
Abstract
Cerebral cavernous malformation (CCM) is a brain vascular disease which can cause stroke, cerebral hemorrhage and neurological deficits in affected individuals. Loss-of-function mutations in three genes (CCM1, CCM2 and CCM3) cause CCM disease. Multiple mouse models for CCM disease have been developed although each of them are associated with various limitations. Here, we employed the Dre-Cre dual recombinase system to specifically delete Ccm genes in brain endothelial cells. In this new series of CCM mouse models, robust CCM lesions now develop in the cerebrum. The survival curve and lesion burden analysis revealed that Ccm2 deletion causes modest CCM lesions with a median life expectance of ∼10 months and Ccm3 gene deletion leads to the most severe CCM lesions with median life expectance of ∼2 months. The extended lifespan of these mutant mice enables their utility in behavioral analyses of neurologic deficits in adult mice, and allow the development of methods to quantify lesion burden in mice over time and also permit longitudinal drug testing in live animals.
Introduction
CCM is a brain blood vessel disease that leads to hemorrhage, stroke and neurological deficits with ∼0.5% prevalence in the population.1–3 Human genetic studies have identified loss-of-function mutations in three genes (CCM1, CCM2 and CCM3) and gain-of function mutations in PI3K or MEKK3 signalling pathways as the cause of CCM disease.2,4–7 A series of Cre-LoxP mouse models has been established that induce CCM gene deletion in neonatal mice where the formation of CCM lesions are similar to that of human lesions. The Cre recombinase expressing mouse lines used for these models include Tie2-CreERT2, Cdh5-CreERT2, Pdgfb-CreERT2, Slco1c1-CreERT2, Mfsd2a-CreERT2, and neuronal deletion model.8–16
Among the various mouse models that have been developed in the past few years, the most robust model for CCM research is to conditionally delete Ccm genes with tamoxifen in the Cdh5-CreERT2 or Tie2-CreERT2 newborn pups at postnatal day 1 (P1).9,11,17 Vascular malformations were detectable at P6 in the cerebellum as dilated venules in the white matter. By P11, large lesions develop throughout the venous vasculature in the white matter, displaying the phenotypic hallmarks of CCM in human patients.8,11 The neonatal inducible model of CCM has been instrumental in the search for the cause of CCM pathogenesis and was used to identify that the inhibition of MEKK3/KLF2/4 pathway was an effective approach to inhibiting CCM lesion formation and growth.8,10,18 However, these models still have their limitations to recapitulate the human condition, mainly due to the fact that lesions only form in the cerebellum and the early lethality of the induced mice.8,11,13,17 To circumvent these limitations, chronic mouse models have been developed by adjustment of the tamoxifen induction time point and dosage12,15,16 or induction of gene deletion with recombinase driven by brain specific promoters, such as Slco1c1-CreERT2 and Mfsd2a-CreERT2 line.4,13,14 Slco1c1-CreERT2 can drive robust lesion genesis when crossed with the Ccm2fl/fl mice. 14 Additionally, Mfsd2a-CreERT2 mice have been used to crossed with the Ccm3fl/fl mice to generate a robust chronic model of CCM 13 although Mfsd2a expression is not limited to brain endothelial cells and can be detected in hepatocytes.
In recent years, a Dre-Cre dual deletion system has been developed to more precisely target a subpopulation of cells in a specific organ. 19 The combination of Tie2-Dre and Mfsd2a-CrexERT2 has been developed to drive gene deletion in brain endothelial cells. 19 In this study, we employed Tie2-Dre/Mfsd2a-CrexER dual recombinases (BEC-Cre) deletion system 19 to precisely delete Ccm1, Ccm2, or Ccm3 genes in brain endothelial cells and assessed the natural history of CCM lesion development in these models. In these new models, CCM lesions can form in both the cerebrum and cerebellum and these mice survive longer than most of previous developed models. In the current model, CCM lesions continue to form and grow in mature mice. The ongoing development of CCM lesion in mature mice can be assessed in vivo with MRI and the behavioural deterioration examination. Similar to that found in humans, Ccm3 deletion confers most aggressive CCM lesions among these three genes. Thus, this new non-inducible deletion system provides a faithful robust model for CCM pathogenesis studies and drug testing.
Materials and methods
Mice
All experiments were conducted under the guidelines/regulations of Tianjin Medical University, in compliance with the ARRIVE guidelines and the guidelines of National Research Council.20,21 The Institutional Animal Care and Use Committee (IACUC) of Tianjin Medical University approved all animal ethics and protocols. Tie2-Dre, Mfsd2a-CrexER, Cdh5-CreERT2, Krit1fl/fl, Ccm2fl/fl, Pdcd10fl/fl and RosaRFP animals have been previously described.8,12,19,22 Experimental animals were maintained on a 129/C57BL/6J mixed genetic background. Both female and male mice were used in the experiments. The data points for female and male are noted in the figures or figure legends. All experiments were performed with mice from at least three different litters using littermate as controls. The Ccm1BECKO, Ccm2BECKO and Ccm3BECKO represents Tie2-Dre;Mfsd2a-CrexER;Krit1fl/fl, Tie2-Dre;Mfsd2a-CrexER;Ccm2fl/fl, and Tie2-Dre;Mfsd2a-CrexER;Pdcd10fl/fl mice, respectively.
Histological analysis
Mouse tissues were fixed in 4% formaldehyde overnight, gradually dehydrated in 30%, 50%, 70%, 95% and 100% ethanol and embedded in paraffin. Paraffin sections were stained for H&E staining using standard protocols. For immunostaining, deparaffinized sections were subjected to rehydration, followed by antigen retrieval by heating in citrate buffer (Beyotime, P0083) for 20 minutes. After blocking in 10% normal donkey serum (Jackson ImmunoResearch) +1% BSA in 0.1% PBS-T for 1 hour, the sections were incubated with primary antibodies overnight at 4°C. The secondary antibodies were incubated for 1 hour. The sections were washed with PBS-T and mounted with mounting solution containing DAPI (Vector Laboratories). The following primary and secondary antibodies were used: anti-CD31 (Dianova clone SZ31, Dianova, Hamburg, Germany,1:300 dilution) and anti-RFP (Rockland, 600-401-379, 1:400 dilution). Secondary antibodies ImmPRESS HRP reagent kit:anti-Rat IgG, mouse-adsorbed (Vector Laboratories, cat no. MP-7444), goat anti–rabbit IgG (H + L) Alexa Fluor 488 conjugate (A11059; Invitrogen; 1:500 dilution). Imaging was performed using a Ni-U (Nikon, Japan) microscope.
Micro-CT scan and analysis
CCM lesion burden was analyzed using micro-CT imaging techniques, as previously described. 23 The brain samples were scanned by a blinded operator and the micro-CT images were analyzed by a different blinded person. According to the CCM lesion volume of different time points, we classified CCM lesions into three size groups: small lesion (volume ≤106 µm3), medium lesion (volume = 106–107 µm3) and large lesion (volume ≥107 µm3).
In vivo retinal fundus imaging
Imaging was performed on anesthetized mice (1.2% avertin) using a Micron IV camera (Phoenix Research Laboratories, San Ramon, CA). To dilate the pupils, eyes were moistened using drops containing tropicamide (Shenyang Xingqi Eye Medicine Co., Ltd., China). A lubricating gel (Viscotears, Novartis Pharmaceuticals, Australia Pty Limited, North Ryde, NSW, Australia) was placed on the microscope objective to prevent drying of the cornea. Then, 20-second videos were captured for both eyes in bright field and fluorescence modes before fluorescein angiography was performed, for which 20 μL of 10% sodium fluorescein (Alcon Australia) were injected subcutaneously.
Retina dissection, processing and staining
Mice were sacrificed and eyes were enucleated in PBS. The eyes were fixed in 4% PFA/PBS for 1 hour at 4°C. Retinas were dissected, washed with PBS and permeabilized with PBS containing 0.3% Triton X-100 and 1%BSA overnight at 4°C. Then, the retinas were incubated with Alexa Fluor 597-conjugated isolectinGS-IB4 solution (1:250) at 4°C for 8 hours. After washing, flat-mounted retinas were analyzed using a Zeiss Axio-Imager LSM-800 confocal microscope.
