
Abstract
Post-stroke intra-infarct repair promotes peri-infarct neural reorganization leading to functional recovery. Herein, we examined the remodeling of extracellular matrix proteins (ECM) that constitute the intact basal membrane after permanent middle cerebral artery occlusion (pMCAO) in mice. Among ECM, collagen type IV remained localized on small vessel walls surrounding CD31-positive endothelial cells within infarct areas. Fibronectin was gradually deposited from peri-infarct areas to the ischemic core, in parallel with the accumulation of PDGFRβ-positive cells. Cultured PDGFRβ-positive pericytes produced fibronectin, which was enhanced by the treatment with PDGF-BB. Intra-infarct deposition of fibronectin was significantly attenuated in pericyte-deficient Pdgfrb+/− mice. Phagocytic activity of macrophages against myelin debris was significantly enhanced on fibronectin-coated dishes. In contrast, laminin α2, produced by GFAP- and aquaporin 4-positive astrocytes, accumulated strongly in the boundary of peri-infarct areas. Pericyte-conditioned medium increased the expression of laminin α2 in cultured astrocytes, partly through TGFβ1. Laminin α2 increased the differentiation of oligodendrocyte precursor cells into oligodendrocytes and the expression of myelin-associated proteins. Peri-infarct deposition of laminin α2 was significantly reduced in Pdgfrb+/− mice, with attenuated oligodendrogenesis in peri-infarct areas. Collectively, intra-infarct PDGFRβ-positive cells may orchestrate post-stroke remodeling of key ECM that create optimal environments promoting clearance of myelin debris and peri-infarct oligodendrogenesis.
Introduction
Reperfusion therapy with intravenous administration of recombinant tissue-plasminogen activator or catheter-mediated endovascular thrombectomy has been established as the most effective neuroprotective and neurorestorative therapy for acute ischemic stroke.1,2 A potential therapeutic target for acute ischemic stroke is enhancement of neurorestoration with endogenous functional recovery during subacute phase. Although functional recovery occurs with rehabilitative therapy in most stroke patients until approximately 3 months after onset, its extent varies among individuals with comparable infarct size and localization. Evidence suggests that macrophage-mediated clearance of intra-infarct debris is an important factor promoting post-stroke functional recovery.3,4 We have demonstrated that microvascular platelet-derived growth factor receptor β (PDGFRβ)-positive pericytes crucially participate in post-stroke restoration of blood flow and subsequent recruitment of monocytes/macrophages within infarct areas through the production of chemokines such as monocyte chemoattractant protein-1 (MCP1)/C–C motif chemokine ligand 2 (CCL2). 3 Recruited macrophages remove myelin debris (MD) with the aid of neighboring pericytes, while promoting intra-infarct fibrotic responses mediated by PDGFRβ-positive cells. The clearance of debris and tissue repair within infarct areas, achieved by the close interaction of pericytes and macrophages, enhances peri-infarct neural reorganization, including astrogliosis and oligodendrogenesis, facilitating functional recovery.3,5–7 Thus, creation of an optimal environment to support these intercellular interactions and functions in intra-infarct and peri-infarct areas is necessary to promote post-stroke functional recovery.
Extracellular matrix proteins (ECM) can modulate cellular functions through direct interaction with adhesive molecules expressed on cellular membranes. 8 In the brain, endothelial cells, pericytes, and astrocytes produce ECM, such as collagens, laminins, fibronectin, and perlecan, and constitute the basal membrane (BM), a crucial component of the blood–brain barrier (BBB).9–12 Since BBB is disrupted by degradation of ECM in ischemic areas, they have to be suitably remodeled to promote tissue repair and functional recovery.13,14 However, it is not clear how the production of ECM is regulated after acute ischemic stroke for post-stroke tissue repair and functional recovery. 14 In this study, we examined the remodeling of ECM and their roles in tissue repair and functional recovery using a permanent middle cerebral artery occlusion (pMCAO) model in mice.
Materials and methods
Animals
Animal experiments were conducted in accordance with the Guidelines for Proper Conduct of Animal Experiments by the Science Council of Japan (June 1, 2006) (http://www.scj.go.jp/ja/info/kohyo/pdf/kohyo-20-k16-2e.pdf). The Animal Care and Use Review Committee of Kyushu University approved the experimental procedures (Fukuoka, Japan) (A29-153) and the study has been reported according to the ARRIVE guidelines. 3 129S Pdgfrb+/− mice were purchased from Jackson Laboratory (Bar Harbor, ME, USA; https://www.jax.org/strain/007846) and were backcrossed ten times with C57BL/6 mice to produce C57BL/6 Pdgfrb+/− mice. 5 We used male and female Pdgfrb+/− mice and their wild-type littermates aged 8–15 weeks, weighing 20–30 g of male and 17–22 g of female mice (supplemental Table I). Mice were housed two per cage in the animal facility of Kyushu University, which was maintained at 21°C, 65% humidity, 12-h light–dark cycle, with ad libitum access to food and water. 3
Mouse stroke model
Mice were randomly assigned to the animal surgeon, anesthetized by inhalation of 2% isoflurane in air, and maintained on anesthesia using 1.5% isoflurane. Rectal temperatures were maintained at 35–37°C using a heating lamp. Cerebral blood flow before and during ischemia was measured at the ipsilateral parietal bone (2 mm posterior and 4 mm lateral to bregma) using a laser Doppler flowmeter (PeriFlux System 5000, PERIMED, Stockholm, Sweden). Focal cerebral ischemia was induced by pMCAO using a laser-induced photochemical reaction as described previously. 5 The number of mice used in each experiment is shown in Supplemental Table I.
Behavioral tests
Neurological tests were performed pre-MCAO and 1, 7, 14, 21, and 28 days after MCAO. Motor coordination was evaluated in a quantitative manner using a beam balance test. The apparatus was the surface of an elevated wooden beam ((90 cm (L) × 2 cm (W) × 2 cm (H)) at a height of 10 cm from the ground. The number of times that each animal fell off the beam was recorded, and the performance was rated on a scale of 0 to 6 (Supplemental Table II). Three trials were performed on each animal with an interval of 5 min between attempts. The final beam balance score was defined as the mean of the scores received in the three trials.
Quantitative polymerase chain reaction
Total RNA was isolated from the ischemic area or cultured cells using TRIzol reagent (Thermo Fisher Scientific #15596018, Waltham, MA, USA). Reverse-transcription polymerase chain reaction (PCR) and quantitative real-time PCR were performed as described previously. 5 The mRNA copy numbers were standardized using 18 s ribosomal RNA as internal control. Primer sequences are presented in Supplemental Table III.
Immunoblot analysis
Immunoblot analysis was performed as described previously. 5 Briefly, membranes were incubated at 25°C for 1 h in ECL Advance blocking reagent (GE Healthcare #RPN418, Chicago, IL, USA) and probed overnight at 4°C with primary antibodies [p-PDGFRβ (1:500; Cell Signaling Technology (CST) #2227, Boston, MA, USA), PDGFRβ (1:1000; CST #3169), p-STAT3 (1:1000; CST #9145), STAT3 (1:1000; CST #4904), fibronectin (1:500; BD Biosciences #610077, Franklin Lakes, NJ, USA), laminin α2 (1:500; Santa Cruz Biotechnology #sc55605, Dallas, TX, USA), and anti-β-actin (1:2,000; Sigma-Aldrich #A5441, St. Louis, MO, USA)]. Membranes were then washed and incubated with secondary antibodies (1:50000; CST #7074 or #7076) for 45 min at 25°C. Antibodies used are shown in Supplemental Table IV.
