
Abstract
Voluntary asphyxia imposed by static apnea challenges blood-brain barrier (BBB) integrity in humans through transient extremes of hypertension, hypoxemia and hypercapnia. In the present study, ten ultra-elite breath-hold divers performed two maximal dry apneas preceded by normoxic normoventilation (NX: severe hypoxemia and hypercapnia) and hyperoxic hyperventilation (HX: absence of hypoxemia with exacerbating hypercapnia) with measurements obtained before and immediately after apnea. Transcerebral exchange of NVU proteins (ELISA, Single Molecule Array) were calculated as the product of global cerebral blood flow (gCBF, duplex ultrasound) and radial arterial to internal jugular venous concentration gradients. Apnea duration increased from 5 m 6 s in NX to 15 m 59 s in HX (P = <0.001) resulting in marked elevations in gCBF and venous S100B, glial fibrillary acidic protein, ubiquitin carboxy-terminal hydrolase-L1 and total tau (all P < 0.05 vs. baseline). This culminated in net cerebral output reflecting mildly increased BBB permeability and increased neuronal-gliovascular reactivity that was more pronounced in NX due to more severe systemic and intracranial hypertension (P < 0.05 vs. HX). These findings identify the hemodynamic stress to which the apneic brain is exposed, highlighting the critical contribution of hypoxemia and not just hypercapnia to BBB disruption.
Introduction
The neurovascular unit (NVU) comprises the interactive cellular network responsible for the regulation of cerebral blood flow (CBF) and blood-brain barrier (BBB) integrity that synergistically preserve neuronal, glial and vascular homeostasis. 1 The NVU is integral to establishing efficient clearance of carbon dioxide (CO2) and other waste products while continuously supplying oxygen (O2)/glucose to which the human brain has evolved exquisite sensitivity given its disproportionately high mass-specific energy demands, limited energy stores and almost exclusive reliance on aerobic metabolism. 2 This renders the brain especially vulnerable to asphyxia, with substrate depletion and progression towards rapid and largely irreversible neuronal death. 3
The competitive sport of maximal static apnea performed by ultra-elite breath-hold (BH) divers provides a unique clinical opportunity to study extreme (voluntary) asphyxia. The current world record with face immersion is 11 min 35 s; this can be further extended to a remarkable 24 min 33 s following prior hyperoxic hyperventilation with divers capable of achieving arterial O2 tensions <20 mmHg and CO2 tensions >70 mmHg at apnea breakpoint. 4 Yet despite profound asphyxia, the cerebral delivery of oxygen (CDO2) remains well preserved due to a marked increase in CBF. 5 These hemodynamic challenges provide a unique opportunity to investigate the structural and functional ‘bandwidth’ of NVU integrity in unanesthetized and otherwise healthy humans and mechanisms underlying cerebral substrate conservation during the most severe extremes of O2-CO2 stress recorded to date.
In this work, we sought to evaluate the transcerebral exchange kinetics of a comprehensive and ultrasensitive panel of NVU proteins via the Fick principle, combining volumetric assessment of CBF with concurrent sampling of arterio-jugular venous concentration gradients (a-vD). This allows a quantitative evaluation of indicators of BBB permeability and brain damage under normal or dysregulated conditions after apnea, albeit in the absence of complimentary neuroimaging approaches (see Limitations). Our primary aim was to examine the impact of apnea on the structural integrity of the NVU with a specific focus on the BBB. Our secondary aim was to distinguish between hypoxemia and hypercapnia as the dominant vasoactive stimulus, although we were not in a position experimentally to fully control for the potentially confounding effects of different BH times and degree of peripheral/central acidosis, that likely impact biomarker exchange originating from the brain and/or other organs.
The present study enrolled ultra-elite BH divers to perform two maximal dry, static apneas following: [1] normoxic normoventilation (NX) resulting in severe apneic hypoxemia-hypercapnia and [2] hyperoxic hyperventilation (HX) to alleviate apneic hypoxemia while compounding hypercapnia. We hypothesized that: [1] apnea is generally associated with a net cerebral release or output of NVU proteins (v > a) specifically S100B, glial fibrillary acidic protein (GFAP), neuron-specific enolase (NSE), neurofilament light-chain (NF-L), ubiquitin carboxy-terminal hydrolase L1 (UCH-L1) and total tau (T-Tau) collectively reflecting increased BBB permeability and neuronal-gliovascular reactivity and [2] despite considerably shorter BH times, NVU protein output would be more pronounced during hypoxemic compared to hyperoxemic-hypercapnic stress (NX>HX) as a consequence of higher systemic and intracranial hypertension (IH). The conceptional framework of this working hypothesis is illustrated in Figure 1(a).