Seizure video record and velocity measurement
Ten-hour video recording was used to monitor the changes in epileptic behavior for 4 days. Seizure intensity was graded by using Racine's standard five-stage scale 24 as follows: I - immobility and staring; II – rigid posture; III – repetitive movements and head bobbing; IV – rearing and myoclonic twitching; V – severe tonic-clonic seizures. We extracted the severe tonic-clonic seizures section and rigid posture and myoclonic twitching sections. Each second of these two videos was divided to 25 frames. The velocity was calculated using the Image-J measurement of the displacement.
Rotarod testing
The duration to balance on the rod was recorded at different rotational accelerations using a rotarod device (76-0771, Panlab Harvard Apparatus, Barcelona, Spain). For the rotational acceleration, the rotarod device was set to accelerate from 1 rpm to 20 rpm within 60 s. Five trials were measured in each mouse with a 15 min interval between trials.
Gait balance testing
Gait analysis was conducted using a clear corridor apparatus (65 cm × 5 cm × 15 cm), which was lined with a pre-cut piece of white paper. Animals were trained to run to the enclosed darkened box at the end of the corridor by placing the mouse at the far end of the corridor and encouraging them to move towards the goal box at the end. Training was conducted twice for each mouse until the animal readily ran to the end box without encouragement. For testing, the paws of the animal were painted with non-toxic ink (Shanghai Fine Stationery Co., Ltd., China); red was used for the front paws and blue for the hind paws. The animals were then placed at the near end of the apparatus and ran to the enclosed goal box at the far end of the apparatus, leaving a print of the associated foot prints on the paper at the base of the apparatus. The paper print was then allowed to air dry to enable analysis for stride length and width for both the front and hind paws of each animal to provide gait measurements.
Ponatinib and dexamethasone treatment
Control and CcmBECKO mice from a same litter were random assigned to vehicle or ponatinib or dexamethasone treatment group. Both ponatinib [Selleck, 10 mg/ml dilution made in 25 mM citrate buffer (pH 2.75)] and dexamethasone [MCE, 0.5 mg/ml dilution made in 5% sterile glucose solution] were administrated intragastrically using gavage. Ponatinib was administrated every other day for 2 weeks with a dosage of 45 mg/kg for first two treatments and followed by five administrations of 30 mg/kg. Control animals were given vehicle [25 mM citrate buffer (pH 2.75)]. Dexamethasone was administrated every other day for 2 weeks at a dose of 2 mg/kg for 7 times. Control animals were given vehicle [5% sterile glucose solution].
MRI imaging and analysis
MRI scans was performed on anaesthetized mice using an 7.0 T Magnetic Resonance Imaging system (Biospec 70/20USR, Bruker, Germany). A gradient recoiled spin echo sequence T2WI was used to acquire coronal slices spanning the whole brain and the axial of spinal cord. Sequence parameters were as follows: repetition time, 3080 ms; echo time, 41 ms; FoV read, 30 mm; FoV phase, 80.0%; slices 18; slice thickness, 0.5 mm; voxel size, 0.125 × 0.094 × 0.500 Rel. Lesion area and whole brain area were quantified by using ImageJ software. The MRI index = total lesion area of 18 images/whole brain area of 18 images.
Statistical analysis
All survival curves were analysed with Mental-cox test in Prism. All data distributions were confirmed with Shapiro-Wilk normality test in Prism. All the other statistical analysis in the study was done using Student’s t test or One-Way ANOVA in GraphPad Prism statistical software. Values are presented as Mean ± SD. Statistical significance was considered when P ≤ 0.05.
Results
Deletion of Ccm2 with BEC-Cre confers CCM lesion in cerebrum, cerebellum and retina
Ccm genes are essential for cardiovascular development, and deletion of Ccm genes in endothelium causes early embryonic lethality in mice. 2 To circumvent the embryonic lethality issue, mouse models of CCM disease have been developed by inducing Ccm gene deletion in pan-endothelial cells or brain endothelial cells in postnatal mice.8–14 The efficacy of lesion generation is highly sensitive to the induction time point within the limited induction time window when using the neonatal mouse models.11,12,15 In order to generate a robust CCM mouse model with less dependency on the induction time window and its associated variability, we employed the recently reported Tie2-Dre and Mfsd2a-CrexER(Dre-Cre)duo recombination system 19 to drive Ccm gene deletion in brain ECs. Tie2 is expressed in all endothelial cells from initial cell type specification, Mfsd2a is highly expressed in hepatocytes, multiple cell types in the brain and the skin (sFig. 1). 25 The expression of Mfsd2a in brain endothelial cells initiates from late gestation state (E15.5) in mice. 26 We reasoned the temporal and spatial overlap of Tie2 and Mfsd2a expression patterns will allow this Dre-CrexER dual recombinase system to be used to drive Ccm gene deletion specifically in endothelial cells of the central nerve system (CNS) and induce CCM lesion development without the requirement of tamoxifen induction, and bypass some of the limitations in models driven by mouse lines expressing inducible pan-endothelial or Mfsd2a-CreERT2 lines. We first generated the Tie2-Dre;Mfsd2a-CrexER;RosaRFP mice to test the recombination activity of this system in CNS endothelial cells. The RFP reporter from RosaRFP allele demonstrated Tie2-Dre; Mfsd2a-CrexER (thereafter BEC-Cre) can specifically and efficiently drive recombination of the floxed fragment in the Rosa allele in endothelial cells of cerebrum, cerebellum and retina (sFig. 2), but not in peripheral organs such as liver and testis (sFig. 3 A–B). We then introduced the Dre-Cre dual recombinase system with Ccm2 floxed mice to generate the Tie2-Dre; Mfsd2a-CrexER;Ccm2fl/fl (thereafter, Ccm2BECKO) mice (Figure 1(a)). The Ccm2BECKO mice appear grossly normal and with normal body weight up to 5 months of age (sFig. 4 A). Micro-CT imaging of P30 Ccm2BECKO brains revealed CCM lesions in both the cerebellum and cerebrum (Figure 1(b)). This contrasts with previous CCM model driven by Cdh5-CreERT2 in which CCM lesions were only found in the cerebellum.8,11 The presence of lesions were confirmed with H&E staining of dissected brain tissues (sFig. 4B). Consistent with the robust gene recombination in retina vessels (sFig. 2C), CCM lesions also developed at peripheral of retina in venous track (Figure 1(c)). The dilated and torturous venous vessel can be imaged with Retinal Fundus Imaging in live mice with intravital injection of Fluorescein Sodium dye (Figure 1(d)), although the CCM lesions at peripheral of retina were not visible due to its localization. In consistent with the gene recombination activity of Tie2-Dre;Mfsd2a-CrexER, no vascular malformation were detected in peripheral organs, including liver and testis (sFig. 4 C-E), which has been shown to develop vascular malformation in tamoxifen induced Cdh5cre;Ccm1fl/fl. 4 These results suggest the BEC Dre-Cre system can be used to generate CCM mouse model by driving efficient Ccm2 gene deletion and cause CCM lesion formation throughout the brain.
