
Abstract
Background
Ribbing disease, or multiple diaphyseal sclerosis, is a rare benign bone dysplasia.
Purpose
To systematically review the literature to determine the clinical and radiological presentation of patients with Ribbing disease as well as the effects of attempted treatments.
Material and Methods
We considered individual patient data of patients diagnosed with Ribbing disease derived from patient reports and patient series. All stages of the review were performed by two reviewers independently. Standard descriptive statistics were used for quantitative analyses and mixed model analyses were used when appropriate
Results
The literature search yielded 420 unique hits of which 23 studies were included, covering a total of 40 patients of whom 29 had bilateral involvement. The mean age at diagnosis was 35 years and the mean time between diagnosis and onset of symptoms, mostly pain, was five years (range = 1–16 years). The tibial diaphysis was the most commonly involved bone in 35 of 36 patients. Non-surgical treatment consisted of non-steroidal anti-inflammatory drugs (NSAIDs), prednisone, and bisphophonates with mixed results. Surgical treatment consisted of intramedullary reaming and fenestration and was very effective to reduce pain.
Conclusion
The clinical presentation and imaging findings of patients with Ribbing disease are becoming more apparent. However, there is paucity of evidence on the natural disease progression and effectiveness of treatment modalities.
Keywords
Introduction
Ribbing disease, or multiple diaphyseal sclerosis, is a rare benign bone dysplasia first described by Ribbing in 1949 (1). It is characterized by sclerosing bone lesions in the diaphyses of long bones in adult patients (1–4). The lower extremities are most often affected and the disease is usually asynchronous when multiple bones are involved. Some authors estimate that in the whole literature only 20–30 cases have been reported (3,4). Since its occurrence is so rare and due to lack of knowledge of this infrequent disease, the diagnosis is often delayed and may be confused with other sclerosing bone dysplasias, metabolic diseases, or even osteomyelitis (3–5). Hence, most of the time Ribbing disease is diagnosed by exclusion. Presently, no formal systematic review on Ribbing disease exists. The purpose of this study was to systematically review the literature to determine the clinical and radiological presentations of patients with Ribbing disease as well as the effects of attempted treatments.
Material and Methods
We performed a systematic review on individual patient data of patients diagnosed with Ribbings disease (i.e. multiple diaphyseal sclerosis) derived from patient reports and patient series. During all stages of the review process, a referee (PD), professor of orthopedic surgery with over 16 years of experience in musculoskeletal oncology, was available for consultation. The reporting of this systematic review is in accordance with the PRISMA guidelines (6).
Literature search
A thorough literature search was performed together with a medical librarian (JS), experienced in the field of orthopedics, in order to increase the likelihood of retrieving all relevant studies (7). The following bibliographies were searched up to November 2015: PubMed, MEDLINE, Embase, Web of Science, COCHRANE, CENTRAL, CINAHL, Academic Search Premier, ScienceDirect, Wiley, LWW, HighWire, PubMedCentral, and Google Scholar. References of included articles were screened for relevant studies. Articles in English, French, Italian, Spanish, Dutch, and German were considered. The search strategy consisted of the following components, each defined by a combination of controlled vocabulary and free text terms: (i) Ribbing disease; and (ii) multiple diaphyseal sclerosis.
See Appendix 1 for more details on the strategy and terms.
Inclusion and exclusion analysis
Initial screening on the basis of title and abstract was performed by two reviewers independently and in duplicate (BS and KS) to identify studies of patients diagnosed with Ribbing disease. When the information in the abstract did not suffice or when there was any doubt, the studies remained eligible. The full text of eligible studies was subsequently evaluated in duplicate by two reviewers (BP and KS) independently. Both recorded their findings in a pre-designed electronic database. Any disagreements were resolved by consensus or by consulting a referee. All bibliographic records identified through the electronic searches were collected in an electronic reference database and subjected to the following inclusion criteria: (i) patient report or patient series of patients diagnosed with Ribbing disease/multiple diaphyseal sclerosis; and (ii) clinical data on diagnosis and/or treatment.
Data extraction
Data were extracted independently by two reviewers (BP and KS) using a pre-defined electronic data collection sheet. Data consisted of study characteristics, patient demographics, diagnostic findings, and clinical outcome. The data sheet was designed during the extraction of trial data on a random sample of eligible studies. Any disagreements were resolved by consensus or by consulting a referee.