Immunohistochemistry
Mice were euthanized by intraperitoneal administration of pentobarbital (150 mg/kg body weight) and perfused with 4% paraformaldehyde (PFA) in phosphate buffered saline (PBS) at 4°C. After fixing the brain overnight in 4% PFA, 2-mm coronal slices embedded in paraffin were cut into 4-μm thick sections. The sections were deparaffinized, rehydrated through a graded series of ethanol solutions, and washed in PBS. 6 After blocking with 5% skimmed milk solution for 30 min at 25°C, sections were incubated with primary antibodies [anti-CD31 (1:200; BD Biosciences #550274), laminin α2 (1:500; Santa Cruz Biotechnology #sc59854), laminin α4 (1:500; Santa Cruz Biotechnology #sc130541), fibronectin (1:500; BD Biosciences #610077), anti-collagen type IV (1:100; Millipore #AB769, Burlington, MA, USA), anti-perlecan (1:250; Millipore #AB1948P), vitronectin (1:100; Santa Cruz Biotechnology #sc74484), anti-glial fibrillary acidic protein (GFAP) (1:2; DAKO #IS524, Santa Clara, CA, USA), anti-CD13 (1:200; R&D Systems #AF2335, Minneapolis, MN, USA), anti-F4/80 (1:100; Abcam #ab6640, Cambridge, UK), anti-PDGFRβ (1:200; R&D Systems #AF1042), anti-aquaporin 4 (AQP4) (1:400; Santa Cruz Biotechnology #sc20812), anti-TGFβ1 (1:100; Santa Cruz Biotechnology #sc130348), anti-APC (1:100; Millipore #OP80), and anti-Glutathione S-transferase π (GSTπ) (1:200; MBL #312, Nagoya, Japan)] at 4°C overnight. After washing with PBS/Triton X-100, the sections were incubated with secondary antibodies conjugated to Alexa Fluor dyes (Thermo Fisher Scientific) or stained with 3,3′-diaminobenzidine (Nichirei, Tokyo, Japan). For with 3,3′-diaminobenzidine staining, endogenous peroxidase was inactivated with 0.3% hydrogen peroxide for 30 min before blocking with skim milk solution. EdU (5-ethynyl-2′-deoxyuridine; Thermo Fisher Scientific #A10044) was used to label the proliferating cells to assess oligodendrogenesis. Intraperitoneal EdU injections (50 mg/kg) were administered once daily for 7 days, beginning on day 1 after pMCAO. Mice were sacrificed on day 28 and perfused with 4% PFA. Brains were removed, 2-mm coronal slices were postfixed for an hour in 4% PFA, and cryoprotected in 30% sucrose overnight. Frozen coronal slices were embedded in optimum cutting temperature compound and sectioned at 4 μm. EdU staining was performed using a Click-iTTM EdU imaging kit (Thermo Fisher Scientific #A10338), as described previously. 5 The sections were observed using a BIOREVO BZ-9000 microscope (Keyence, Osaka, Japan) or an inverted confocal microscope (Nikon A1R, Tokyo, Japan). PDGFRβ, CD13, and fibronectin-positive areas within infarct areas and peri-infarct GFAP- and laminin α2-positive areas were obtained in three sections spaced 400 μm apart (from bregma +1.0 mm to bregma –0.2 mm). To determine the density of positive cells, three randomly selected squares (200 × 200 μm) were analyzed. The density of GSTπ-positive cells in the peri-infarct areas was calculated using three sections spaced 400 mm apart (from bregma +1.0 mm to bregma –0.2 mm). To determine the density of positive cells in the peri-infarct areas, four randomly selected squares (400 × 400 mm) were analyzed. Ten to 14 animals per group were analyzed.
Cell culture
Human brain vascular pericytes (#CP1200) were purchased from ScienCell Research Laboratories (Carlsbad, CA, USA) (https://www.sciencellonline.com/human-brain-vascular-pericyte-cell-pellet.html#product_tabs_review_tabbed) and cultured in pericyte medium containing 2% FBS and pericyte growth supplement (#CP1201, ScienCell Research Laboratories). PDGF-BB (10 nM, Wako #16424031), LY364947 (1 µM, Wako #123-05981), and stattic (6 μM, R&D Systems #19983-44-9) 15 were purchased. Poly-l-lysine (PLL)-coated dishes were purchased from IWAKI (Tokyo, Japan). Bone marrow-derived macrophages (BMDMs) were extracted from male C57BL/6J mice as described previously. 3 Astrocytes, oligodendrocyte precursor cells (OPCs), and microglia were extracted from ICR mice (Japan SLC, Inc) on postnatal day 1, as described previously.5,16 Cells were cultured in an incubator with 5% CO2 at 37°C. To investigate the influence of different ECM substrates, culture dishes were coated with a 5 μg/mL solution of fibronectin (Sigma-Aldrich #F1141) or laminin 211 (BioLamina AB, Sundbyberg, Sweden), diluted in PBS, overnight at 4°C.
Immunocytochemistry
Cells were fixed in 4% PFA and processed for immunostaining. They were then incubated for 30 min at 25°C in a blocking solution (5% FBS and 0.3% Triton X-100). The antibodies used to analyze the marker expression profiles were: anti-F4/80 (1:100; Abcam #ab6640), anti-IBA1 (1:500; Abcam #ab5076), anti-MBP (1:500; BioLegend #808401, San Diego, CA, USA), and anti-OLIG2 (1:200; R&D Systems #AF2418). Macrophage phagocytic activity was evaluated using Oil red O (ORO) staining. 3 Fixed cells were dehydrated in 100% propylene glycol for 5 min and stained with 0.5% ORO solution (Sigma-Aldrich #O0625) at 25°C for 15 min. Samples were processed in 85% propylene glycol for 5 min and rinsed with distilled water three times. Stained cells were visualized using an inverted confocal microscope (Nikon A1R, Tokyo, Japan). Cell counting was performed in a blinded manner by randomly selecting four to six fields in each dish.
Oxygen and glucose deprivation
Oxygen glucose deprivation (OGD) was performed using an airtight hypoxia chamber. Cells were washed once with glucose-free Dulbecco's Modified Eagle Medium (DMEM), then subsequently filled with glucose-free DMEM and incubated at 37°C in a humidified atmosphere with 5% CO2, 1% O2, and 94% N2 for 24 h.
Preparation of myelin debris
MD was isolated from the brains of 3-month-old mice using sucrose density gradient centrifugation, as described previously. 3 MD was used to stimulate BMDMs and microglia at a concentration of 1 mg/mL myelin protein in all experiments.
Preparation of conditioned medium
BMDM-conditioned medium (MCM) was prepared by first treating the cells for 72 h with DMEM supplemented with 15% conditioned medium from L929 cells and 5% FBS, containing PBS or MD (1 mg/mL). Subsequently, debris was removed from the medium by centrifugation at 200 × g for 5 min. Pericytes were treated with MCM and DMEM at a ratio of 1:2 for 10 min or 24 h. Control medium was prepared using DMEM supplemented with 15% conditioned medium from L929 cells and 5% FBS depleted of BMDMs and DMEM at a ratio of 1:2. Pericyte-conditioned medium (PCM) was prepared by incubating pericytes in minimum pericyte medium containing PBS or platelet-derived growth factor-BB (PDGF-BB) and no growth supplement for 48 h. Subsequently, PCM was collected (referred to as PCM/PBS and PCM/PDGF-BB, respectively). Astrocytes were treated with PCM and DMEM, without serum, at a ratio of 1:1 for 24 h. Control medium was prepared using pericyte medium depleted of cultured pericytes and DMEM at a ratio of 1:1.
Adhesion assay
Cell adhesion assays were performed according to a previously established method. 11 A 96-well cell culture plate was coated with the adhesion molecule overnight at 4°C and blocked with 1% heat-denatured bovine serum albumin at 25°C for 30 min. Cells were preincubated by rotating gently with anti-integrin β1 blocking antibody (BD Biosciences #555003) or control antibody (Armenian Hamster IgM, BD Biosciences #553958) at 50 μg/mL at 37°C for 1 h. Cells were spread at a final concentration of 4 × 104 cells/well and incubated at 37°C in 5% CO2 incubator for 30 min. After washing twice with 0.1% bovine serum albumin in DMEM, cells were fixed with 4% PFA and stained with 0.5% crystal violet (Sigma-Aldrich). The dye was extracted with 2% SDS and the optical density was measured at 590 nm.