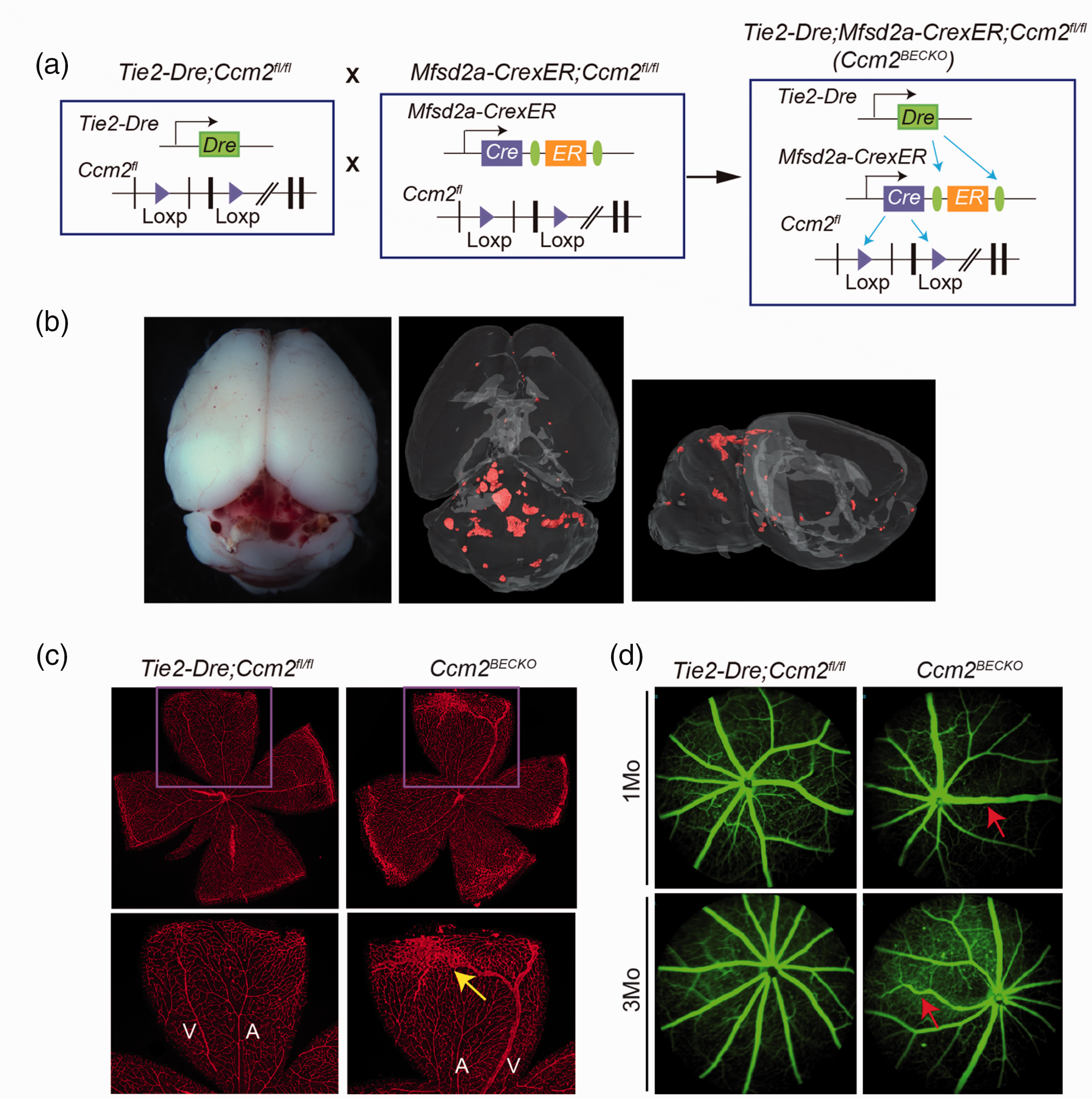
Brain and retina vascular malformations in Ccm2BECKO mice. (a) Schematic of the crosses used to generate the brain endothelial cell (EC) specific Ccm2-deficient mice. (b) White field image of dissected brain and µCT imaging of CCM lesions in the Ccm2BECKO mice at 1 month (mo) of age. (c) Whole-mount IsoB4 staining of the retina vasculature in the Ccm2BECKO mice and littermates at P15. “A” indicates artery; “V” indicates vein; The yellow arrow indicates the malformed vascular plexus in the peripheral of retinal and (d) Fundus fluorescein angiography of the retinal vasculature of the Ccm2BECKO mice and littermates at 1 mo and 3 mo of age. Images were obtained using a Micro IV Retinal imaging Microscope containing a barrier filter. The red arrows indicate the malformed vessels.
Natural history of CCM lesion development in the CCM2BECKO model
The neonatal Ccm2 mouse model driven by Cdh5-CreERT2 mice bears pan-endothelial gene deletion and has several associated limitations, such as the limited induction window that is limited within the first week of postnatal life, the lethality of mice before weaning, and restriction of CCM lesion development in the cerebellum and not in the cerebrum. A chronic Ccm2 mouse model were developed with Slco1c1-CreERT2 line. These model mice have median survival of 183 days after tamoxifen induction at P1. 14 Our Ccm2BECKO mice are all viable up to 7 months of age with complete lethality observed by 1 year of age, and there is no survival difference between female and male Ccm2BECKO mice (Figure 2(a)), the body weight of both female and male Ccm2BECKO mice start to decrease from 6 mo of age (sFig. 4 A). Gross and micro-CT images of CCM lesion development in the Ccm2BECKO mice from P6 to 5 months of age demonstrate gradual development of CCM lesions in both the cerebrum and cerebellum as the mice aging (Figure 2(b)). Lesion burden as calculated by CCM lesion volume (Figure 2(c)) and lesion number (Figure 2(d)) is more severe in the cerebellum than in the cerebrum of the Ccm2BECKO mice from P15 onwards. The lesion volume in the cerebellum show a faster lesion growth rate between P6 and P15, whereas the lesion volume in cerebrum increased the greatest between two to three months of age (Figure 2(c)). We grouped the CCM lesions into three classes according to the lesion volume (small, ≤106 µm3; medium, 106–107 µm3; large, ≥107 µm3). Quantification of lesion numbers reveal there was an exponential emergence of small CCM lesions between 2–3 months of age while the presence of medium-sized CCM lesions significantly amplified at 3–5 months of age in both the cerebrum and cerebellum (Figure 2(d)). Notably, the earliest accumulation of large lesions was observed from P6 to P15 in the cerebellum compared to 2 to 3 month in the cerebrum (Figure 2(d)). The significant increase in large lesions correlated with the sharp increase in lesion volumes in cerebellum from P6 to P15 and in cerebrum from 2 to 3 month (Figure 2(c)). Together, these data suggest that lesion growth in cerebellum occur from P6-P15, and an active lesion genesis window between 2-3mo in both cerebrum and cerebellum for Ccm2BECKO mice. The 2–3 months age period hence could be an important time window of this model for drug treatment and mechanistic studies. Thus, we have generated an effective chronical mouse model of CCM for downstream studies.
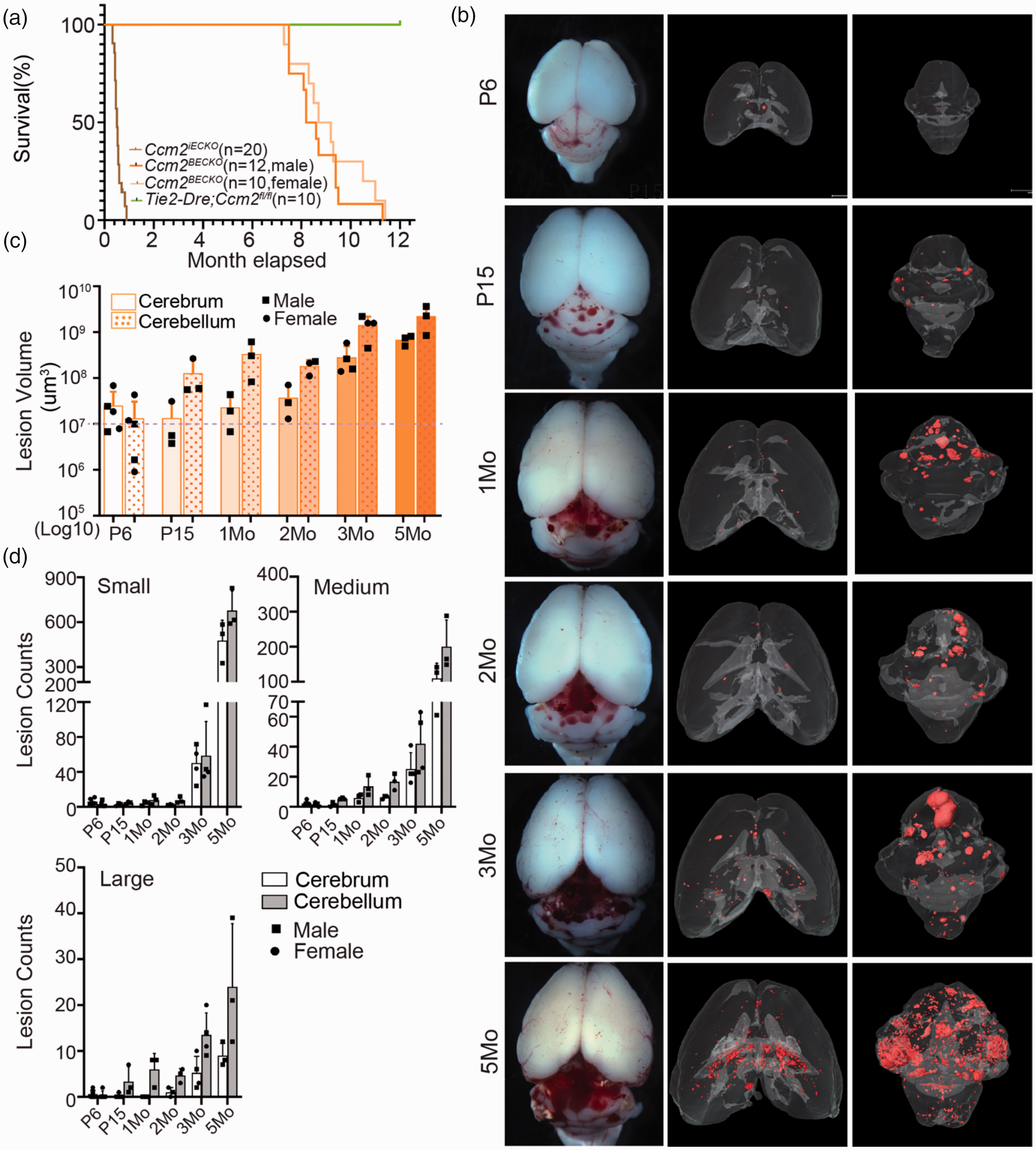
Natural history of CCM lesion development in Ccm2BECKO mice. (a) Survival curve of control, Cdh5-CreERT2;Ccm2fl/fl (Ccm2iECKO) and Ccm2BECKO (male and female) mice. (b) µCT images of CCM lesions in the Ccm2BECKO mice at different ages. (c) Log10 scale quantification of CCM lesion volumes of the cerebrum and cerebellum in the Ccm2BECKO mice at different ages and (d) Quantification of the lesion counts of small (volume ≤106 µm3), medium (volume = 106–107 µm3) and large (volume ≥107 µm3) sized lesions in the Ccm2BECKO mice at different ages. N ≥ 3 for each time point. Each dot represents one mouse. The circle and square symbols indicate datapoints from female and male respectively.