Data synthesis and analysis
Since this systematic review deals with individual patient data from patient reports and patient series standard, descriptive statistics were used for quantitative analyses and mixed model analyses were used when appropriate (8). We checked for duplicate patients by comparing gender, age, bones effected, authors, country, and treatment on a case-by-case basis.
Results
The literature search yielded 420 unique hits of which 23 studies were included, covering a total of 40 patients (eight women, 32 men) (Fig. 1, Suppl. Table 1) (1–4,9–27). There were no duplicate patients identified. Nine studies originated from North America comprising 15 patients (2,9,11,19,20,22,24,25,27). Seven studies originated from Asia, comprising 11 patients (3,10,12–14,21,26). Six studies originated from Europe, comprising 13 patients (1,4,15–18). One study originated from South America, comprising one patient (23).
Prisma flow chart.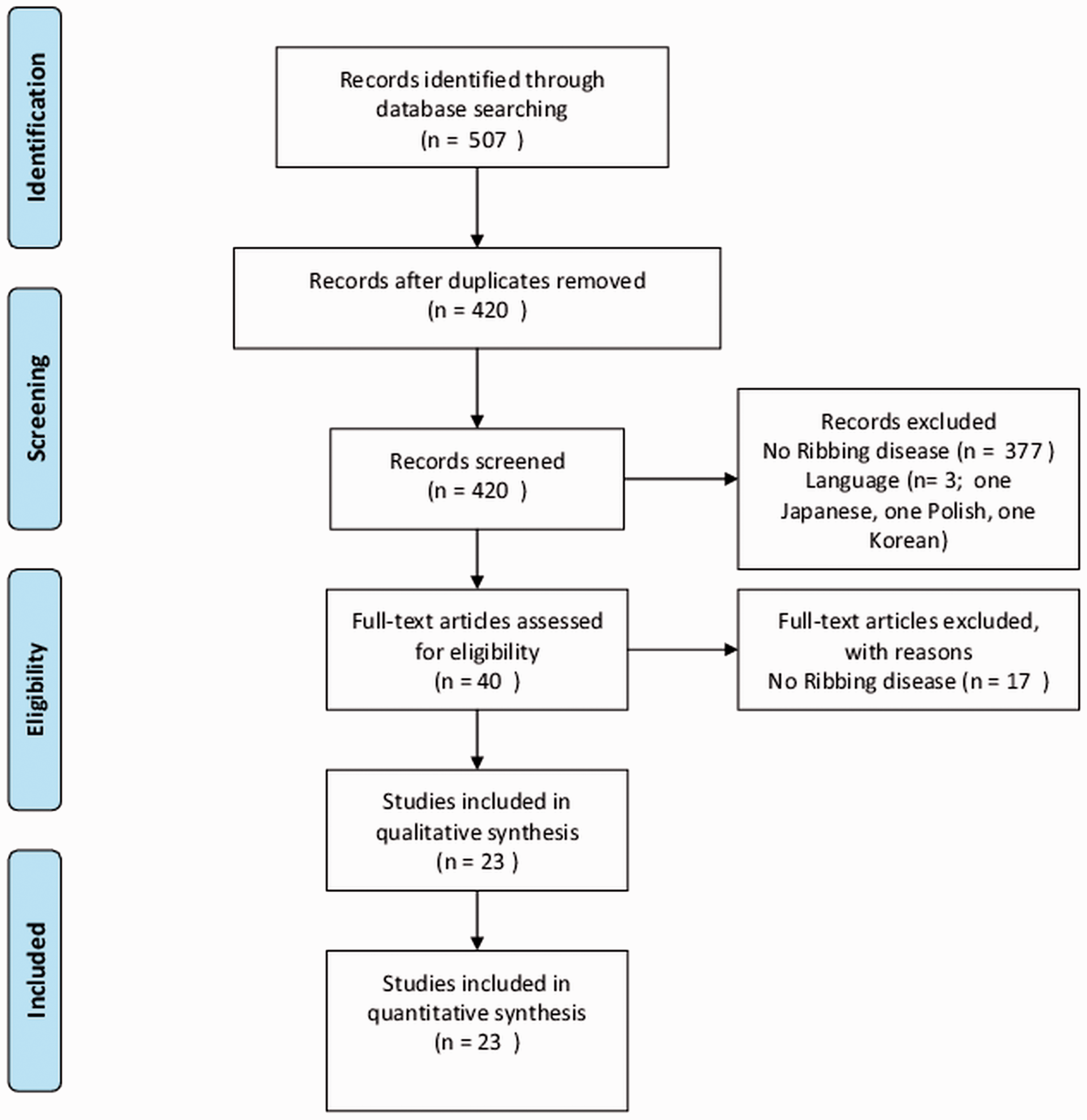
Clinical presentation
In 30 of 33 patients, pain (diaphyseal) was the presenting sign. Five of eight patients also suffered from fatigability and seven of 15 patients experienced muscle weakness. The mean age at diagnosis was 35 years (SD = 13 years). The mean age at which the symptoms began was 30 years (SD = 12 years). The mean time between diagnosis and onset of symptoms was five years (range = 1–16 years). For 27 patients, the family history was reported. The family history was negative for (diaphyseal) musculoskeletal complaints or Ribbing disease in 13 patients and positive in 14 patients.