In vitro OPC differentiation assay
Primary OPCs were cultured with OPC differentiation medium (N2 medium containing 50 ng/mL of both T3 and T4, Sigma-Aldrich #T2877 and #T2376) on PLL with or without laminin 211 (5 μg/mL)-coated dishes for 5 or 7 days. The medium was replaced every two days.
Experimental design and statistical analysis
Statistical analyses were performed using Prism 8 (GraphPad Software). The Kolmogorov–Smirnov normality test was initially performed on all datasets. The statistical analyses were performed using Student’s t-tests (for two-group comparisons) and one-way analysis of variance, followed by a post-hoc Bonferroni’s comparison test (for multiple group comparisons), as appropriate. All data are presented as mean ± standard deviation. All p values <0.05 were considered significant.
Results
Post-stroke remodeling of basement membrane ECM proteins
We examined post-stroke remodeling of ECM that constitute the BM in the intact brain, such as collagen type IV, perlecan, fibronectin, vitronectin, and laminin α2 and α4 in male mice.9,10 We confirmed that these ECM were localized in the BM surrounding CD31-positive endothelial cells of the intact brain (Figure 1A and Supplemental Figure I). Following pMCAO, collagen type IV (Figure 1(b) and (c)) and perlecan (Supplemental Figure I) remained localized on small vessel walls surrounding CD31-positive cells within infarct areas and in intact areas (Figure 1(d)), while fibronectin (Figure 1(b) and (c)) and vitronectin (Supplemental Figure I) redistributed gradually within the infarct areas from peri-infarct areas towards the ischemic core, and eventually occupied the entire infarct areas over 28 days. Their localization was very similar to that of PDGFRβ, a marker of pericytes, pericyte-derived fibroblast-like cells, and smooth muscle cells (Figure 1(d)). In contrast, perivascular laminin α2 gradually decreased within infarct areas; instead, it deposited strongly at the boundary between infarct and peri-infarct areas on day 28 (Figure 1(b) and (c)). Laminin α4 was localized in infarct areas and peri-infarct areas (Supplemental Figure I).
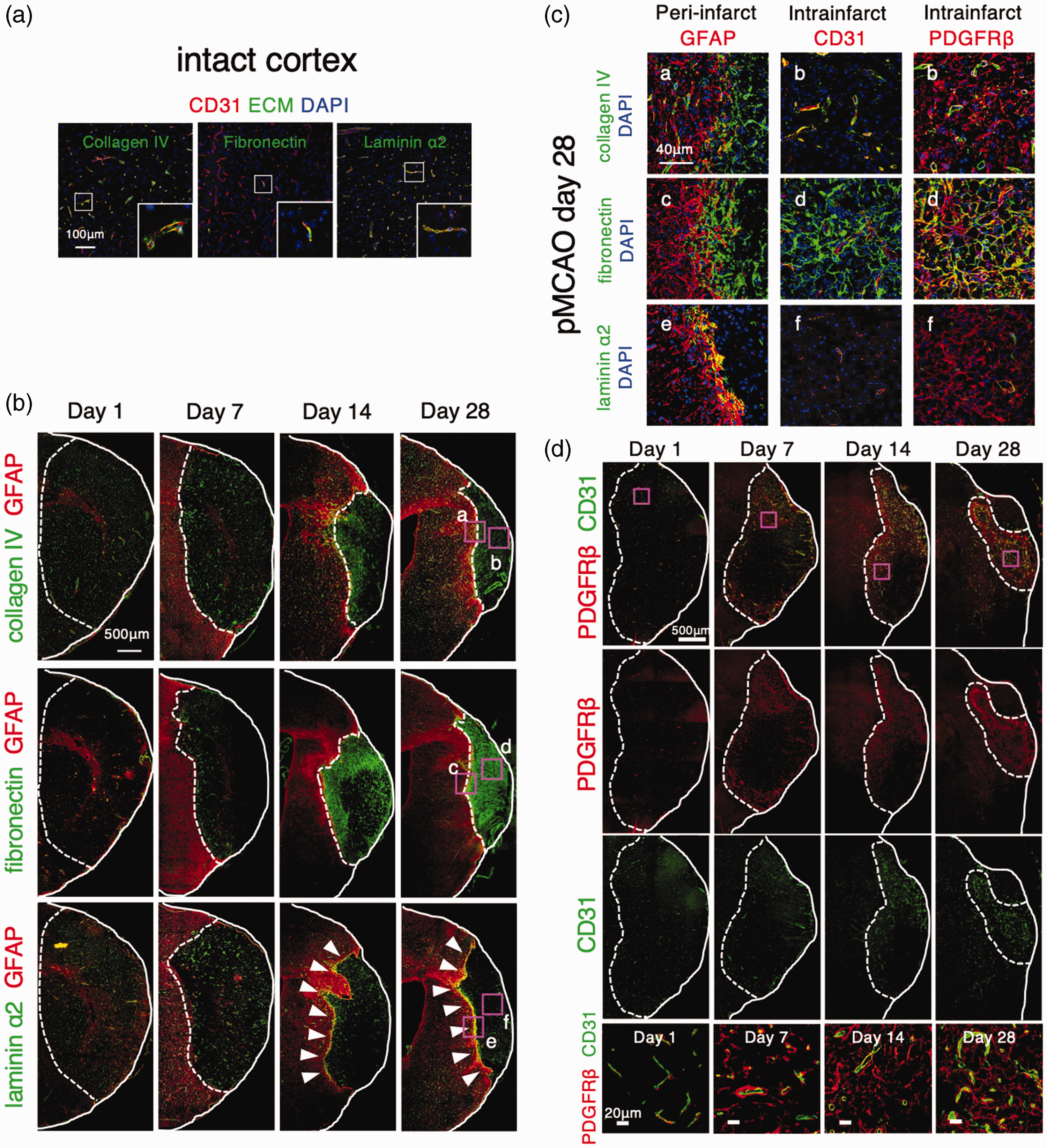
Remodeling of extracellular matrix (ECM) proteins after acute ischemic stroke. (a) Immunofluorescent triple labeling of CD31 (red), ECM proteins, either collagen type IV, fibronectin, or laminin α2 (green), and DAPI (4′,6-diamidino-2-phenylindole; blue) in the intact brain of male wild-type mice (scale bar, 100 μm). Magnified images in the squares are shown in the inset. (b) Immunofluorescent double labeling of GFAP (glial fibrillary acidic protein; red) and ECM, either collagen type IV, fibronectin, or laminin α2 (green), on day 1, 7, 14, and 28 after permanent middle cerebral artery occlusion (pMCAO) in male wild-type mice (scale bar, 500 μm). Magnified images of the square in the ischemic border areas (a, c, e) and the ischemic core (b, d, f) on day 28 are shown in (c) Arrowheads show laminin α2 accumulation in the boundary between infarct areas and peri-infarct astrogliosis. c, Immunofluorescent triple labeling of GFAP (red), collagen type IV (a), fibronectin (c), or laminin α2 (e) (green), and DAPI (blue) in peri-infarct area on day 28 after pMCAO in male wild-type mice (scale bar, 40 μm). Immunofluorescent triple labeling of CD31 or PDGFRβ (red), collagen type IV (b), fibronectin (d), or laminin α2 (f) (green), and DAPI (blue) in the ischemic core on day 28 after pMCAO in male wild-type mice (scale bar, 40 μm) and (d) Immunofluorescent double labeling of CD31 (green) and PDGFRβ (red) on day 1, 7, 14, and 28 after pMCAO in male wild-type mice (scale bar, 500 μm). Magnified images in the squares (magenta) are shown at the bottom (scale bar, 20 μm). PDGFRβ, platelet-derived growth factor receptor β.
