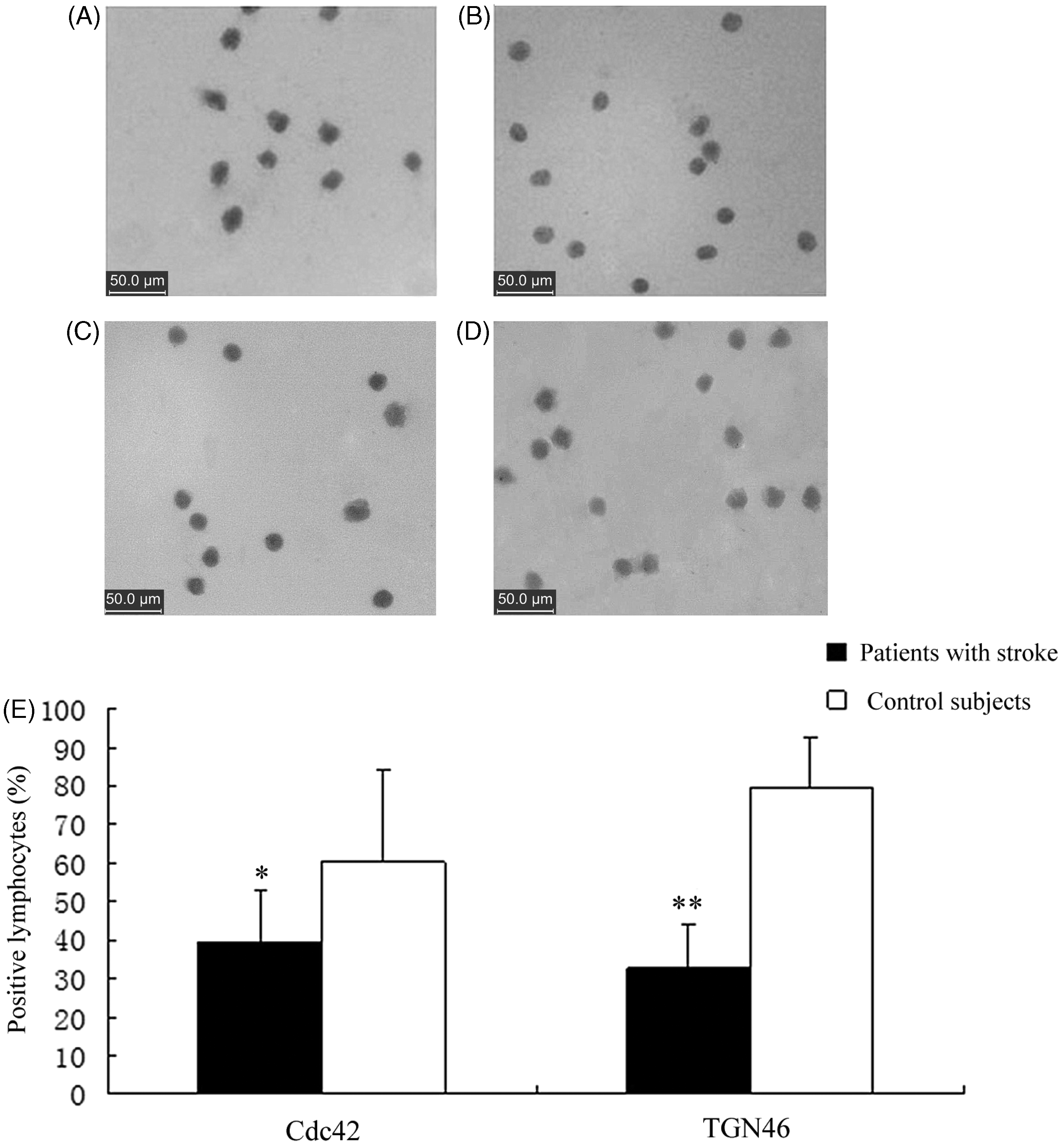


Discussion
The present study measured levels of Cdc42 protein in peripheral lymphocytes from patients in the acute stage following ischaemic stroke, using immunocytochemical staining and Western blotting. Immunofluorescent staining demonstrated that Cdc42 protein was mainly located in the cytoplasm and around the nucleus in lymphocytes, which is consistent with previous findings. 25 During the acute phase of ischaemic stroke (i.e. within 24–72 h of the onset of focal neurological symptoms), both the proportion of Cdc42-positive lymphocytes and the levels of Cdc42 protein by Western blot analysis in peripheral lymphocytes were decreased, compared with similar findings in healthy control subjects. These findings support the hypothesis that decreased levels of Cdc42 protein in peripheral lymphocytes may be involved in the pathogenesis of acute ischaemic stroke.