Imaging
X-ray examinations showed increased bone density (sclerosis) at the diaphysis with cortical endosteal and periosteal thickening (Figs. 2 and 3). In 25 patients, a bone scan (Technetium 99) was performed, which had an increased uptake in 24 patients and normal in one patient. In 11 patients, a computed tomography (CT) scan was performed, which showed periosteal and endosteal thickening with narrowing of the intramedullary canal (2,3,11,12,14,16–18,20,27). In addition to the CT scans, magnetic resonance imaging (MRI) was performed in 13 patients, which showed endosteal marrow edema and no soft tissue involvement of the lesion (2,4,10–14,17,27).
Anteroposterior X-ray of lower leg, CT lower leg coronal image, and MRI lower leg coronal images T1 and T2 showing typical presentation of Ribbing disease. Lateral X-ray of lower leg, CT lower leg axial image, and MRI lower leg axial image showing typical presentation or Ribbing disease.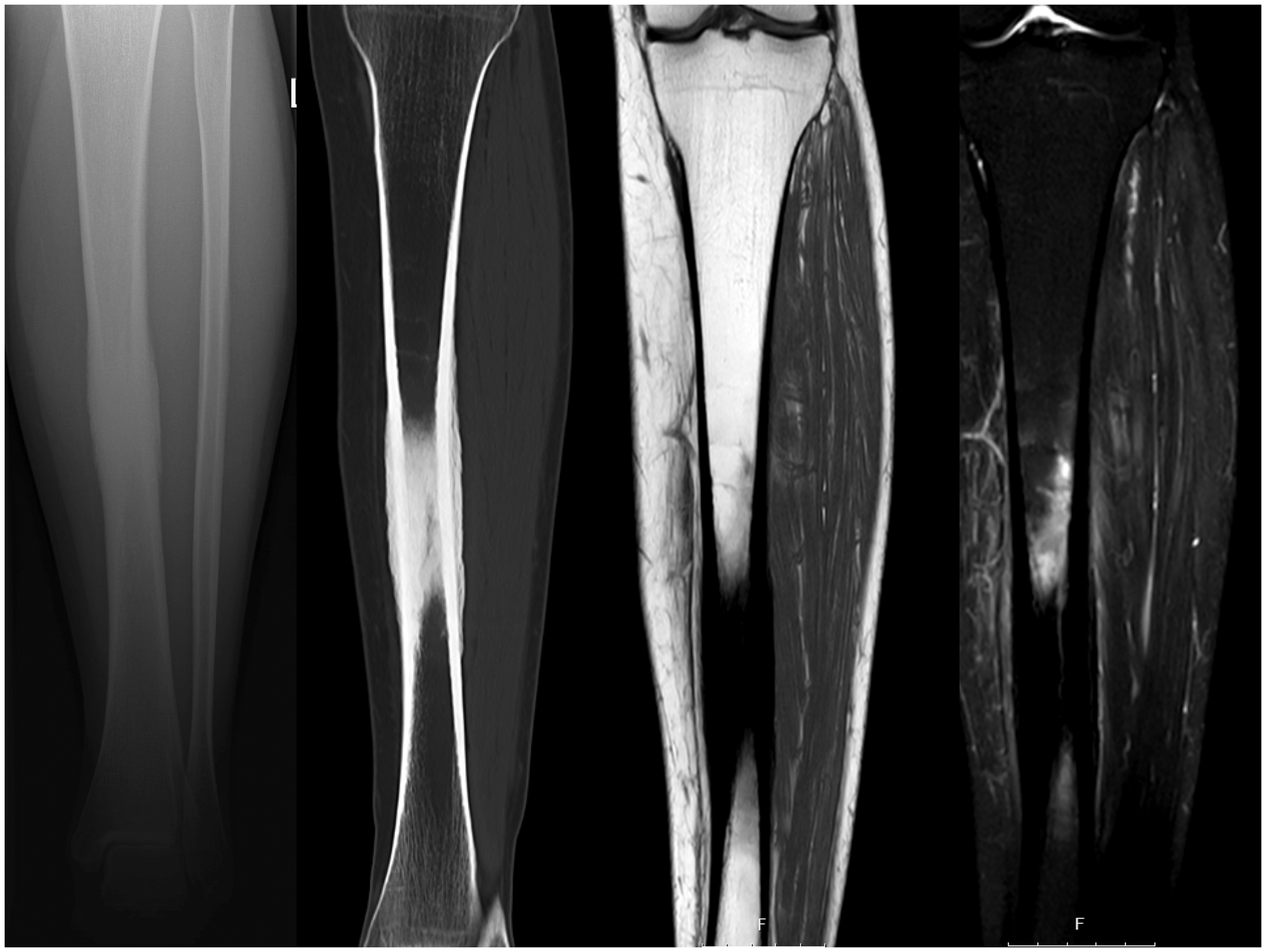
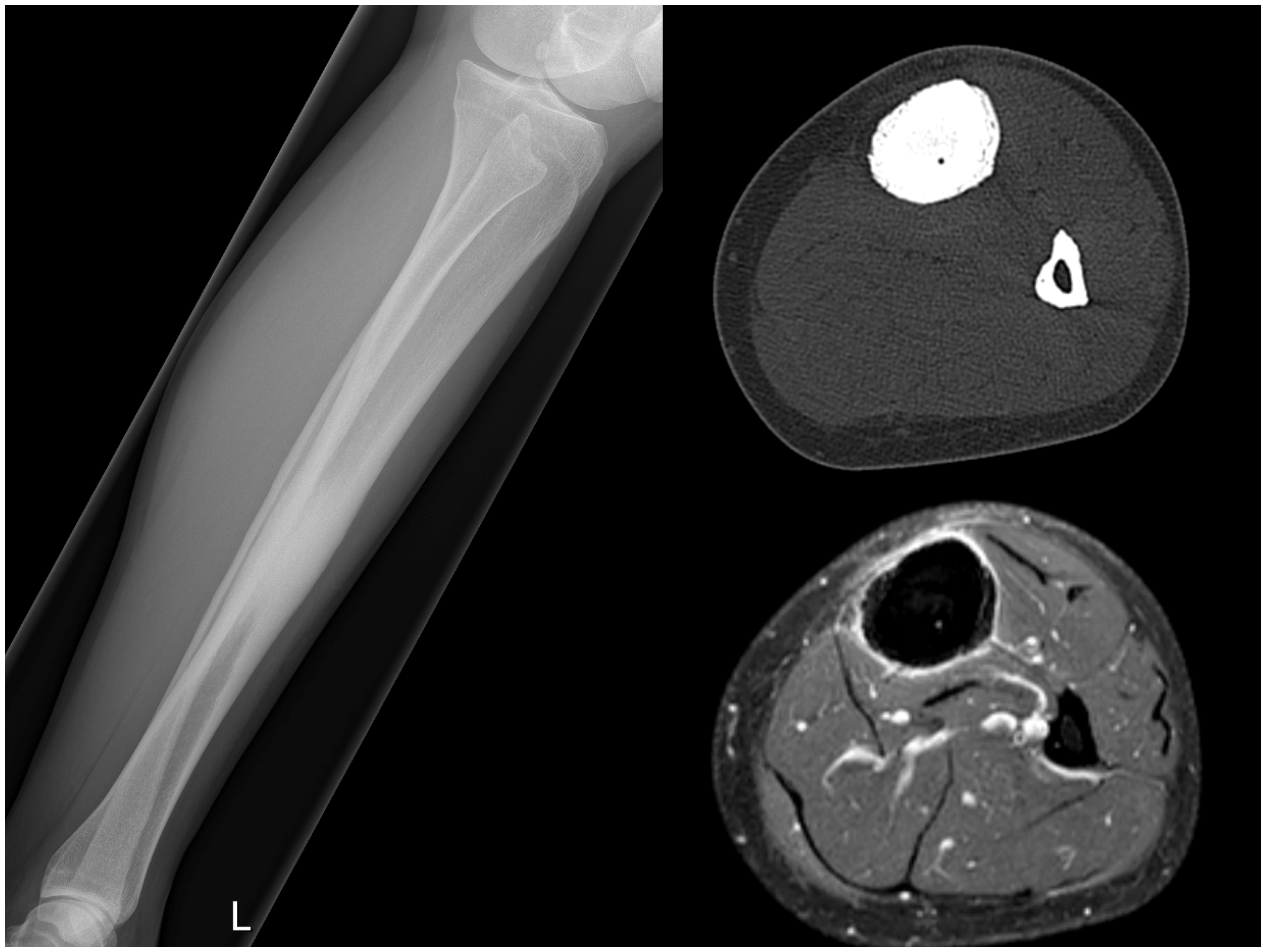
Laboratory findings
In 17 patients, laboratory findings of whole blood were reported. In 16 of 17 patients, the erythrocyte sedimentation rate (ESR) was normal. In 16 of 16 patients, white blood cell count was normal. In 13 of 14 patients, alkaline phosphatase was normal. Cultures of microorganisms were negative in all reported cases except in one (25) where S. Epidermidis was grown and considered contamination because subsequent cultures were negative and no wound infection occurred (2,3,12,15–17,19,20,25).