A comparison of CCM lesion development in the CCM1BECKO, CCM2BECKO and CCM3BECKO mice
While mutations in any CCM genes can cause CCM disease, the severity of CCM lesion burden in patients differ depending on which CCM gene is affected. 27 We thus generated the Ccm1BECKO and Ccm3BECKO mice and compared lesion development with the Ccm2BECKO model (Figure 2). Micro-CT analysis show the presence of CCM lesions in both the cerebrum and cerebellum in both the Ccm1BECKO and Ccm3BECKO and as seen in the Ccm2BECKO mice (Figure 3(a) and (b)). However, lesion burden was increasingly more severe in the Ccm3BECKO mice compared to the other lines (Figure 3(a) and (b)). The median survival for Ccm1BECKO, Ccm2BECKO, and Ccm3BECKO were 116, 253, and 56 days, respectively (Figure 3(c)). Similar to that of Ccm2BECKO mice, no survival difference were observed between female and male Ccm1BECKO mice or Ccm3BECKO (sFig. 5). CCM lesion volume and number of lesions were comparable between Ccm1BECKO and Ccm2BECKO mice. Additionally, similar to the Ccm2BECKO mice, P6-P15 was the window that led to significant increases in lesion volume and the number of large lesions in the cerebellum in Ccm1BECKO mice (Figure 3(d) and (e)). Differences in CCM lesion development between the Ccm1BECKO and Ccm2BECKO mice was most noticeable in the cerebellum of the Ccm1BECKO mice where lesion volume significantly increased from P6 brains and remained unchanged after P15 while lesion volume in the Ccm2BECKO cerebellum increased over time (Figure 3(d)). Quantification of lesion number shows that a dramatic increase in the number of small lesions started between 1–3 months, whereas the significant jump in medium and large lesion counts occurred between 3–5 months (Figure 3(e)). The Ccm3BECKO mice appeared to have a delayed CCM lesion burden, where the volume of lesions at P6 was significantly lower than those seen in the Ccm1BECKO and Ccm2BECKO mice (Figure 3(f)). While there was a slow initiation of lesions, lesion formation was most aggressive in the Ccm3BECKO mice where lesion volumes reached a scale of 109 µm3 and 1010 µm3 in both cerebrum and cerebellum at P15 and 2 months of age, respectively (Figure 3(f)). In contrast to Ccm1BECKO and Ccm2BECKO mice, the lesion burden (both volume and number) was more severe in the cerebrum than in the cerebellum in the Ccm3BECKO mice (Figure 3(f) and (g)). These results suggest that while the Ccm3BECKO mice have a milder lesion burden phenotype in the initial stages of lesion formation, the rate of lesions development was most rapid and aggressive such that lesions observed in a P15 Ccm3BECKO brain was comparable to that of Ccm1BECKO mice at 3 months and Ccm2BECKO at 5 months (Figures 2(d), 3(e) and (g)). The exponential increases of the number CCM lesion in Ccm3BECKO brains at 1–2 months of age is consistent with CCM3 patients presenting with more aggressive lesions. These three knockout mouse models represent important tools to investigate the mechanism of each of the gene’s contribution to CCM lesion formation, particularly in understanding the mechanism causing the aggressiveness of CCM lesion growth in CCM3 mutants.
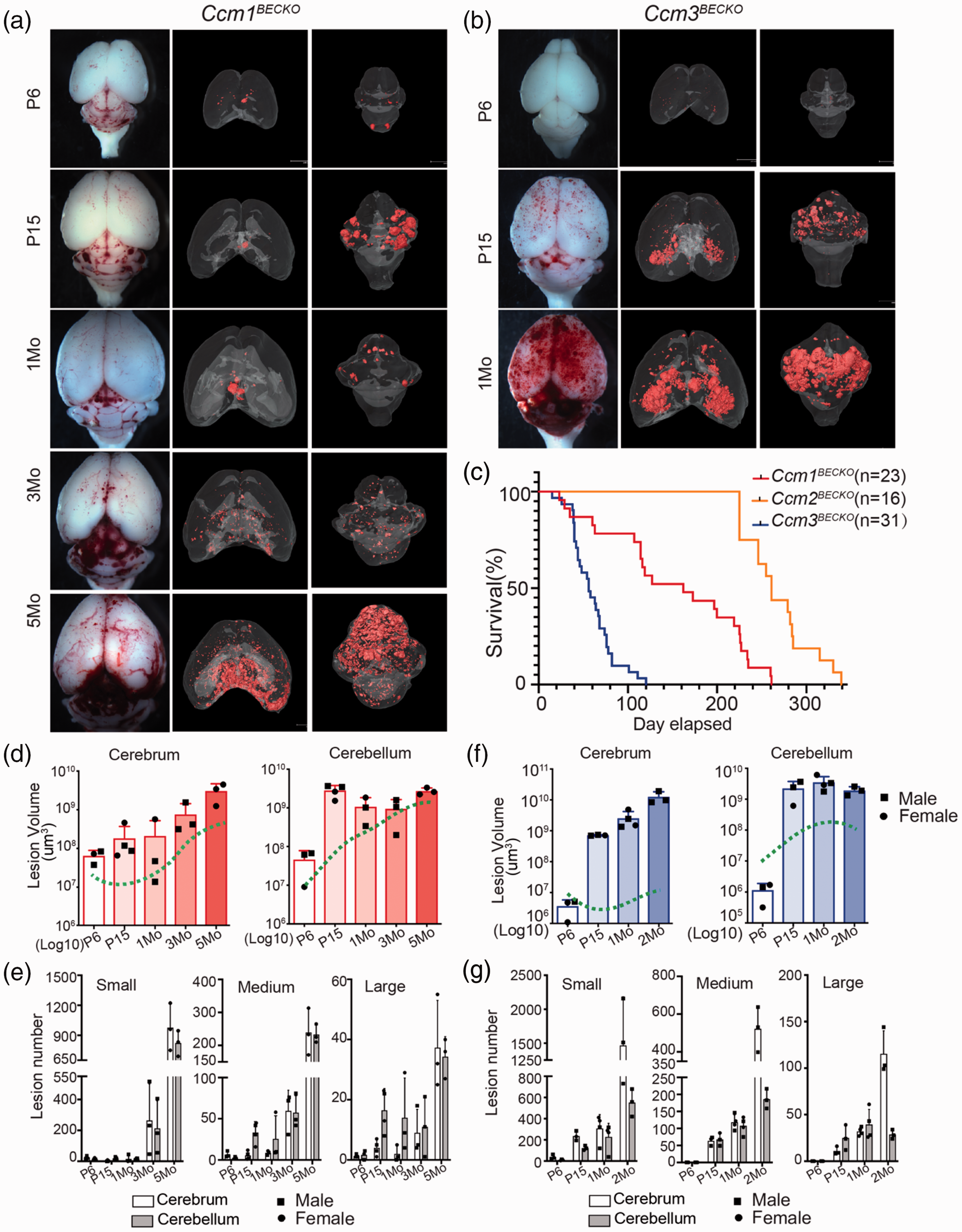
CCM lesion development in Ccm1BECKO and Ccm3BECKO mice. (a) µCT images of CCM lesions in the Ccm1BECKO at different time points. (b) µCT images of CCM lesions in the Ccm3BECKO at different time points. (c) Postnatal survival curve of Ccm1BECKO, Ccm2BECKO and Ccm3BECKO mice. (d) Log10 scale quantification of CCM lesion volumes in cerebrum and cerebellum of in Ccm1BECKO mice. The green dashed lines indicate the trend observed in the Ccm2BECKO mice. (e) Quantification of the lesion counts of small (volume ≤106 µm3), medium (volume = 106-107 µm3) and large (volume ≥107 µm3) sized lesions in the Ccm1BECKO mice. (f) Quantification of CCM lesion volumes in the cerebrum and the cerebellum of the Ccm3BECKO mice. The green dash lines indicate the trend observed in the Ccm2BECKO mice and (g) Quantification of the lesion counts of small (volume ≤106 µm3), medium (volume = 106–107 µm3) and large (volume ≥107 µm3) sized lesions in the Ccm3BECKO mice. N ≥ 3 for each time point. Each dot represents one mouse. The circle and square symbols indicate datapoints from female and male respectively.