PDGFRβ–positive cells may primarily be responsible for intra-infarct deposition of fibronectin with the aid of neighboring macrophages after pMCAO
We recently demonstrated that PDGFRβ-positive pericytes and macrophages play pivotal roles in the clearance of MD and tissue repair within infarct areas through reciprocal interaction (Supplemental Figure II). 3 We therefore examined the expression of fibronectin in cultured BMDMs and pericytes. Quantitative PCR revealed that fibronectin expression was significantly higher in cultured pericytes than cultured BMDMs at baseline (Figure 2(a)). While OGD did not affect the expression of fibronectin in BMDMs, it significantly increased the expression of fibronectin in cultured pericytes (Figure 2(a)). Furthermore, cultured pericytes showed significantly increased expression of fibronectin in response to PDGF-BB treatment (Figure 2(a)). Macrophages phagocytosing MD increased the expression of Pdgfb and basic fibroblast growth factor (Fgf2), an up-regulator of PDGFRβ 17 (Figure 2(b)). Consistently, MCM treated with MD showed increased phosphorylation of PDGFRβ and expression of fibronectin in cultured pericytes (Figure 2(c)). We confirmed that MD did not directly affect the phosphorylation of PDGFRβ and the expression of fibronectin in cultured pericytes (supplemental Figure III). Immunohistochemistry demonstrated that intra-infarct deposition of fibronectin following pMCAO was significantly attenuated in Pdgfrb+/− mice, wherein post-stroke expression of PDGFRβ and CD13, another marker of pericytes 18 , was significantly attenuated within infarct areas compared with wild-type mice (Figure 2(d)). Using quantitative PCR, we confirmed that the increased expression of fibronectin in ischemic areas on day 28 after pMCAO was significantly attenuated in Pdgfrb+/− mice (Figure 2(d)).
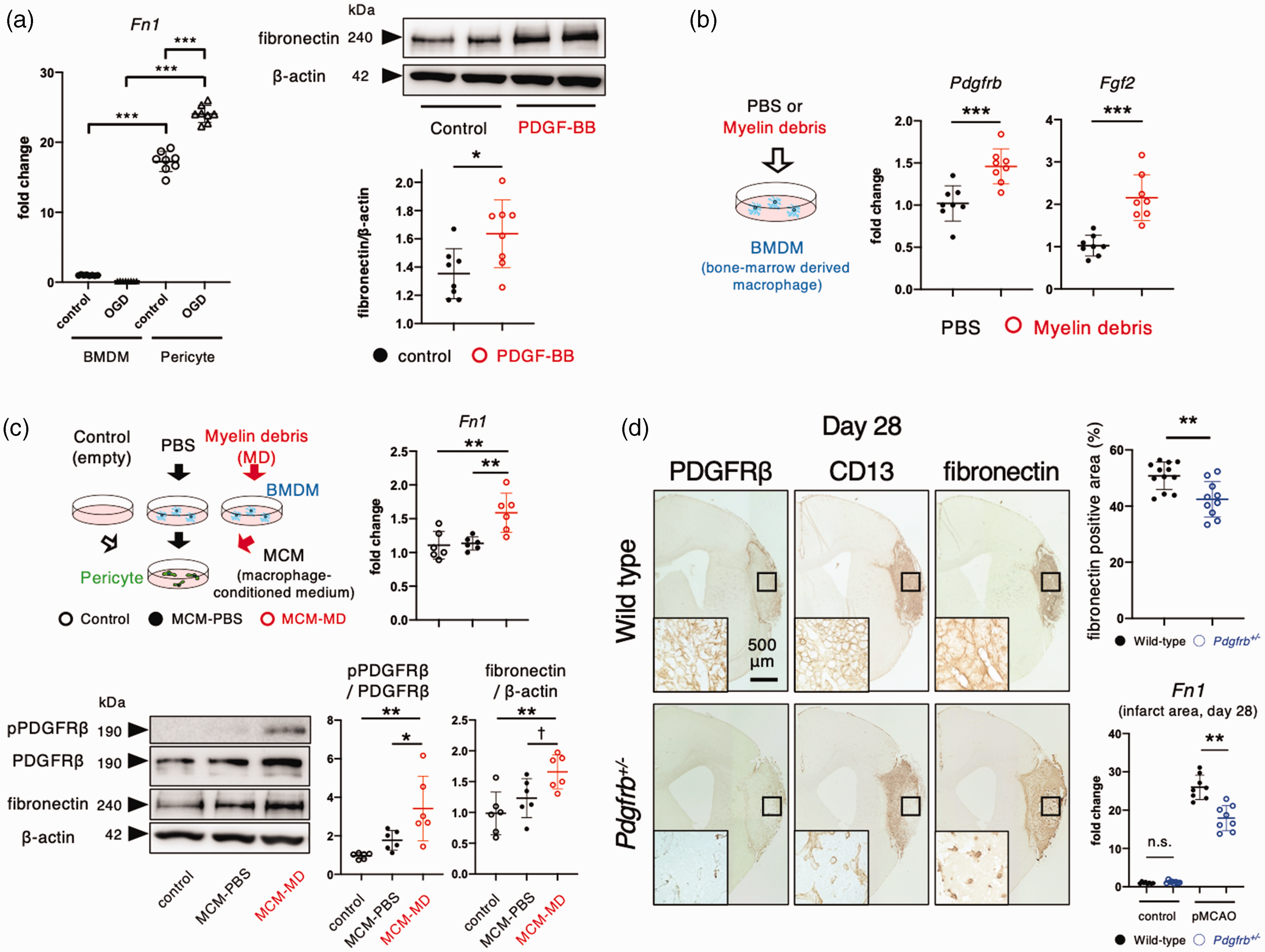
PDGFRβ (platelet-derived growth factor receptor β)-positive pericytes primarily produce fibronectin with the aid of macrophages within infarct areas. (a) Quantitative polymerase chain reaction (PCR) for Fn1 (fibronectin) in cultured pericytes and bone marrow-derived macrophages (BMDMs) at baseline and after oxygen glucose deprivation for 24 h is shown on the left. Immunoblot analyses of fibronectin and β-actin in cultured pericytes after treatment with PBS or PDGF-BB (10 ng/mL) for 24 h (n = 8, each group) is shown on the right. (b) Effects of myelin debris (MD) for 72 h on the expression of Pdgfb and Fgf2 in BMDM (n = 8). (c) Effects of macrophage-conditioned medium (MCM) treated with PBS (MCM-PBS) or MD (MCM-MD) for 72 h on PDGFRβ signaling for 10 min (n = 6) and expression changes of Fn1 mRNA and its protein for 24 h (n = 6) in cultured pericytes and (d) Immunohistochemistry of PDGFRβ, CD13, and fibronectin on day 28 after permanent middle cerebral artery occlusion (pMCAO) in male wild-type and Pdgfrb+/– mice (scale bar, 500 mm) (n = 12). Magnified images of ischemic core (black square) are shown in the inset. Quantitative PCR for Fn1 in the intact brain and in infarct areas on day 28 after pMCAO in male wild-type and Pdgfrb+/– mice (n = 8). Data are shown as mean ± standard deviation. a, b, d, *p < 0.05, **p < 0.01, and ***p < 0.001; unpaired t-test. c, †p < 0.1, *p < 0.05, and **p < 0.01; one-way analysis of variance followed by Bonferroni’s post-hoc test.