Pathology
In 24 cases, histopathologic examinations were performed, which described osteosclerosis and foci with woven bone, mild osteitis, chronic osteoperiostitis, thickened trabeculae of lamellar bone with various sizes of the Haversian system, cortical thickening with fibrosis, new bone formation with non-specific changes, new bone with unusually wide trabeculae, and reactive cortical thickening (1–4,9,10,12–17,19,20,22,25).
Osseous involvement
In all patients, lesions were restricted to the diaphyses: there was no involvement of the metaphyses or epiphyses or progression to the metaphyses or epiphyses reported. In 29 of 37 patients, there was bilateral involvement of the bones. In 31 of 37 patients, more than one bone was effected. In 30 of these 31 patients, the stages of the disease were asynchronous. On average 2.8 bones were affected by the disease (range = 1–8 bones). Supplementary Table 1 shows a breakdown on anatomical location. The tibia was the most commonly involved bone in 35 of 36 patients. The femur was the second most involved bone with 14 of 33 patients. The humerus was the least commonly involved bone in two of 28 patients.
Treatment
Non-surgical treatment consisted of non-steroidal anti-inflammatory drugs (NSAIDs), prednisone and bisphophonates. In three of 12 patients, NSAIDs were effective. In two of three patients, prednisone was effective. In two of eight patients, bisphophonates were effective. Surgical treatment consisted of intramedullary reaming and fenestration. Four studies reported on intramedullary reaming, comprising four patients and five bones: four tibias and one femur (2,3,15,17). All the patients were pain-free at last follow-up at mean of 3.4 years (range = 1–5 years). One complication was reported of perforating the tibial cortex (false route from intramedullary reaming), which was treated with a non-weight-bearing cast for six weeks (2). Two studies reported on surgical fenestration, comprising seven patients (12,25). Seeger et al. reported on treatment of six patients with few details on anatomical location, outcome, and follow-up (25). Zhang et al. reported on one patient who underwent fenestration of the femur (12). This patient was pain-free at the eight-month follow-up.
Exploratory analysis
There were significantly more bones effected by the disease if the upper extremity was involved (4 versus 2.6 bones, P = 0.013). With the numbers available, we found no associations between age, gender, time to diagnosis, and total bones effected.