Neurological defects in CCMBECKO chronical disease model
In humans, CCM can cause epilepsy, seizure and neurological deficiency.2,3,28 These behavioural characteristics have not been tested in CCM mouse models to date mostly due to the early lethality of previously used CCM model mice. Since our newly generated CcmBECKO mice can survive to adulthood, we were able to monitor their behavioral symptoms. The Ccm3BECKO mice developed epilepsy according to Racine's standard five-stage seizure scale 24 (supplemental video 1). The epilepsy frequency can be monitored based on the mouse movement within the cages (Figure 4(a)). Over a 10 hour continuous recording period, 2-month-old Ccm3BECKO mice experienced seizures that fluctuated between a Racine score of III–IV (approximately 28 times with an average duration of 13 s) and score of V on average ∼22 times with longer average seizure duration (average duration of 23 s) (Figure 4(a)).
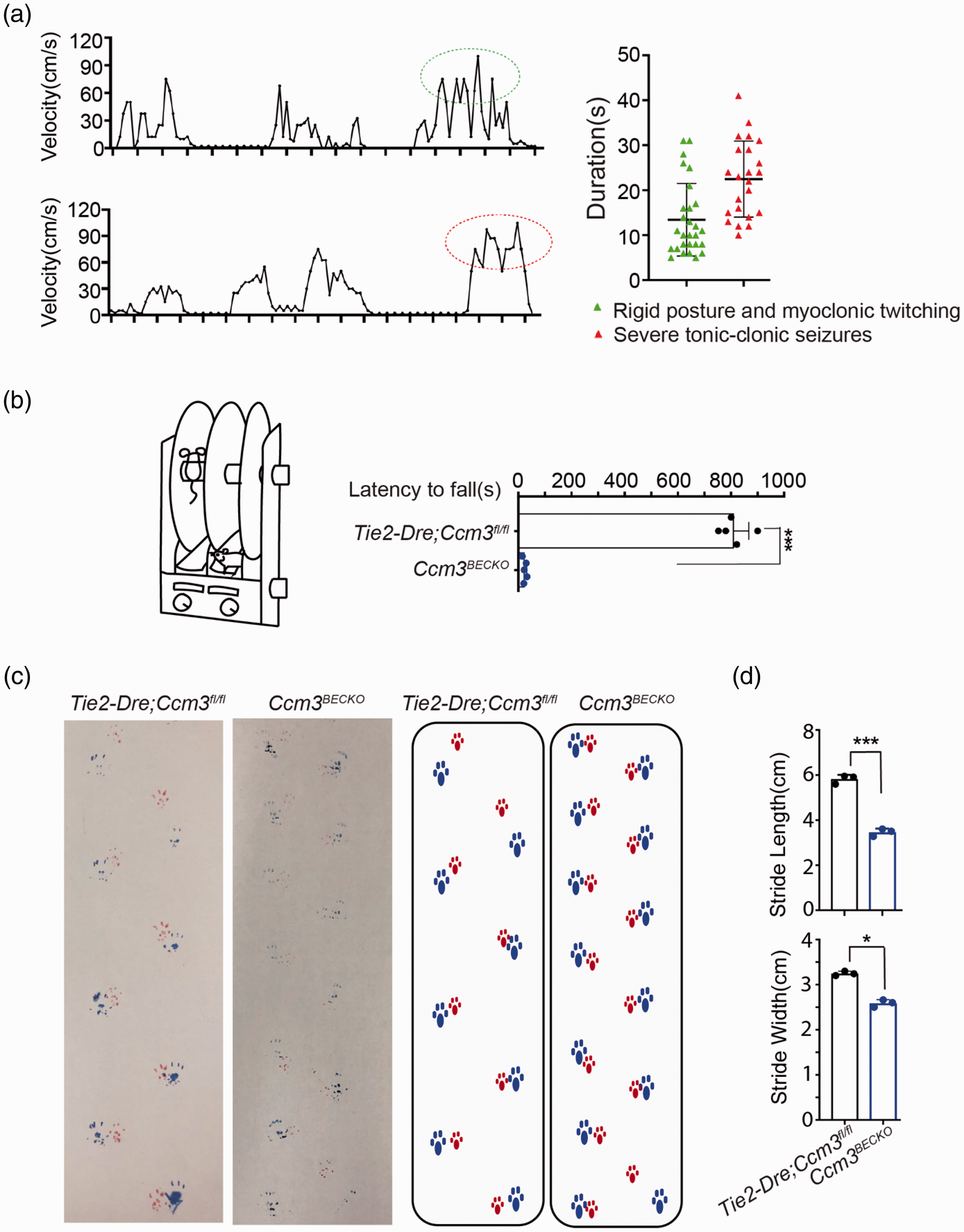
Seizure and movement defects in the Ccm3BECKO mice. (a) Representative velocity plot and duration of seizing episodes in the Ccm3BECKO mice. Tonic-clonic seizures(in red circle)and rigid posture and myoclonic twitching(in green circle) were observed and recorded. (b) Rotarod performance showed as the latency to fall of the Ccm3BECKO and littermate control mice at 2 mo of age. The data represent an average of five animals per group. (c–d) Gait analysis of the Ccm3BECKO and littermate control mice. Representative footprint sequence (two panels on the left) and the corresponding schematic map (two panels on right) (c), and the quantification of the stride length and stride width and (d) are presented. Data in the quantitative plots are presented as mean ± SD and significance determined using unpaired Student’s t-test. N ≥ 3 mice per group, ***P < 0.001; *P < 0.05.
The accelerating rotarod assay was used to assess motor co-ordination and balance function. Ccm3BECKO mice displayed a severely decreased latency to fall from the rotarod in comparison to that of the control animals (Figure 4(b)). Ccm3BECKO mice also presented with shorter stride length and narrower stride width using the gait test (Figure 4(c) and (d)) that indicate weakness in limb muscle control. This is correlated with progressive hemiplegia of the limbs observed in the Ccm2BECKO at 11 months of age (supplemental video 2).