Lymphocyte infiltration is essential for the inflammatory response that follows ischaemic stroke, and plays an important role in the development of the ischaemic attack. 3 Increasing evidence suggests that Cdc42 – a member of the Rho GTPase family related to the integrin family of cell adhesion molecules – has important effects on lymphocyte infiltration. 26 Leucocyte function-associated antigen (LFA)-1, defined as a member of the β-integrin family, consists of two chains: CD11a and CD18. 27 LFA-1 is found on the surface of lymphocytes and binds to intercellular adhesion molecule-1 on endothelial cells, thereby playing a key role in lymphocyte infiltration. 27 Studies have demonstrated that levels of LFA-1 protein on peripheral lymphocytes from acute stroke patients were upregulated, which contributed to cell infiltration and migration.25,28,29 Research has demonstrated that Cdc42 signals inhibited the affinity of LFA-1 on lymphocytes by regulating their downstream effectors, such as phospholipase D1 and phosphatidylinositol-4-phosphate 5-kinase isoform 1γ. 17 The finding from our current study, that levels of Cdc42 protein were decreased in peripheral lymphocytes from patients experiencing acute stroke, suggests that this change might then result in increased expression of LFA-1 and, thus, a increased facilitation of lymphocyte infiltration during the acute stage of ischaemic stroke.
In addition to facilitating lymphocyte infiltration, the decreased levels of Cdc42 protein in peripheral lymphocytes might also affect immune-synapse formation and production of lymphokines and antibodies. An immune synapse is formed when antigen-presenting cells present specific antigens to T lymphocytes. 30 It is believed that during immune synapse formation, Cdc42 acts as a crucial signal that is involved in controlling actin polarity and cytoskeletal dynamics.31,32 A deficiency or reduction in the levels of Cdc42 protein in lymphocytes might result in impaired immune synapse formation. Hug et al. 19 demonstrated that Cdc42-deficient B lymphocytes produced fewer related antibodies, and lower levels of T lymphocyte-related IgG and IgM. Reduced production of antibodies and T lymphocyte-related IgG and IgM may lead to immune deficiency in patients experiencing acute stroke: a phenomenon that corresponds with clinical findings, which demonstrate that patients in the acute stage following ischaemic stroke often have low levels of resistance to infection. 33 Therefore, decreased levels of Cdc42 protein in lymphocytes may be one of the factors that lead to a heightented rate of infection, in patients with acute stroke.
The Golgi apparatus is crucial to the activation of members of the Rho GTPase family.20–22 As one of the major organelles involved in cell metabolism, the Golgi apparatus is responsible for the processing, trafficking and secretion of some proteins (such as cytokines and antibodies), and also for cellular signal transduction. 23 Research has established that dysfunction of the Golgi apparatus could lead to the abnormal secretion of Ras protein (another member of the Rho GTPase family) in peripheral lymphocytes from patients with systemic lupus erythematosus, thus causing immunological abnormalities. 22 Like Ras, Cdc42 can also be affected by Golgi apparatus function. The cellular location of Cdc42 establishes that Cdc42 is the first member of the Rho GTPase family to be found in the Golgi apparatus, which is regulated by Golgi apparatus-related proteins, such as ‘Arf GAP with Rho GAP domain, ankyrin repeat, and PH domain 1’ (ARAP-1) and ‘A-kinase anchor protein 450’ (AKAP-450).20– 22 In our current study, the function of the Golgi apparatus in peripheral lymphocytes was found to be impaired in patients in the acute stage following ischaemic stroke, as demonstrated by both the decreased proportion of TGN46-positive lymphocytes and the decreased levels of TGN46 protein by Western blot analysis. Furthermore, the decreased levels of Cdc42 protein in lymphocytes correlated positively with the decreased levels of TGN46 protein, indicating that the reduction of Cdc42 protein might result from the impaired function of the Golgi apparatus in peripheral lymphocytes. Whether impaired Golgi apparatus function causes a decrease in Cdc42 protein levels in lymphocytes through ARAP-1, AKAP-450 or other pathways following ischaemic stroke remains to be established, however.
In conclusion, this current study demonstrated that: (i) the percentage of Cdc42-positive and TGN46-positive lymphocytes, and levels of Cdc42 and TGN46 proteins, were significantly decreased in peripheral lymphocytes collected from patients during the acute stage of ischaemic stroke compared with healthy control subjects; (ii) that there was a significant positive correlation between levels of Cdc42 protein and levels of the Golgi apparatus-related protein TGN46 during this process. These observations suggest that cerebral ischaemia downregulates Cdc42 production, which is possibly caused by structural or functional modifications in the Golgi apparatus, thus contributing to cell infiltration and protein production. In order to elucidate the role of Cdc42 in cerebral ischaemia, further studies are required to determine how the Cdc42 protein interacts with the Golgi apparatus-related proteins, and how it affects lymphocyte activity and function of lymphocytes. These findings are expected to contribute to the development of Rho GTPase-targeted drugs, which might have therapeutic benefits in patients with acute ischaemic stroke.
Footnotes
Declaration of conflicting interest
The authors declare that there are no conflicts of interest.
Funding
This work was funded by the National Natural Sciences Foundation of China (no. 81171239).
Acknowledgement
We give our sincere thanks to Dr Xiao Han and Dr Wang Peng (Department of Neurology, Second Xiangya Hospital, Central South University, Changsha, Hunan Province, China) for their work on evaluating the numbers of positively-stained cell numbers, using immunocytochemical methods.