Discussion
The results of the systematic review showed that the characteristic patient with Ribbing disease is a 35-year-old woman with symptoms for five years including diaphyseal pain in the lower extremities. The X-rays show bilateral, asynchronous increased and typical sclerosis at the diaphysis with cortical endosteal and periosteal thickening of on average 2.8 bones. The bone scintigraphy has an increased uptake. ESR, whole blood count, and alkaline phosphatase are within normal ranges. Cultures for microorganisms are negative and histology has ruled out malignancy.
The clinical presentation of Camurati-Engelmann disease resembles that of Ribbing disease (1,3,4). However, contrary to Ribbing disease, Camurati-Engelmann disease involves osteosclerosis of the skull base (56.5% of cases), the mandible (25% of cases), symmetry of bone involvement, and the symptoms may start during childhood (1,3,4,28). Unlike Ribbing disease, Camurati-Engelmann disease may show progression into the metaphyses (29). Camurati-Engelmann disease is associated with physical disability due to gait and neurological abnormalities (1,3,4). Furthermore, Camurati-Engelmann is continuously progressive whereas Ribbing disease may become static (1,3,4). There seems to be some genetic overlap: Savoie et al. reported two patients with a known missense mutation in exon 2 of TGFβ1 (4). This mutation has also been found in patients with Camurati-Engelmann (30). Makita et al. identified the Ribbing disease phenotype in a three-generation Japanese family with Camurati-Engelmann or progressive diaphyseal dysplasia and subsequently proposed that Camurati-Engelmann and Ribbing disease represent phenotypic variation of the same disorder (21).
There are also other differential diagnoses that should be considered, such as the group of sclerotic bone dysplasias that are more centered around the skull, but that may involve the peripheral skeleton (van Buchem’s disease, Worth disease, Nakamura disease, Truswell-Hansen, craniodiaphyseal dysplasia, Bakwin-Eiger syndrome), diaphyseal dysplasia with anemia (Ghosal hemato type), Gaffey disease, osteopetrosis group, overlap syndromes, multifocal periostitis, prostaglandin-induced hyperostosis, Fluorosis, hypervitaminosis A, intramedullary sclerosis, osteomyelitis, chronic recurrent multifocal osteomyelitis (CRMO), osteosarcoma, osteoid osteoma, and stress fracture, among others (25).
There is a paucity of evidence regarding treatment. NSAIDs, prednisone, bisphosphonates, and surgical treatment all have been attempted with few data available for each treatment modality. There is also the potential of publication bias and reporting bias as non-effective treatments may not be published or reported. It is therefore likely that the “effect” of the treatment is overestimated and that we are actually looking at the natural disease progress. Nevertheless, it appears that elevated intramedullary pressure may contribute to the experience of pain since surgical relief of the pressure by either reaming or fenestration has an immediate effect on pain with dramatic reduction reported from visual analogue scale for pain (VAS) preoperative of 9 to VAS postoperative of 0 (2,15).
The above clearly illustrates that more evidence is needed on the effectiveness of treatments for patients with Ribbing disease. Improved reporting could be helpful by including pre-intervention and post-intervention VAS pain scores.
Another area for improvement in treating patients with Ribbing disease is reducing the time to diagnosis. Patients suffer from a long period of uncertainty as it takes a mean of five years from the onset of symptoms to make the diagnosis, and can even take 16 years in the most extreme case (4,10,12,20). Delay in diagnosis in turn delays proper treatment which could affect quality of life. A bone scan is paramount to determine the number of affected bones and to determine the best location to obtain tissue samples and microbiological cultures to help differentiate between malignancy and osteomyelitis.
The fact that there were significantly more bones affected by the disease if the upper extremity was involved suggests more advanced disease progression when the upper extremity is involved, i.e. it starts with the lower extremities and may progress to the upper extremities. If a random distribution of the disease was assumed, then no difference between number of involved bones would have been found.
We should consider some limitations. This review comprises 40 patients which a small number. Nevertheless, this number is significantly more than 20–30 cases as estimated by some recent studies (3,4). Also, publication bias and reporting bias could affect the findings particularly regarding treatment.
In conclusion, the clinical presentation and imaging findings of patients with Ribbing disease are becoming more apparent. However, there is a paucity of evidence on the natural course of disease regarding the progression and effectiveness of treatment modalities. Future studies could therefore benefit from improved reporting with emphasis on treatment effects.
Footnotes
Acknowledgments
The authors thank the reviewer for critically revising the manuscript, especially regarding the differential diagnosis.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.