Fibronectin enhances the phagocytic activity of macrophages
We next examined whether fibronectin modulated macrophage functions. Cultured macrophages adhered strongly to fibronectin-coated dishes compared with non-coated ones (control) (Figure 3(a)). Pretreatment with a blocking antibody for integrin β1 abrogated the adhesion of macrophage to fibronectin (Figure 3(a)). Macrophages cultured on fibronectin-coated dishes showed significantly increased expression of Il1b, Il6, Il10, Mmp9, Tgfb, and Msr1, related to their phagocytic activity and remodeling of ECM. Upregulation of these molecules was significantly attenuated by pretreatment with integrin β1 antibody (Figure 3(b)). Macrophages cultured on fibronectin-coated dishes showed enhanced phagocytic activity against MD, as assessed by ORO staining (Figure 3(c)), accompanied by increased phosphorylation of STAT3, an indicator of the phagocytic activity of macrophages 19 (Figure 3(d)). Fibronectin-mediated enhancement of phagocytosis was significantly attenuated by stattic, a STAT3 inhibitor 20 (Figures 3(c) and (d)). We further demonstrated that cultured microglia adhered strongly to fibronectin-coated dishes, where they exhibited enhanced phagocytic activity against MD compared with that of microglia on non-coated dishes (control) (Figure 3(e)).
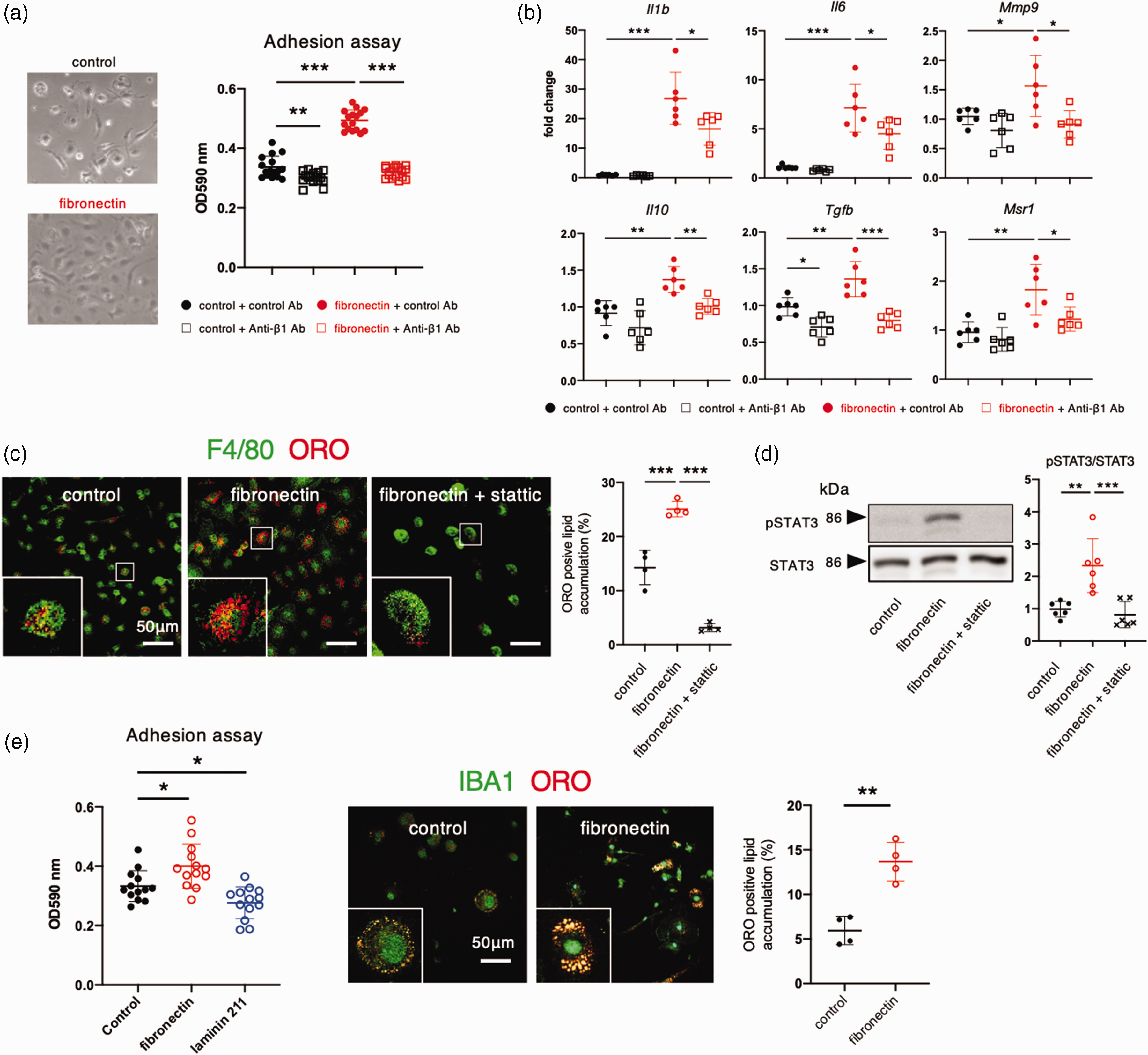
Fibronectin enhances the activity of macrophages within infarct areas. (a) Representative images of bone marrow-derived macrophages (BMDMs) cultured on non-coated (control) and fibronectin-coated dishes. Adhesion of BMDMs to fibronectin is attenuated by co-treatment with an anti-integrin β1 antibody (50 mg/mL). (b) Quantitative polymerase chain reaction for Il1b, Il6, Mmp9, Il10, Tgfb, and Msr1 in BMDMs cultured on control or fibronectin-coated dish with or without an anti-integrin β1 antibody for 6 h (n = 6). (c) Immunofluorescent assessment of phagocytotic activity of BMDM for myelin debris (MD). Immunofluorescent double labeling with F4/80 (green) and Oil Red O (ORO) (red) in BMDMs cultured on control, fibronectin-coated, or fibronectin-coated dish with stattic (6 μmol/L) for 24 h. Quantification of ORO-positive (red) foamy macrophages is shown on the right (n = 4). Magnified images are shown in the inset: F4/80-positive macrophages (green) took up MD and became ORO-positive (red) foamy macrophages in 24 h. (d) Representative immunoblot analyses of total STAT3 (signal transducer and activator of transcription 3) and phosphorylated STAT3 in BMDMs cultured on control, fibronectin-coated, and fibronectin-coated dishes with stattic (6 μmol/L) (n = 6) and (e) Adhesion of cultured microglia on non-coated (control), fibronectin-coated, and laminin α2-coated dishes (n = 13). Assessment of phagocytotic activity of cultured microglia for MD. Immunofluorescent double labeling with F4/80 (green) and ORO (red) in microglia cultured on control and fibronectin-coated dishes for 24 h. Data are shown as mean ± standard deviation. *p < 0.05, **p < 0.01, and ***p < 0.001; one-way analysis of variance followed by Bonferroni’s post-hoc test.
Intra-infarct PDGFRβ-positive cells promote astrocytic production of laminin α2 in peri-infarct areas while decreasing their own production of laminin α2
In contrast to fibronectin, laminin α2 was strongly deposited in the boundary between infarct and peri-infarct areas after pMCAO (Figures 1(b) and (c) and 4(a)). Immunofluorescent labeling demonstrated that peri-infarct laminin α2 was co-localized with GFAP (Figure 1(c)) and AQP4, an astrocyte end-feet protein (Figure 4(a)), but not with PDGFRβ (Figures 1(c) and 4(a)). We further examined the expressional regulation of laminin α2 in pericytes and astrocytes. Treatment with PDGF-BB or conditioned medium of macrophages phagocytosing MD (MCM-MD) significantly decreased the expression of laminin α2 in cultured pericytes (Figure 4(b)) in contrast to fibronectin (Figure 2). Conversely, PCM treated with PDGF-BB significantly increased the expression of laminin α2 in cultured astrocytes (Figure 4(c)). Peri-infarct deposition of laminin α2 was significantly attenuated in Pdgfrb+/− mice, compared with wild-type mice, parallel with the extent of peri-infarct accumulation of GFAP-positive astrocytes (Figure 4(c)). These findings suggest that intra-infarct PDGFRβ-positive cells crucially regulate the deposition of laminin α2 produced by GFAP-positive astrocytes in peri-infarct areas through humoral factors.