Working hypothezes (a) and experimental design (b). a. Hypothesized impact of hypoxemic-hypercapnia and hyperoxemic-hypercapnia on molecular-hemodynamic biomarkers underlying neurovascular unit (NVU) structure and function. Predicted differences between trials highlighted by red circles. O2, oxygen; CO2, carbon dioxide; CBF, cerebral blood flow; MAP, mean arterial pressure; ICP, intracranial pressure; BBB, blood-brain barrier. Note typical human experimental setup highlighting arterial-jugular venous (transcerebral) sampling of blood combined with volumetric assessment of CBF (upper middle left inset). Note also lateral cervical spine radiograph illustrating correct positioning of the jugular venous catheter above the lower border of the C1 vertebra (upper middle right inset). b. Two submaximal (practice) apneas preceded both normoxia (NX) and hyperoxia (HX) trials following prior NX normoventilation (21% O2). The first practice was performed at functional residual capacity (FRC) for 6 involuntary body movements (IBMs) and the second practice at total lung capacity (TLC) for 10 IBMs. Both maximal apneas were performed at TLC and the hyperoxia apnea was preceded by 15 min of controlled hyperventilation with 100% O2. Measurements were performed at baseline (BL) and timed to coincide with the end of each maximal apnea that included volumetric assessment of CBF combined with synchronous sampling of radial arterial and jugular venous blood to assess transcerebral exchange kinetics of NVU proteins.
Materials and methods
Ethics
Experimental procedures were approved by the University of Split Ethics Committee (#H14-00922). All procedures were carried out in accordance with the most (7th) recent amendment of the Declaration of Helsinki of the World Medical Association (with the exception that it was not registered in a publicly accessible database prior to recruitment) with verbal and written informed consent obtained from all participants.
Participants
Given the need for extended apneas and inability to safely ‘recreate’ such extremes of hypoxemia-hypercapnia in ‘normal’ (i.e. non-BH control) participants, we recruited ten ultra-elite BH divers (6♂, 4♀) from the Croatian and Slovenian National Apnea Team. They were 33 (mean) ± 9 (SD) years old with a body mass index of 23 ± 2 kg/m2 and forced vital capacity of 6.40 ± 1.48 L. Six of them were considered world-class given that they were among the world’s top 10 BH divers in their competitive careers. One female diver set a new official world record in dynamic apnea while another male recently set the world record for apnea following hyperoxia (24 min 33 s). All divers had been training specifically for diving apnea four to five times a week for at least three years prior to the study. A medical examination confirmed that all participants were free of cardiovascular, pulmonary and cerebrovascular disease and were not taking any nutritional supplements including over-the-counter antioxidant or anti-inflammatory medications. They were instructed to refrain from physical activity, caffeine and alcohol and to follow a low nitrate/nitrite diet 24 h prior to formal experimentation. 6 Participants attended the laboratory following a 12 h overnight fast.
Design
Parts of this study including select blood gas variables and aspects of cerebral hemodynamic function have previously been published as part of a wider investigation that included a separate focus on the link between hypercapnia and cerebral oxidative metabolism during apnea. 7 Thus, although the present study adopted an identical experimental design, it constitutes an entirely separate question complemented by de novo experimental measures. Participants were required to perform three maximal apneas in normoxia (NX) [1] with and [2] without prior hyperventilation in a counter-balanced fashion followed by [3] hyperventilation in hyperoxia (HX), with each trial separated by 30 min of recovery. Herein, we focus specifically on the two maximal apneas performed ‘without’ hyperventilation in NX and after hyperventilation in HX. The order of trials was non-randomized given the long-lasting carryover effects of hyperoxia and fatigue associated with a more prolonged apnea. 7 Data were collected at baseline and as close as possible to the point of apnea termination (Figure 1(b)).
Procedures
All apneas were completed on a single day with the Croatian national apnea coach (Dr I Drvis) present to motivate divers and ensure true maximal efforts. Each apnea was preceded by 30 min of supine rest followed by two standardized preparatory (practice) apneas designed to maximize the experimental apneas 8 (see below). The first preparatory apnea was performed at functional residual capacity (FRC) until seven involuntary breathing movements (IBMs) were attained. Two minutes later, the second preparatory apnea was performed at total lung capacity (TLC) lasting for ten IBMs. Participants then rested quietly for 6 min prior to two ‘maximal’ apneas that were each performed at TLC, with the extent of glossopharyngeal insufflation (lung packing) standardized across trials:
Trial 1: Prior normoxic normoventilation (NX)
Participants performed a maximal apnea in room air (normoxia).
Trial 2: Prior hyperoxic hyperventilation (HX)
Participants performed a maximal apnea preceded by 15 min of standardized hyperventilation of 100% O2 while receiving auditory feedback from the coach to achieve an end tidal PCO2 (PETCO2) of ∼20 mmHg.