Treatment with ponatinib and dexamethasone inhibit CCM in CCM2BECKO chronical mouse model
The CCM lesion development in chronic CCM models closely resemble human CCM conditions and provide longer time window to test drug treatment regimen and efficacy than previous models. We have previously found ponatinib, a kinase inhibitor, could effectively inhibit CCM lesion initiation and progression in the Cdh5-CreERT2 driven neonatal CCM models. 18 To determine whether the CcmBECKO chronic models can be used for preclinical drug testing, we first adopted our previous treatment protocol in neonatal mouse model by treating the most aggressive Ccm3BECKO model at P6 and analyzed lesion burden at P11. A single treatment with ponatinib (10 mg/kg) at P6 reduced CCM lesion volume by >90% and CCM lesion number by >60% in both the cerebrum and cerebellum (sFig. 6). These data confirm our previous work demonstrating ponatinib as a highly effective inhibitor of CCM lesion, and that it is also effective in preventing lesion formation in the CcmBECKO mice at the neonatal stages. We next treated Ccm2BECKO mice with ponatinib (10 mg/kg) every other day for 2 weeks starting at 2 months of age, a time point when there are well-established CCM lesions and yet new lesions are still actively forming. Analysis of CCM lesion burden at 5 months of age show ponatinib treatment reduced CCM lesion volume by 74.2% and 70.8% in cerebrum and cerebellum, respectively (Figure 5(a) to (c)). The total number of CCM lesions in ponatinib treated group reduced by 60.5% and 65.1% in cerebrum and cerebellum, respectively. And the average lesion number for each size group of lesions were decreased (cerebrum lesion, small: from 577.6 ± 173.5 to 229.8 ± 68.8; medium: from 121.6 ± 61.7 to 48.25 ± 15.9; large: from 13.8 ± 8.1 to 3.5 ± 2.5, cerebellum lesions small: from 447.2 ± 173.8 to 137.8 ± 78.8; medium: from 125.8 ± 64.5 to 38.75 ± 22.8; large: from 15.2 ± 8.1 to 8.0 ± 2.7), although only the reduction of small lesion size group is statistically significant (Figure 5(c)). This suggests that ponatinib is highly effective in inhibiting new lesion formation in this chronical model.
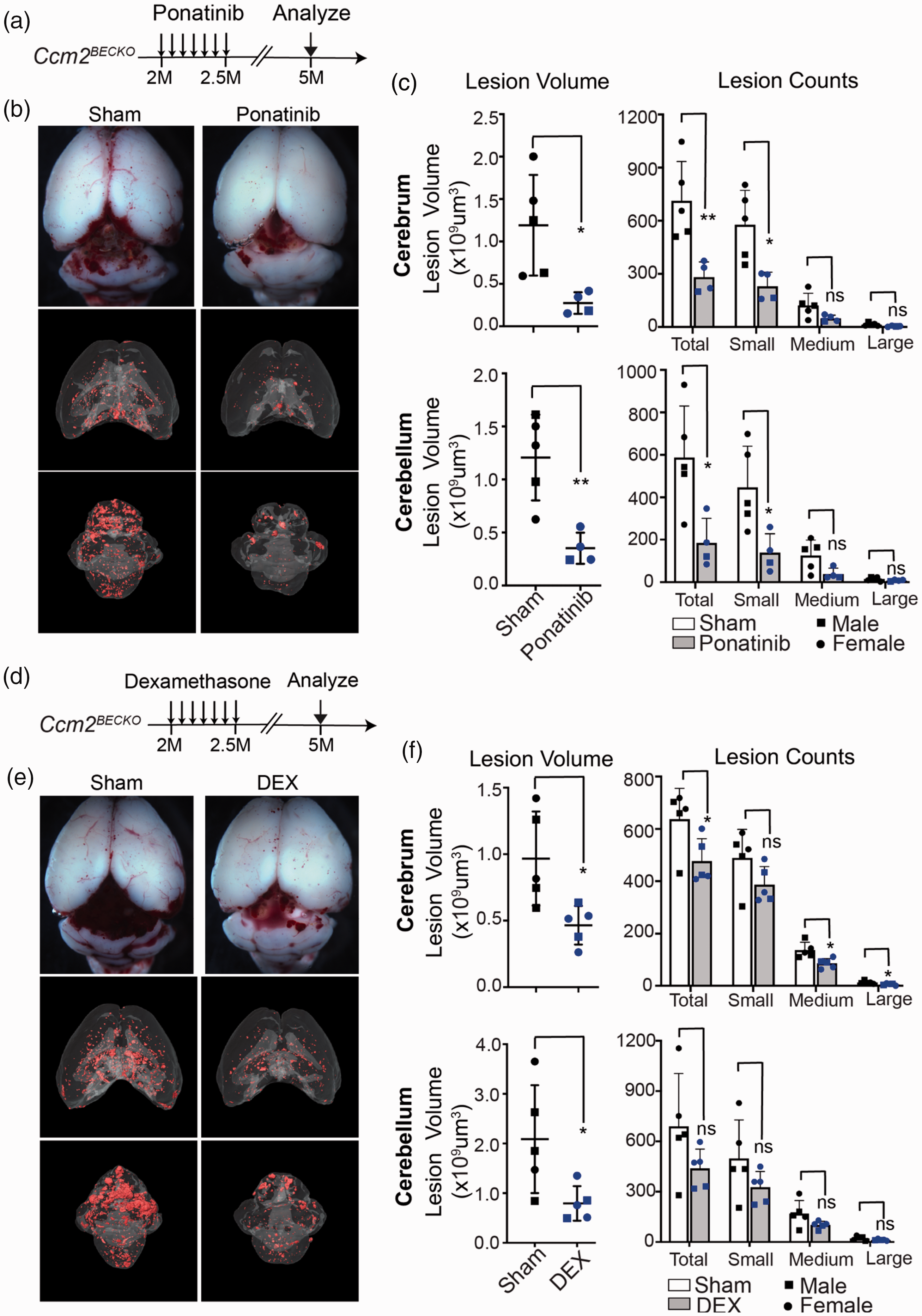
Ponatinib and dexamethasone treatment inhibits CCM lesion growth in adult CCM mouse model. (a) Schematic of ponatinib and vehicle treatment in the Ccm2BECKO mice from 2 months of age for 2 weeks. Brains were collected at 5 months for analysis. (b) µCT images of the Ccm2BECKO mice treated with ponatinib or vehicle. (c) Quantification of CCM lesion volumes (left panels) and lesion counts of different size groups (right panels) in the cerebrum and cerebellum of the control and ponatinib treated Ccm2BECKO mice. (d–e) Two months old adult pups were treated with dexamethasone (DEX) for 2-weeks (d). Whole-mount and µCT images were acquired from dissected brain of control and dexamethasone treated Ccm2BECKO mice at 5-month of age and (f) Quantification of CCM lesion volumes (left panels) and lesion counts of different size groups (right panels) in cerebrum and cerebellum of control and dexamethasone treated Ccm2BECKO mice. N = 5 for the vehicle group, n = 4 for the ponatinib
A previous study has shown inflammatory signals promote CCM lesion formation, and treatment with dexamethasone decrease CCM lesion formation in neonatal model with Cdh5-CreERT2 driven Ccm1 deletion. 10 In the Ccm2BECKO mice, 2 weeks treatment of 2 mg/kg (one dose every other day) starting from 2 months of age reduced CCM lesion volume in cerebrum and cerebellum by 52.1% and 57.5%, respectively (Figure 5(d) to (f)). Dexamethasone (Dex) treatment reduced total lesion number by 25.1% and 36.3%. In cerebrum, the lesion number of medium size and large size were significantly decreased, but not small size group. In cerebellum, lesion numbers for all size groups have a trend of reduction but not significantly reduced (Figure 5(f)). These results suggest Dex mainly affect lesion growth rather than new lesion formation, and Dex is less effective than ponatinib in preventing new lesion formation.
Chronic CCMBECKO model permits drug efficacy analysis in live mice with MRI
Micro-CT has provided the most accurate approach to measure CCM lesion burden to date.8,23 The disadvantage with this assay is that it is terminal so it can only be applied on dissected brains. A method with a capability to measure CCM lesion non-invasively in live animal will be necessary to efficiently assess lesion progression and drug efficacy in animal models. With the establishment of our chronic CCM model where the mice can survive to adulthood, we reasoned that the MRI technology may be able to detect CCM lesion in these mice with sufficient resolution. We use T2 sequence to image CCM lesions in Ccm3BECKO mice at 2 months of age. MRI signal of the lesion (dark area) nicely corresponded to CCM lesions as detected with H&E staining at the comparable section levels of the brain (Figure 6(a)). Because Ccm2BECKO mice have the longest survival time, we assessed the time course of lesion development in Ccm2BECKO mice at the age of 1, 2, and 5 months (Figure 6(b)). Quantifications of CCM lesion burden using the MRI closely reflected the growth of CCM lesions in the Ccm2BECKO mice as measured with micro-CT (Figure 6(c)). In accordance to the limb paralysis phenotype seen in Ccm2BECKO mice (supplemental video 2), we were able to use MRI method to detect numerous cavernous malformations in the spinal cord of the Ccm2BECKO mice that was absent in controls (Figure 6(d)). The cavernous malformations were validated with H&E staining (Figure 6(d)). The robust generation of cavernous malformations in the spinal cord in these CCM model mice provide an additional tissue source to investigate the interaction between endothelial cells and neuronal cells in CCM pathogenesis.