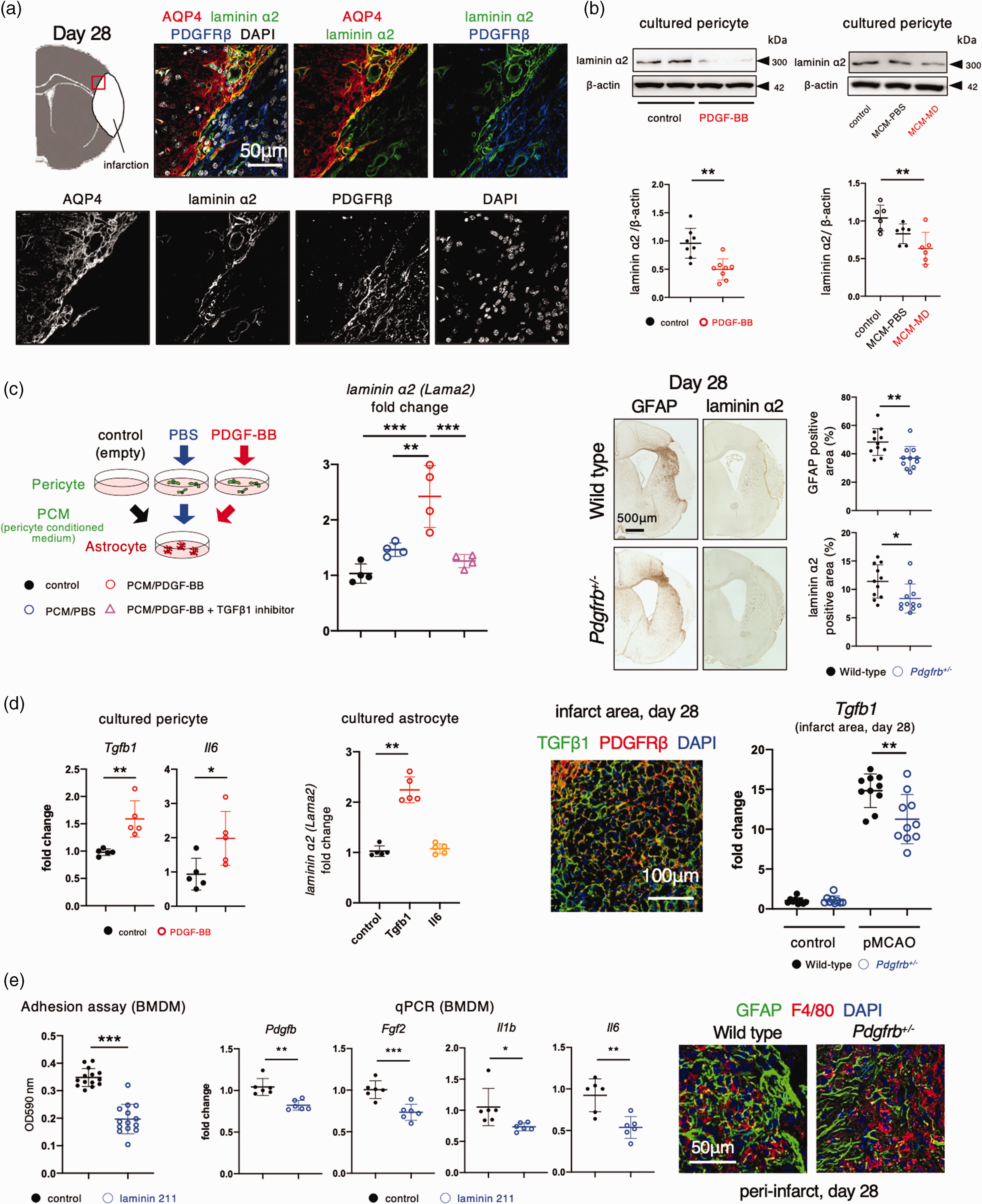
Intra-infarct PDGFRβ (platelet-derived growth factor receptor β)-positive cells promote astrocytic production of laminin α2 in peri-infarct areas. a, Immunofluorescent quadruple labeling with DAPI (4′,6-diamidino-2-phenylindole; white), AQP4 (aquaporin 4, red), laminin α2 (green), and PDGFRβ (blue) in peri-infarct areas on day 28 after permanent middle cerebral artery occlusion (pMCAO) (scale bar, 50 mm). The peri-infarct region (red square) in the top-left panel is magnified. b, Immunoblot analyses of laminin α2 and β-actin in cultured pericytes after treatment with PBS or PDGF-BB (10 ng/mL) for 24 h (n = 8, each group) (left). Effects of macrophage-conditioned medium (MCM) treated with PBS (MCM-PBS) or myelin debris (MCM-MD) on expression changes of laminin α2 in cultured pericytes (n = 6). c, Effects of pericyte-conditioned medium (PCM) (black, control) and PCM treated with PBS (blue, PCM/PBS) or PDGF-BB (10 ng/mL; red, PCM/PDGF-BB) on the expression of laminin α2 in cultured astrocytes. Effect of LY364947 (1 µM), an inhibitor of TGFβ1 receptor, was tested (n = 4) (left). Immunohistochemistry of GFAP (glial fibrillary acidicContinued.protein) and laminin α2 on day 28 after pMCAO in male wild-type and Pdgfrb+/– mice (scale bar, 500 mm). Quantitative data are shown on the right (n = 10). (d) Expression of Tgfb1 and Il6 in cultured pericytes and their expression changes in response to PDGF-BB (n = 5) (left). Effects of TGFβ1 and IL-6 on the expression of laminin α2 in cultured astrocytes (n = 5) (middle). Immunofluorescent triple labeling with TGFβ1 (green), PDGFRβ (red), and DAPI (blue) in infarct areas on day 28 after pMCAO (scale bar, 100 mm). Expression of Tgfb1 mRNA in intact areas at day 0 (control) and that within infarct areas at day 28 after pMCAO in wild-type and Pdgfrb+/– mice (n = 10) and (e) Adhesion of bone marrow-derived macrophages (BMDMs) to non-coated (control) or laminin α2 (laminin 211)-coated dishes (left). Quantitative polymerase chain reaction for Pdgfb, Fgf2, Il1b, and Il6 in BMDMs on control or laminin α2 (laminin 211)-coated dishes (middle). Representative immunofluorescent triple labeling with GFAP (green), F4/80 (red), and DAPI (blue) on day 28 after pMCAO in male wild-type and Pdgfrb+/– mice (scale bar, 50 mm) (right). Data are shown as mean ± standard deviation. (b) *p < 0.05, **p < 0.01, and ***p < 0.001; one-way analysis of variance followed by Bonferroni’s post-hoc test. (c–e) *p < 0.05, **p < 0.01, and ***p < 0.001; unpaired t-test.
We then explored whether PDGFRβ-positive cell-derived molecules could upregulate the expression of laminin α2 in peri-infarct astrocytes. Among all candidates, we focused on transforming growth factor β1 (TGFβ1) and interleukin-6 (IL-6) because these two molecules were expressed in cultured PDGFRβ-positive pericytes and their expression was significantly increased in response to PDGF-BB (Figure 4(d)). Treatment with TGFβ1, but not with IL-6, significantly increased the expression of laminin α2 in cultured astrocytes (Figure 4(d)). Pretreatment with LY364947 (1 µM), a TGFβ1 receptor inhibitor, significantly attenuated the upregulation of laminin α2 induced by PCM (treated with PDGF-BB) in the cultured astrocytes (Figure 4(c)). Consistently, TGFβ1, which was expressed partly in PDGFRβ-positive cells, was markedly increased within infarct areas, but significantly decreased in Pdgfrb+/− mice (Figure 4(d)).