Blood sampling
Catheterization
Participants were placed slightly head down on a hospital bed, and two catheters were inserted retrograde using the Seldinger technique under local anesthesia (1% lidocaine) via ultrasound guidance. A 20-guage arterial catheter (Arrow, Markham, ON, Canada) was placed in the right radial artery (RA) and attached to an in-line waste-less sampling setup (Edwards Lifesciences VAMP, CA, USA) connected to a pressure transducer that was placed at the height of the right atrium (TruWave transducer). A central venous catheter (Edwards PediaSat Oximetry Catheter, CA, USA) was placed in the right internal jugular vein (JV) and advanced towards the jugular bulb located at the mastoid process, approximately at the C1-C2 interspace (Figure 1). Facial vein contamination was ruled out by ensuring that all jugular venous oxyhemoglobin saturation (SO2) values were <75% during resting conditions. All catheters were continuously infused with normal saline (3 mL/h) to maintain patency. Participants rested for at least 30 min following catheter placement prior to the collection of baseline samples.
Collection and storage
Blood samples were drawn without stasis simultaneously from the RA and JV directly into Vacutainers (Becton, Dickinson and Company, Oxford, UK) before centrifugation at 600 g (4 °C) for 10 min. With the exception of blood for determination of blood gas variables, serum samples were decanted into cryogenic vials (Nalgene Labware, Thermo Fisher Scientific Inc, Waltham, MA) and immediately snap-frozen in liquid nitrogen (N2) before transport under nitrogen gas (Cryopak, Taylor-Wharton, Theodore, AL) from Croatia to the United Kingdom. Samples were left to defrost at 37 °C in the dark for 5 minutes prior to batch analysis.
Measurements
Blood gas variables
Whole blood was collected into heparinized syringes, maintained anerobically at room temperature and immediately analyzed for hemoglobin (Hb), hematocrit (Hct), partial pressures of oxygen, carbon dioxide (PO2/PCO2), SO2, pH and glucose (Glu) using a commercially available cassette based analyzer (ABL-90 FLEX, Radiometer, Copenhagen, Denmark). Molar hydrogen ion (H+) concentration was calculated as [H+] = 10−pH. Intra and inter-run coefficients of variation (CVs) for all metabolites were <5%.
NVU proteins
A panel of serum biomarkers were quantified using ultrasensitive analytical platforms. S100B, a calcium-binding protein expressed and released predominantly by astrocytes and Schwann cells found at the perivascular brain space 9 and GFAP, an intermediate filament protein expressed predominantly by astrocytes 10 were employed as biomarkers of BBB permeability and glio-vascular damage. NSE, an intracytoplasmic glycolytic enzyme derived from neuronal cytoplasm and neuroendocrine cells, 11 NF-L, a component of the axonal cytoskeleton expressed primarily in large-caliber myelinated subcortical axons 12 and UCH-L1, a neuron-specific cytoplasmic enzyme concentrated in dendrites 13 were taken to reflect neuronal-axonal damage. T-Tau, a microtubule-associated protein expressed predominantly in short cortical unmyelinated axons 14 was employed as a surrogate for axonal degeneration. The underlying source(s), biochemistry, detection and clinical interpretation of these biomarkers have recently been reviewed. 15 Automated high-sensitivity clinical grade ELISA (LIAISON®, DiaSorin, Saluggia, Italy) was used to measure S100B and NSE. We employed the Neurology 4-Plex assay kit (Quanterix Corp, Lexington, MA, USA) 16 to measure GFAP, NF-L, UCH-L1 and T-Tau proteins on a single molecule array (Simoa) HD-1 Analyzer (Quanterix Corp). Based on singulation of enzyme labelled immune-complex on paramagnetic beads, this digital ELISA assay is considered ∼1,200 fold more sensitive than conventional immunoassays. 17 All samples were analyzed following (4-fold) dilution with the diluent provided in the kit (phosphate buffer with bovine serum and heterophilic blocker solution) to minimise matrix effects. The intra- and inter-assay CVs for all metabolites were <5%.
Cardiopulmonary function
All cardiopulmonary measurements were averaged over 15 s immediately prior to blood sampling. A lead II electrocardiogram (Dual BioAmp; ADInstruments, Oxford, UK) was used to measure heart rate (HR). Intra-arterial ABP was recorded directly via a pressure transducer placed at the level of the heart for determination of MAP. Internal jugular venous pressure
Cerebrovascular function
Intracranial
Blood velocity in the middle cerebral artery (MCAv, insonated through the left temporal window at a depth of ∼1 cm distal to the MCA-anterior cerebral artery bifurcation) and posterior cerebral artery (PCAv, insonated at the P1 segment through the right temporal window) were measured using standardized procedures with a 2 MHz pulsed transcranial Doppler ultrasound (TCD; Spencer Technologies, Seattle, WA, USA). Bilateral TCD probes were secured using a specialized commercial headband (Mark600, Spencer Technologies, Seattle, WA, USA) using standardized search techniques. Between-day CVs for MCAv and PCAv are 3% and 2%, respectively.