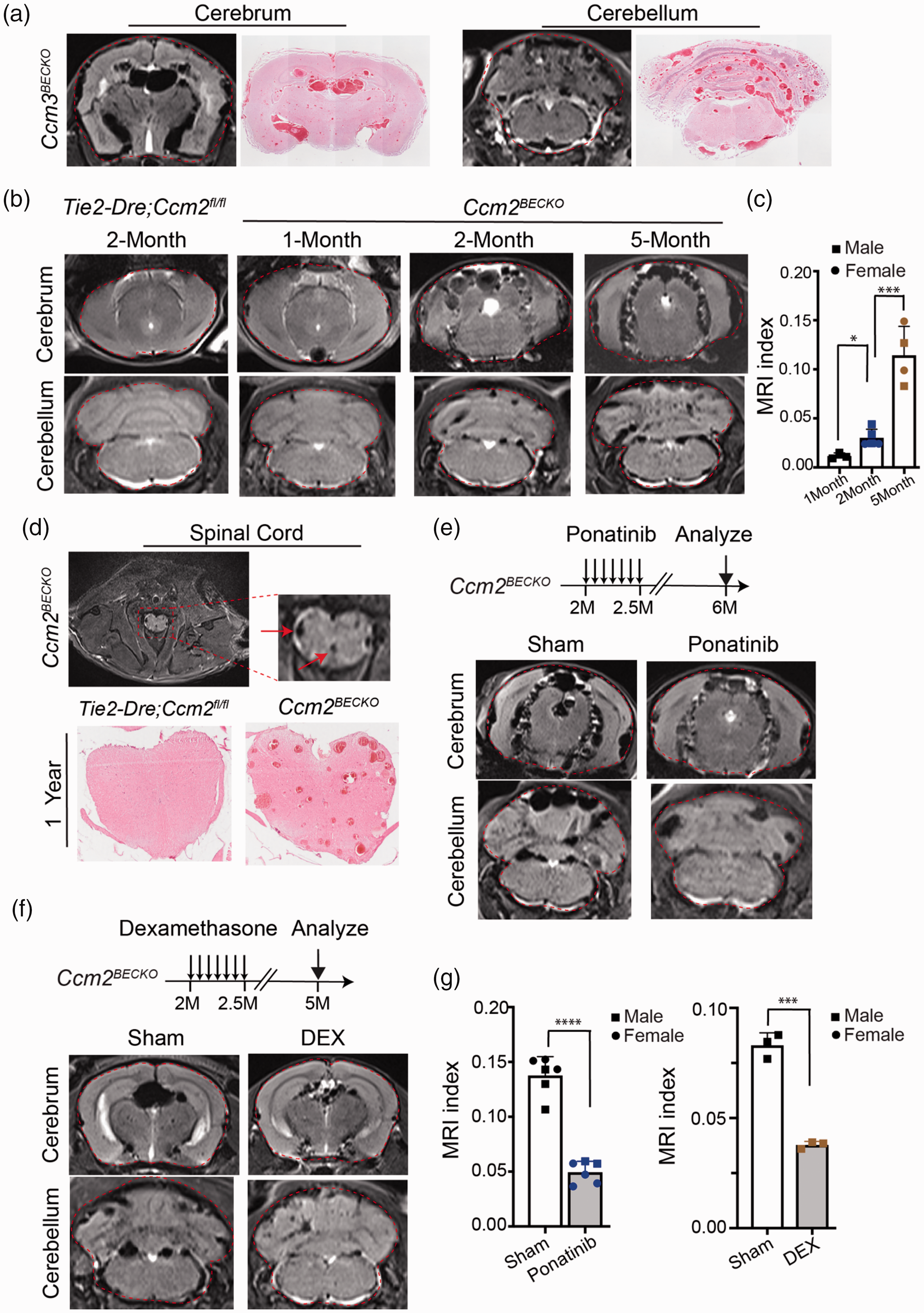
MRI live imaging of CCM lesion in CcmBECKO models. (a) Comparison of MRI images and H&E staining of sections of cerebrum and cerebellum from corresponding Ccm3BECKO mice. The red dashed line indicates the total areas of forebrain or hindbrain. (b) Representative MRI images of control and Ccm2BECKO mice at different ages. MRI index indicates the lesion severity. (c) Quantification of MRI index of Ccm2BECKO mice at different ages. (d) MRI image of the spinal cord of the Ccm2BECKO mouse at 5 month of age (upper panels). Red arrow indicates lesions. H&E staining of the spinal cord of Ccm2BECKO mouse at 12 month of age (lower panels). (e) Representative MRI images of sham or ponatinib treated Ccm2BECKO mouse brains. (f) MRI images of sham or dexamethasone (DEX) treated Ccm2BECKO mouse brains and (g) Quantification of MRI index of CCM lesions in brains of ponatinib or dexamethasone treated Ccm2BECKO mice. Error bars are shown as SD. Data in the quantitative plots are presented as mean ± SD and significance determined using unpaired Student’s t-test. *P < 0.05; ***P < 0.001; ****P < 0.0001. The circle and square symbols indicate datapoints from female and male respectively.
To test whether MRI method can be used to assess the effectiveness of drug treatment on CCM lesion formation non-invasively, MRI was used to scan brains of sham and ponatinib treated animal. Lesion burden, evaluated as an average ratio of lesion area to total brain area, was significantly reduced in the ponatinib and dexamethasone treated mice (Figure 6(e) to (g)). Thus, the combination of adult mouse models of CCM with MRI technology will provide a feasible and efficient resource to screen drugs for CCM treatment.
Discussion
A number of mouse models of CCM have been development in recent years, and they can be classified into three subsets: 1) Ccm gene heterozygosity with loss of function of genome stability genes (p53 and Msh2);29,30 2) inducible Ccm gene deletion driven by pan-endothelial promoters (Cdh5-CreERT2, Tie2-CreERT2, Pdgfb-CreERT2);8,9,11,12,17 and 3) Ccm gene deletion with brain EC selective promoters (Slco1c1-Cre, Slco1c1-CreERT2 and Mfsd2a-CreERT2).10,13,14 The inducible models driven by pan-EC promoters such as Cdh5 and Pdgfb were most widely used in CCM studies, however there are several limitations associated with these models including: 1) the effective induction window is limited and tamoxifen induction introduces large variability in lesion development; 2) CCM lesions only develop in the cerebellum, but not in cerebrum (or in very low numbers); 3) pan-EC deletion cause CCM gene deletion in vascular bed of other organs and cause early premature death of the induced mice. To overcome some of these limitations, optimizing the induction time within the first week of postnatal life has achieved CCM lesion bearing adult mice.12,15 Numerous CCM lesions were observed in both the cerebrum and cerebellum in adult mice of CCM3 models driven by Pdgfb-CreERT2 recombination. 15 Chronic mouse models were also generated by crossing floxed Ccm mouse lines with mouse lines expressing tamoxifen inducible CreERT2 under the control of genes specifically expressed in brain endothelial cells. In the model driven by Slco1c1-CreERT2, the mutant mice had a median life expectancy of 183 days, and CCM lesions were observed in both cerebrum and cerebellum, although the full 3-D distribution of CCM lesion in brain were not shown. 14 In CCM3 model driven by Mfsd2a-CreERT2 with tamoxifen induction at P1-3, most mice survived past 6 months, and numerous CCM lesions were observed in both cerebrum and cerebellum. However, the lesion burden was highly sensitive to induction time. 13 Here, in our Dre-Cre dual recombinase deletion models, we further restrict the deletion domain of Ccm genes in brain endothelial cells, and bypass the induction step requiring tamoxifen, and thus avoid the potential variability associated with the induction procedure. All three models (Ccm1, Ccm2, and Ccm3) has robust CCM lesion formation and survive into adulthood. But our Ccm3BECKO model mice has shorter lifespan than recently reported inducible Ccm3 model driven by the Mfsd2a-CreERT2. 13 This is possibly due to Ccm3 deletion occurred in brain EC earlier and more complete in our non-inducible model than that of inducible model with induction initiation at P1-3.