We further examined the possibility whether peri-infarct laminin α2 can also function as a glial limitans against macrophages. Cultured macrophages did not adhere to laminin α2 (laminin 211)-coated dishes (Figure 4(e)). Macrophages cultured on laminin α2-coated dishes showed reduced expression of Pdgfb, Fgf2, Il1b, and Il6 (Figure 4(e)). Moreover, the adherence of cultured microglia was reduced on laminin α2-coated dishes (Figure 3(e)). In Pdgfrb+/− mice, more F4/80-positive cells infiltrated into GFAP-positive peri-infarct areas, probably due to reduced deposition of laminin α2 at the infarct boundary (Figure 4(e)).
Peri-infarct laminin α2 may promote oligodendrogenesis, a key process for functional recovery
Finally, we examined the effects of laminin α2 on the oligodendrogenesis in peri-infarct areas, a key process promoting post-stroke functional recovery.5,21 In contrast to macrophages, cultured OPCs adhered well to laminin α2 (laminin 211)-coated dishes (Figure 5(a)). Quantitative PCR revealed that OPCs showed increased expression of myelinating proteins, such as Mbp, Mag, and Plp, on laminin α2 (laminin 211)-coated dishes (Figure 5(a)). Consistently, immunofluorescent labeling showed increased expression of MBP in OLIG2-positive OPCs cultured on laminin α2-coated dishes (Figure 5(b)). Immunofluorescent triple labeling demonstrated that some APC-positive oligodendrocytes were EdU-positive close to laminin α2-positive peri-infarct areas, suggesting that the oligodendrocytes were generated from OPCs in these areas (Figure 5(c)). The number of peri-infarct GSTπ-positive oligodendrocytes was significantly lower in Pdgfrb+/− mice than wild-type mice (Figure 5(c)). We demonstrated that post-stroke functional recovery was significantly attenuated in Pdgfrb+/− mice (Figure 5(d)).
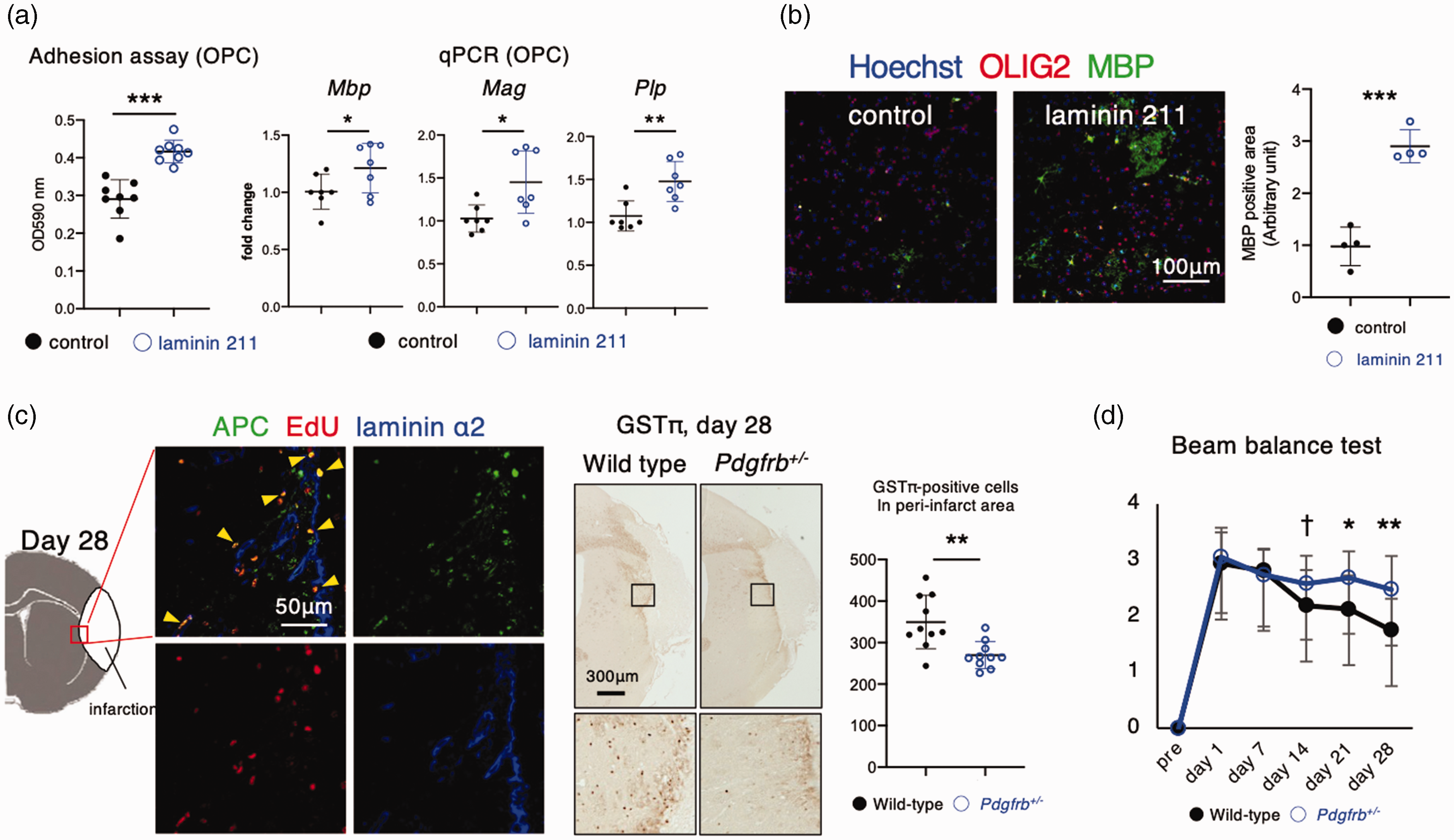
Laminin α2 promotes oligodendrogenesis, a key process of functional recovery, in peri-infarct areas. (a) Adhesion of oligodendrocyte precursor cells (OPCs) to non-coated (control) or laminin α2 (laminin 211)-coated dishes. Quantitative polymerase chain reaction of myelin basic protein (Mbp), myelin-associated glycoprotein (Mag), and myelin proteolipid protein (Plp) in OPCs cultured on control or laminin α2 (laminin 211)-coated dishes for 5 days (n = 7). (b) Representative immunofluorescent triple labeling of Hoechst (blue), MBP (green), and OLIG2 (oligodendrocyte lineage transcription factor 2; red) in OPCs cultured on control or laminin α2 (laminin 211)-coated dishes for 7 days (n = 4; scale bar, 100 μm), and quantification of the MBP-positive areas. (c) Representative immunofluorescent triple labeling with laminin α2 (blue), APC (adenomatous polyposis coli, green), and EdU (5-ethynyl-2′-deoxyuridine, red) in peri-infarct areas (red square) on day 28 after permanent middle cerebral artery occlusion (pMCAO) in male wild-type mice (scale bar, 50 mm) (left). Immunohistochemistry of GSTπ (glutathione S-transferase π) on day 28 after pMCAO in male wild-type and Pdgfrb+/– mice (scale bar, 300 mm) (n = 10) (right) and (d) Neurological function, assessed using the beam balance test, at baseline (Pre) and at days 1, 7, 14, 21, and 28 after pMCAO in male wild-type (black; n = 16) and Pdgfrb+/– mice (blue; n = 17). Data are shown as mean ± standard deviation. †p < 0.1, *p < 0.05, **p < 0.01, and ***p < 0.001; unpaired t-test.
We further confirmed that post-stroke deposition of intra-infarct fibronectin and peri-infarct laminin α2 and OPC differentiation, as assessed by GSTπ, were decreased with worse functional recovery in female Pdgfrb+/− mice (supplemental Figure IV).