Extracranial
Continuous diameter, velocity and blood flow recordings in the right internal carotid and left vertebral arteries (QICA/QVA) with no evidence of asymmetry, were obtained using a 10 MHz, multifrequency, linear array vascular ultrasound (Terason 3200, Teratech, Burlington, MA). Arterial diameter was measured via B-mode imaging, whereas peak blood velocity was simultaneously measured with pulse-wave mode. The ICA was insonated ∼2 cm from the carotid bifurcation, while the VA was insonated at the C5–C6 or C4–C5 space and standardized within participant across trials. The steering angle was fixed to 60° and the sample volume was placed in the centre of the vessel adjusted to cover the entire vascular lumen. All images were recorded as video files at 30 Hz and stored for offline analysis using customized edge detection software designed to mitigate observer bias. 19 Simultaneous measures of arterial diameter and velocity over >12 consecutive cardiac cycles were used to calculate flow. Between-day CVs for QICA and QVA are 5% and 11%, respectively.
Data integration
All cardiopulmonary measurements were sampled at 1 kHz and integrated into PowerLab® and LabChart® software (ADInstruments, Bella Vista, NSW, Australia) for online monitoring and saved for offline analysis.
Calculations
Perfusion
Volumetric blood flow was calculated offline as:
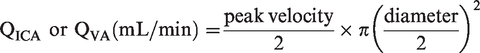
QICA or QVA were averaged for a 1 min baseline and during the last 30 s of apnea.
During the latter half of each apnea, IBMs and corresponding contraction of the sternocleidomastoid prevented reliable blood velocity traces. To circumvent this, QICA and QVA were estimated from changes in MCAv (for ICA flow) and PCAv (for VA flow) respectively using a previously validated approach:
20
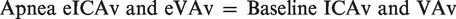
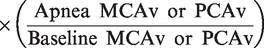
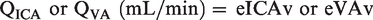
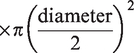
Assuming symmetrical blood flow of contralateral ICA and VA arteries, (brain) mass-normalized global cerebral blood flow (gCBF) was calculated as:
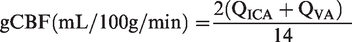
Cerebral perfusion pressure (CPP) was calculated as MAP-IJVP with the latter serving as a validated surrogate of ICP including excellent agreement at <20 mmHg for ICP. 22
Bioenergetics
Arterial and jugular venous O2 content (caO2 and cjvO2) were calculated as:
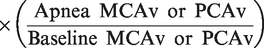
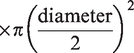
Cerebral O2 and glucose delivery (CDO2/CDGlu) were calculated as:
CDO2 (μmol/100 g/min) = gCBF × caO2
CDGlu (μmol/100 g/min) = gCBF × aGlu
O2 and glucose extraction were calculated as:
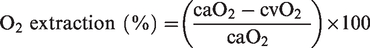
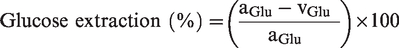
The global cerebral metabolic rate of oxygen (gCMRO2) and glucose (gCMRGlu) were calculated as:
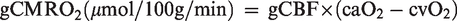
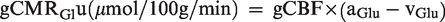
Exchange
gCBF was converted into global cerebral serum (gCSF) flow:
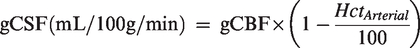
Net exchange (pg/100g/min) = gCBF or gCSF × a-vD
where a-vD represents the arterio-jugular venous concentration difference of each NVU protein. By convention, a positive value refers to net uptake (loss or consumption) whereas a negative value indicates net output (gain or formation) across the cerebrovascular bed.
Statistical analysis
Data were analyzed using the Statistics Package for Social Scientists (IBM SPSS Statistics Version 28.0). Distribution normality was confirmed using Shapiro-Wilk W tests (P > 0.05). Data were analyzed using a combination of two (State: Baseline vs. Apnea × Site: Arterial vs. Venous) and three (Trial: NX vs. HX × State: Baseline vs. Apnea × Site: Arterial vs. Venous) factor repeated measures analyzes of variance. Post-hoc Bonferroni-corrected paired samples t-tests were employed to locate differences following an interaction. Relationships were analyzed using Pearson Product Moment Correlations. Significance was established at P < 0.05 for all two-tailed tests and data presented as mean ± SD.
Results
Apnea duration
BH times increased from 5 m 6 s ± 1 m 2 s (range: 3 m 37 s to 6 m 49 s) in NX to 15 m 59 s ± 3 m 21 s (range: 9 m 39 s to 21 m 2 s) in HX (P = <0.001). No adverse events such as loss of motor control or consciousness were recorded.
Blood gas analysis
The NX apnea induced severe systemic hypoxemia [reduced partial pressures of O2 (PO2), saturation (SO2) and O2 content (cO2)] and profound hypercapnia (elevated PCO2) and metabolic acidosis (reduced pH and elevated
Blood gas variables.