The advantages of our dual-recombinase models include: 1) CCM lesions develop in both cerebrum and cerebellum; 2) no neonatal induction step is needed thus reducing variability in lesion genesis; 3) the precision of gene deletion specifically in brain ECs extend the life expectancy of the mouse and allow the development of neurologic deficits that closely resemble the human condition. The non-inducible, chronic nature of lesion development throughout the brain in these models make them robust models for pre-clinical drug tests. On the other hand, our current non-inducible dual-recombinase system drives gene deletion in brain endothelial cells from embryonic day 15.5. 26 This is different from human conditions, in which loss of function of CCM genes usually occur postnatally after a second hit.31,32 The deletion of gene expression from late-gestation stage could confer potential defects in the maturation of brain microvasculature, thus complicate CCM lesion pathogenesis in these models. To overcome this limitation, an inducible dual-recombinase system will be helpful for the precise modelling of human conditions. In our current Ccm1/2/3BECKO models, we did not observe any obvious defect in vascular patterning or BBB permeability, although a thorough analysis of neovascular unit and BBB integrity are necessary to fully explore the role of Ccm genes in brain vasculature.
It is not clear why CCM lesion only occurs in cerebellum in neonatal model, but can occur in cerebrum in chronic models.13,14 In our Ccm2 model, CCM lesion burden in the cerebrum were one magnitude less than that of cerebellum from P15 throughout the lifespan. In the Ccm3 model, CCM lesion burden in cerebrum was about 10 time less than that of cerebellum at P15, but grown aggressively and reach 10 time more in the cerebrum than that of cerebellum by adulthood. We speculate that CCM lesions form more efficiently in the cerebrum in our models is likely due to earlier deletion of Ccm genes in the cerebrum when the cerebrum is in an active angiogenic stage. Mfsd2a expression appears in brain endothelial cells as early as E15.5 26 the embryonic endothelium in cerebrum maintains more proliferative capacity compared to that of postnatal mice. Recent discovery of gain-of-function mutation of PI3K contributing to CCM lesion formation directly supports this hypothesis.4,5,7 A direct comparison of absolute lesion burden between our model and other chronic model will be helpful to further address this hypothesis.
The extended lifespan of these chronic models has allowed us to monitor and compare the natural history of CCM diseases with regard to particular CCM gene mutation. In this BECKO system, Ccm1, Ccm2 and Ccm3 deletion all result in CCM lesions in both the cerebrum and the cerebellum that were detected as early as P6. While Ccm3BECKO brains show the least CCM lesion burden at P6, progression and development of lesion burden was most aggressive in this mouse strain compared to other genotypes. This nicely recapitulated the human disease, where loss of function of CCM3 results in the most severe CCM lesions in patients. Looking at lesion development time course in our models, Ccm1BECKO and Ccm2BECKO has a burst of new CCM lesion generation between 2–3 months of age, but different from Ccm2BECKO, Ccm1BECKO mice has an early increase phase of large lesions in cerebellum from P6 to P15, suggesting aggressive lesion growth during this period. The Ccm3BECKO mice have an initial burst of lesion genesis between P6 and P15. This suggest loss of Ccm3 may reprogram the gene expression profile in BECs and prime the endothelial cells to be more susceptible or aggressive to adopt a CCM phenotype. In these chronic models, the aggressive lesion genesis between 2–3 months is an interesting discovery. Elucidating the molecular events underlining this transition may provide intervention targets for CCM patients after first diagnosis of CCM. A blockage of the permissive signal to inhibit new lesion genesis will be a valuable treatment approach. Similarly, these chronic models are also suitable to be used to screen drugs to prevent hemorrhage and the development of neurological deficits. The CCM lesions in the Ccm3BECKO mice usually cause loss of mobility of hindlimb at around 40 days of age, and severe neurologic defects and death at 2 months. Similar symptoms were observed in the Ccm2BECKO mice at 10-12 months of age. The broadness and severity of CCM symptom spectrum allow these mice models to be a more suitable and accurate assay to mimic the human condition and for use in drug discovery studies.
Sex differences in cerebral microvascular gene/protein expression profiles in rodents has been reported in recent studies.33,34 In our study, we analysed CCM lesion development in both female and male of our model mice, the number of datapoints for lesion volume and count in each sex are not sufficient to statistically analyse the difference of lesion burden between female and male, though the datapoints for female and male mice appear to evenly distributed with the data range. The statistical analysis of the survival curves indicated there is no sex difference of long-term outcome of lesion burdens in all three mouse models.
One major technical hurdle for the use of mouse model in the study of molecular and chemical intervention of CCM disease progress is the quantification of CCM lesion burden. Histological sections of brain tissues could be assessed to represent CCM lesion burden, but due to the randomness of lesion location in brain, it is difficult to generate an accurate assessment of lesion burden. A few years ago, we developed a micro-CT method to quantify CCM burden in mouse models.8,23 Micro-CT can give accurate assessment of CCM lesion volume, number and localization, however, a major weakness of this method is the requirement for fixed samples, which mean the mouse has to be sacrificed. A better approach to assess lesion dynamics would be to monitor lesion burden in live mice non-invasively that will allow the monitoring of an individual lesion or overall lesion number and growth over time. We have now developed an MRI protocol to monitor CCM lesion in live mouse. Using our current protocol we are able to achieve the resolution needed to assess CCM burden in live mice. Importantly, this technical is also less time consuming (∼15 min per mouse) compared to ∼2 hours needed for X-ray µCT. As a proof of principle, we show we are able to detect the effect of ponatinib treatment on CCM lesion burden in treated and untreated groups in live animals. We are currently optimizing the protocol to generate images with higher contrast to facilitate quantification, and to improve the sensitivity to detect smaller lesions. These include the implementing of T2*, T2-space and SWI sequences to reach a most effective parameter settings for a selected sequence to facilitate future preclinical studies. The establishment of these new models and the MRI method provide an essential platform for drug discovery research in preclinical studies. An ultimate goal is to create high resolution 3-D rendering of MRI images for accurate quantification.
An interesting question emerging from this study is why Ccm3 deficient mice have less CCM lesions in the initiation stage, but aggressively increase lesion burden in a relatively short time window. Transcriptomic, proteomics and cell behavior studies of ECs at different disease stages from the Ccm3BECKO mice model compared to that of Ccm1BECKO or Ccm2BECKO mice may reveal the novel molecular and/or cellular events that contribute this rapid growth of CCM lesions. An interesting area will be to test whether the clonal expansion and wildtype EC recruitment mechanism of CCM35,36 are differential regulated between CCM1, CCM2 and CCM3 models. Recent studies identified gain-of-function PI3K signaling facilitates CCM lesion formation.4,5,7 It is worth exploring how the classic CCM signaling in combination with unique activation of PI3K signaling may contribute to CCM lesion formation.
Supplemental Material
sj-pdf-1-jcb-10.1177_0271678X221105995 - Supplemental material for Cerebral cavernous malformation development in chronic mouse models driven by dual recombinases induced gene deletion in brain endothelial cells
Supplemental material, sj-pdf-1-jcb-10.1177_0271678X221105995 for Cerebral cavernous malformation development in chronic mouse models driven by dual recombinases induced gene deletion in brain endothelial cells by Xi Yang, Zifeng Dai, Caixia Gao, Yongqiang Yin, Changbin Shi, Renjing Liu, Qichuan Zhuge, Yue Huang, Bin Zhou, Zhiming Han and Xiangjian Zheng in Journal of Cerebral Blood Flow & Metabolism
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: These studies were supported by National Key Research and Development Program of China (grants 2019YFA0802003), National Natural Science Foundation of China grants 81771240 and 821770300 to XZ.
Acknowledgements
We thank Dr. Dean Li for the generous gifts of Ccm1fl/fl and Ccm3 fl/fl mice, Dr Mark Kahn and the members of Kahn Lab for constructive discussions on this project, Dr Shaowu Li at Tiantan Hospital of Capital Medical University for technical assistance of MRI analysis, Drs Chengyong Shen at Zhejiang University and Sijun Zhu at SUNY Upstate Medical University for advices on mouse movement analysis.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
XY, ZD and XZ designed and performed most of the experiments. CG, YY and ZH provided technical and facility support for micro-CT analysis. RL and YH provided support for MRI analysis. CS and QZ helped with clinical analysis of CCM models. BZ provided Tie2-Dre and Mfsd2a-CrexER mouse lines. XY, RL, ZH and XZ analyzed the data and wrote the manuscript.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.