Discussion
We demonstrated that ECMs constituting the BM in the intact brain were drastically remodeled after acute ischemic stroke: some ECMs, such as collagen type IV and perlecan, remained in the BM surrounding residual endothelial cells within infarct areas, 11 while others were remodeled to infarct areas in parallel with accumulation of PDGFRβ-positive cells (fibronectin and vitronectin) 22 or to peri-infarct areas (laminin α2). 23 Among them, we focused on fibronectin and laminin α2 in the present study. Fibronectin, chiefly produced by PDGFRβ-positive cells with the aid of macrophages within infarct areas, enhanced macrophage-mediated clearance of MD and tissue repair within infarct areas. In contrast, laminin α2 may demarcate the infarct border and function as a promoter of oligodendrogenesis in peri-infarct areas. Since the peri-infarct deposition of laminin α2 was reduced in Pdgfrb+/− mice, intra-infarct PDGFRβ-positive cells may organize the post-stroke remodeling of these ECM.
PDGFRβ-positive cell-derived fibronectin is a key ECM promoting macrophage-mediated clearance of debris within infarct areas
Fibronectin can be produced by various cell types including pericytes/fibroblasts, 22 astrocytes, 24 and microglia/macrophages. 25 However, the present study clearly demonstrated that PDGFRβ-positive cells may primarily be responsible for the production of fibronectin within infarct areas, based on the following findings: (1) fibronectin was deposited gradually within infarct areas in parallel with intra-infarct accumulation of PDGFRβ-positive cells following pMCAO3; (2) fibronectin deposition was significantly reduced in Pdgfrb+/− mice; and (3) quantitative PCR demonstrated that the expression levels of fibronectin were much higher in cultured PDGFRβ-positive pericytes than macrophages, another major cell type within infarct areas. 3 Infiltrating macrophages contribute to the PDGFRβ-positive cell-mediated production of fibronectin, which enhances macrophage activity, such as adhesion, gene expression, and clearance of MD. Thus, fibronectin may mediate close interaction between PDGFRβ-positive cells and macrophages, thereby completing the clearance of debris within infarct areas. 3 Reportedly, fibronectin plays an important role in mediating axonal regeneration.24,26 Additionally, we and others have demonstrated recently that effective intra-infarct tissue repair promotes peri-infarct astrogliosis and oligodendrogenesis leading to better functional recovery.3,5 Collectively, fibronectin produced by PDGFRβ-positive cells may be a key ECM promoting post-stroke functional recovery through enhancement of macrophage-mediated debris clearance within infarct areas. In this context, we suppose that attenuated deposition of intra-infarct fibronectin and peri-infarct laminin α2 in Pdgfrb+/− mice are attributable chiefly to phenotypic changes of pericytes and pericyte-derived fibroblast-like cells5,7,27,28; however, we could not exclude the possibility that vascular smooth muscles and/or non-pericyte-derived fibroblast-like cells also contribute to the altered ECM deposition because these cells can also express PDGFRβ.
In addition to BMDMs infiltrating into infarct areas, resident microglia may also play roles in the clearance of MD in ischemic areas and post-injury functional recovery, since cultured microglia also adhered strongly to fibronectin-coated dishes, thereby enhancing their phagocytic activity against MD. 29
PDGFRβ-positive cells regulate the production of astrocytic laminin α2 in peri-infarct areas
Laminin α2 can be produced by pericytes and astrocytes and is a key ECM14,23,30 that restricts the extravasation of blood-derived monocytes/macrophages into the brain parenchyma in the BBB. 31 Within infarct areas, (1) astrocytes are lost; (2) PDGFRβ-positive pericytes decrease the expression of laminin α2 in response to PDGF-BB or MCM; and (3) infiltrating macrophages, along with pericytes, 32 produce MMP9, a metalloproteinase preferentially degrading laminin α2.33,34 Therefore, the BM laminin α2 is reduced within infarct areas, as shown in the present study. In contrast to reduced expression of laminin α2 within infarct areas, it was produced by peri-infarct GFAP-positive astrocytes and deposited strongly in the boundary between infarct and peri-infarct areas. Since pericyte-conditioned media increased the production of laminin α2 in cultured astrocytes and the deposition of laminin α2 was significantly attenuated in Pdgfrb+/− mice, intra-infarct PDGFRβ-positive cells may crucially regulate the laminin α2 production in peri-infarct astrocytes probably through humoral factors. In this context, we identified TGFβ1 as a candidate molecule produced by PDGFRβ-positive cells, which could increase the expression of laminin α2 in peri-infarct GFAP-positive astrocytes (Figure 4(d)). However, to prove this in vivo, we should examine whether post-stroke deposition of laminin α2 in peri-infarct areas would be decreased in mice with PDGFRβ-positive cell-specific deletion of Tgfb1.
Astrocytic laminin α2 may function as a promotor of oligodendrogenesis in peri-infarct areas
Peri-infarct astrocytic laminin α2 may have important roles after ischemic stroke. It may promote peri-infarct oligodendrogenesis/remyelination, leading to functional recovery. We have recently demonstrated that effective intra-infarct repair promotes peri-infarct oligodendrogenesis and astrogliosis. 5 Furthermore, various molecules produced by astrocytes promote oligodendrogenesis;5,21,35 laminin α2 may be one such factor as it functions as a scaffold for OPCs for differentiation into oligodendrocytes in peri-infarct areas. Previous reports have demonstrated that the deficiency of laminin α2 causes developmental abnormality of OPC differentiation, thereby leading to myelination defects in both humans 36 and animals. 37 Reportedly, pericytes directly stimulated OPC differentiation and remyelination through the production of laminin α2 in a mouse demyelination model using Pdgfrb-deficient mice. 38 However, at least in acute ischemic stroke, PDGFRβ-positive pericytes may indirectly stimulate peri-infarct OPC differentiation through astrocyte-mediated production of laminin α2.
Limitations
This study has certain limitations. First, we did not examine post-stroke remodeling of all ECM components in the brain. There may be more interesting ECM, besides fibronectin and laminin α2, that crucially affect post-stroke tissue repair and functional recovery. Second, we should confirm the in vivo role of fibronectin and laminin α2 using mice with pericyte-specific deletion of fibronectin or astrocyte-specific deletion of laminin α2, although Yao et al. demonstrated that deficiency of astrocytic laminin causes instability of the BBB in vivo. 23 Finally, we only used young mice. Aged mice or those with risk factors for stroke should be used to examine the effects of PDGFRβ deficiency on post-stroke remodeling of ECM. Bell et al. demonstrated that pericytic dysfunctions become greater with aging in Pdgfrb+/− mice 39 ; therefore, we speculate that post-stroke ECM remodeling may be impaired more in aged Pdgfrb+/− mice.
In conclusion, intra-infarct PDGFRβ-positive cells organize post-stroke remodeling of ECM, like fibronectin and laminin α2, thereby promoting tissue repair and functional recovery following acute ischemic stroke. These ECM can be therapeutic targets for post-stroke functional recovery.
Supplemental Material
sj-pdf-1-jcb-10.1177_0271678X221145092 - Supplemental material for PDGFRβ -positive cell-mediated post-stroke remodeling of fibronectin and laminin α 2 for tissue repair and functional recovery
Supplemental material, sj-pdf-1-jcb-10.1177_0271678X221145092 for PDGFR
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by Grants-in-Aid for Scientific Research (B 16H05439), (B 20H03791) (TK and TA), (C 20K09373) (TA), (C 26462163), (GAG9K09530) (YW), and (C GAG9K09511) (KN) from the Ministry of Education, Culture, Sports, Science and Technology, Japan; a grant from Mochida Memorial Foundation for Medical and Pharmaceutical Research (KN); a grant from SENSHIN Medical Research Foundation, Japan (TA and KN); a grant from the Smoking Research Foundation (TA); and research grants from Boehringer Ingelheim, Bristol-Myers Squibb, Daiichi Sankyo, Eisai, and Takeda (TK and TA).
Acknowledgements
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
TS, KN, YW, and TA contributed to the conception and design of the study; TS, MS, KY, MT HT, and MH contributed to the acquisition and analysis of data; TS and TA contributed to drafting the manuscript; and TS, KN, YW, TK, and TA contributed to revising the manuscript.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.