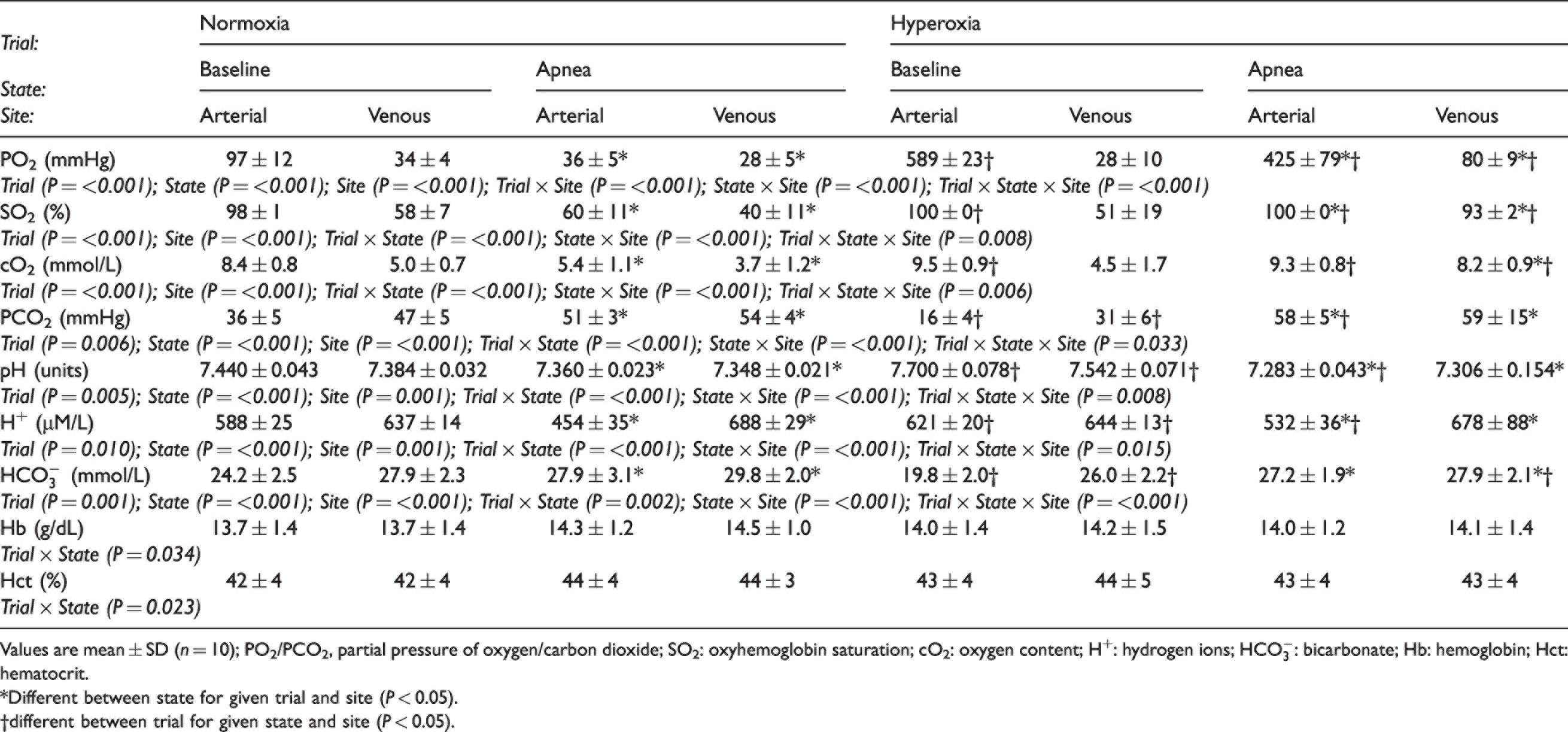
Values are mean ± SD (n = 10); PO2/PCO2, partial pressure of oxygen/carbon dioxide; SO2: oxyhemoglobin saturation; cO2: oxygen content; H+: hydrogen ions;
*Different between state for given trial and site (P < 0.05).
†different between trial for given state and site (P < 0.05).
Cardiopulmonary function
Both SV and
Cardiopulmonary function.
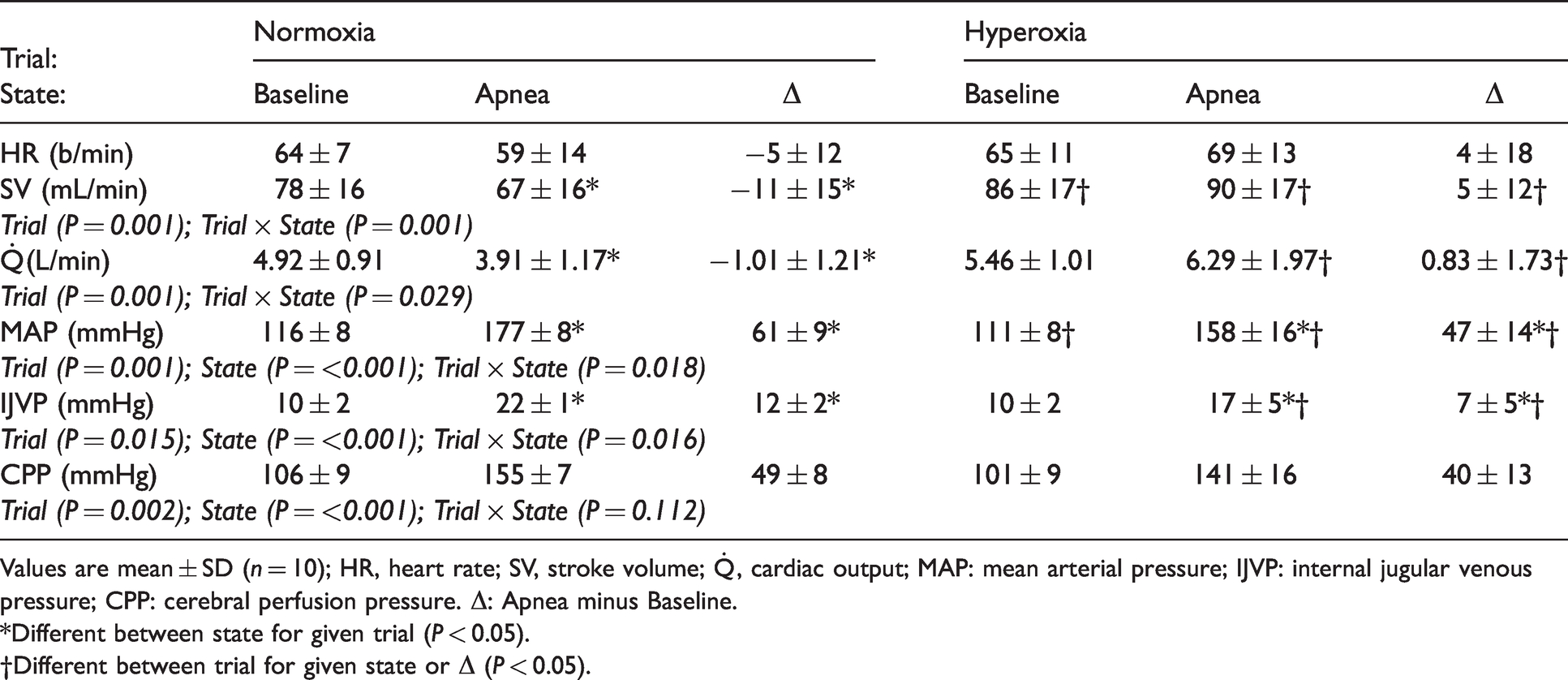
Values are mean ± SD (n = 10); HR, heart rate; SV, stroke volume;
*Different between state for given trial (P < 0.05).
†Different between trial for given state or Δ (P < 0.05).
Cerebrovascular function
Figure 1(a) to (i) summarises the cerebrovascular responses to both apneas highlighting the variability associated with those individual participants with the shortest (S, NX: 3 m 37 s and HX: 9 m 39 s) and longest (L, NX: 6 m 49 s and HX: 21 m 2 s) BH times. Apnea generally increased gCBF given equivalent increases in QICA and QVA (Figure 1(a) to (c)). While maximal gCBF was not different between trials, the relative increase in perfusion was greater in HX (206 ± 52% vs. 83 ± 22%) due to consistently lower perfusion at baseline, coinciding with hyperventilation-induced hypocapnia (Table 1). Corresponding increases in global O2 and glucose delivery (Figure 2(d) and (e)) and reciprocal reductions in extraction (Figure 2(f) and (g)) during apnea were also more pronounced in HX. Apnea generally decreased gCMRO2 and gCMRGlu (Figure 2(h) and (i)) with no differences between NX and HX. An inverse relationship was observed between the elevation in PaCO2 and the reduction in gCMGlu in NX (r = −0.679, P = 0.031). No other relationships were observed between BH time, apnea-induced reduction in PaO2 or elevation in PaCO2 and any of the cerebrovascular metrics in NX (r = −0.590 to 0.345, P = 0.072 to 0.983) or HX (r = −0.470 to 0.555, P = 0.090 to 0.891).
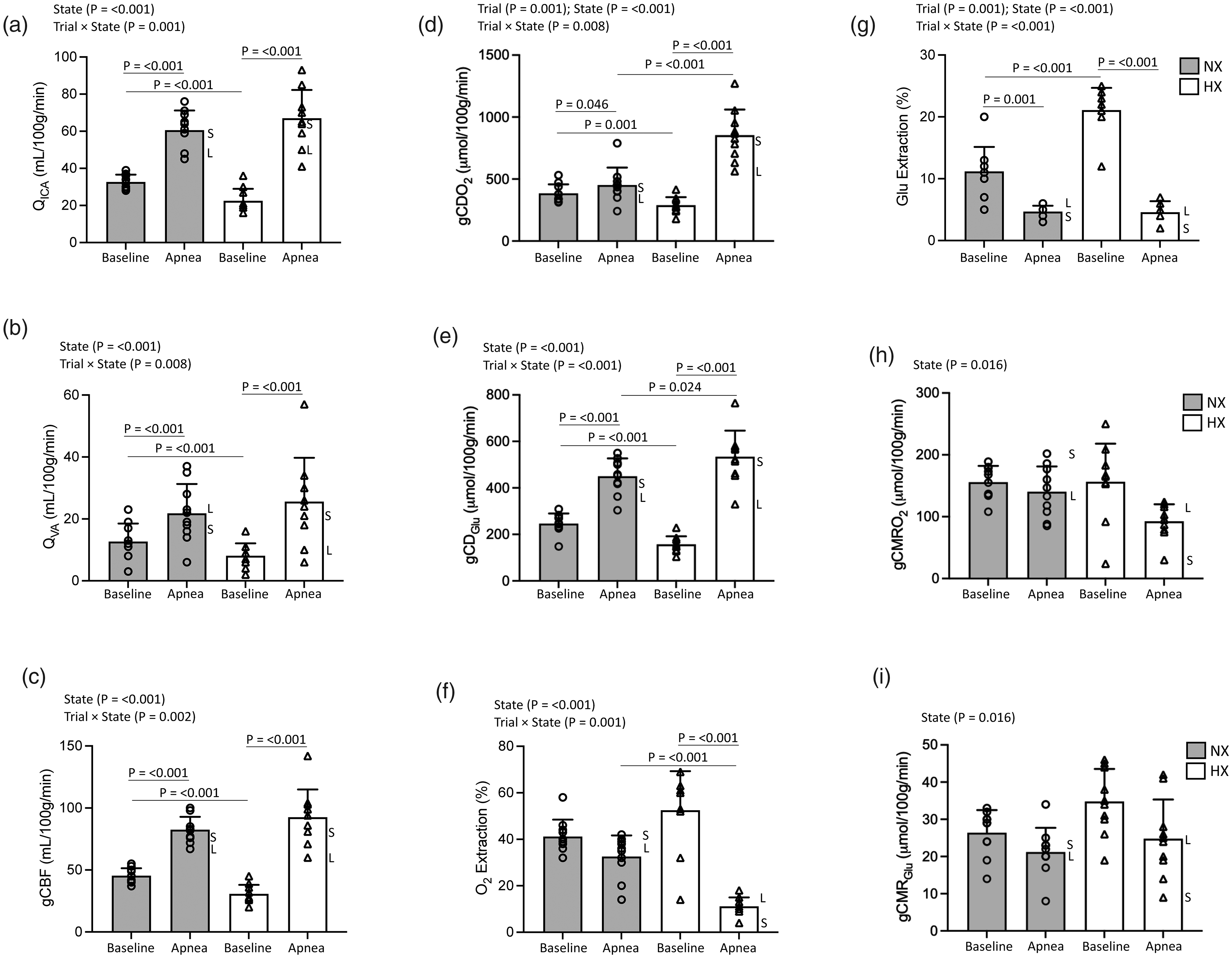
Cerebrovascular function. Impact of maximal apnea(s) on the integrated regulation of cerebral bioenergetics. Values are mean ± SD (n = 10); a, QICA (internal carotid/vertebral artery blood flow ×2); b, QVA (vertebral artery blood flow ×2); c, gCBF (global cerebral blood flow); d, gCDO2, global cerebral delivery of oxygen; e, gCDGlu, global cerebral delivery of glucose; f, O2 (oxygen) extraction; g, Glu (glucose) extraction; h, gCMRO2 (global cerebral metabolic rate of oxygen); i, gCMRGlu (global cerebral metabolic rate of glucose). NX: normoxia trial; HX: hyperoxia trial. S and L annotations for each apnea highlight individual participants with shortest (S, NX: 3 m 37 s and HX: 9 m 39 s) and longest (L, NX: 6 m 49 s and HX: 21 m 2 s) breath-hold times.
NVU proteins
Figure 3(a) to (f) summarizes the transcerebral exchange of NVU proteins during both apneas further highlighting the variability associated with those individual participants with the shortest (S) and longest (L) BH times. Apnea markedly increased the net cerebral output (v > a) of S100B, GFAP, UCH-L1, NF-L and T-Tau and was more pronounced in NX (Table 3, Figure 3(a) and (b), (d) to (f)). In contrast, apnea reversed NSE output (v > a) to uptake (a > v) with no differences between NX and HX (Table 3, Figure 3(c)). Linear relationships were observed between the apnea-induced reductions in PaO2/SaO2 and increase in NF-L gradient in NX (r = 0.770/0.557, P = 0.009/0.045) and elevation in PaCO2 and decrease in GFAP gradient in HX (r = −0.638, P = 0.047). A relationship was observed between the elevation in PaCO2 and decrease in NSE gradient in HX (r = 0.739, P = 0.015). A relationship was observed between the apnea-induced elevation in IJVP and the increase in the gradient for S100B in NX (r = −0.579, P = 0.049). No other significant relationships were observed between BH time, apnea-induced reduction in PaO2 or elevation in PaCO2 and gradients for any of the NVU proteins in NX (r = −0.615 to 0.460, P = 0.058 to 0.989) or HX (r = −0.567 to 0.291, P = 0.087 to 0.979).
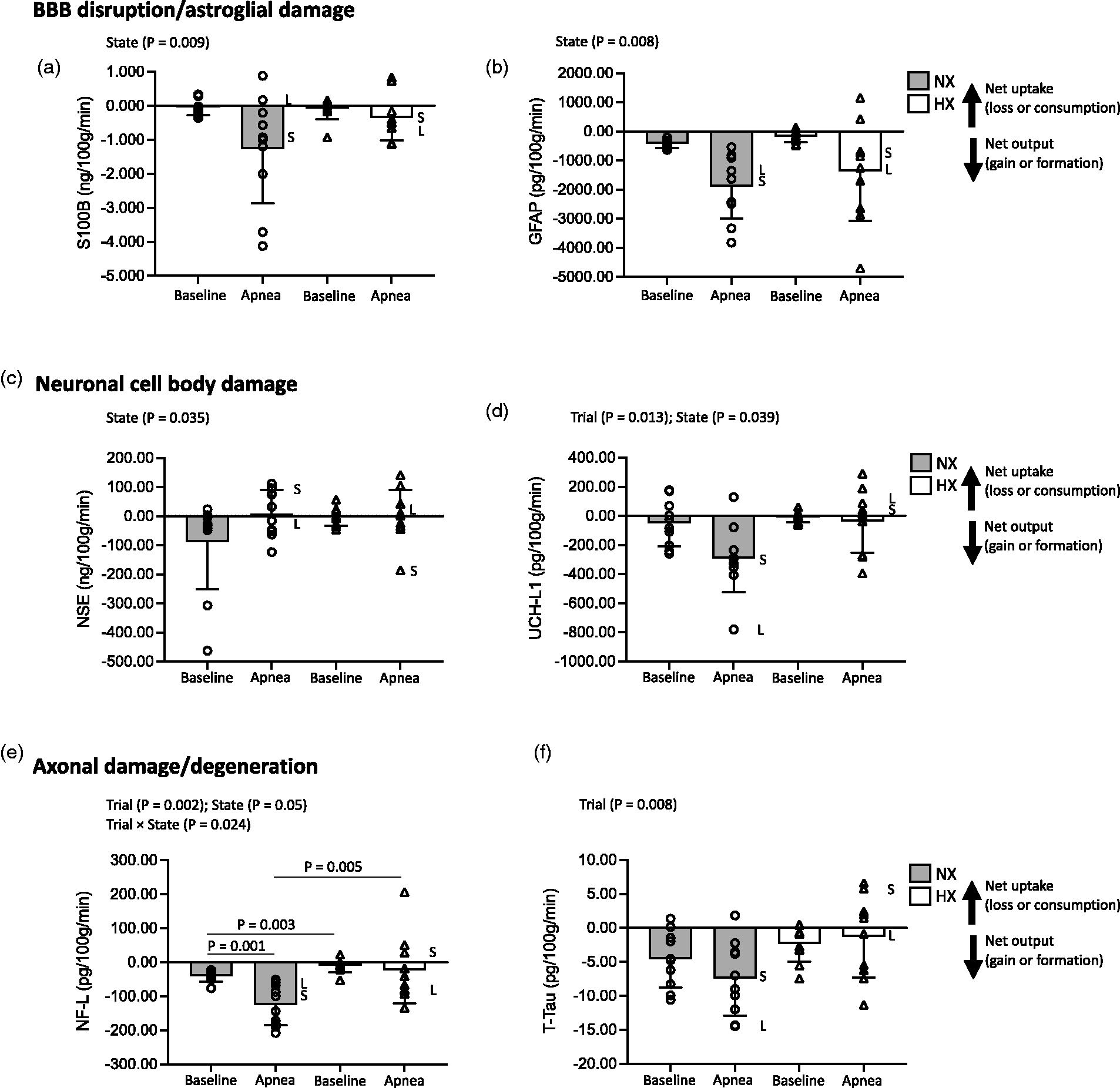
Transcerebral exchange kinetics of neurovascular unit proteins. Changes in the transcerebral exchange of an ultrasensitive panel of select proteins specific to the neurovascular unit in response to maximal apnea(s). Values are mean ± SD (n = 10). a, S100B; b, GFAP (glial fibrillary acidic protein); c, NSE (neuron-specific enolase); d, UCH-L1 (ubiquitin carboxy-terminal hydrolase-L1); e, NF-L (neurofilament light-chain); f, T-Tau (Total Tau). NX: normoxia trial; HX: hyperoxia trial. Exchange calculated by the Fick principle as the product of global (volumetric) cerebral blood flow and radial arterial to internal jugular venous protein concentration gradients. Positive or negative exchange value reflects net cerebral protein uptake (loss or consumption) or output (gain or formation) as indicated. S and L annotations for each apnea highlight individual participants with shortest (S, NX: 3 m 37 s and HX: 9 m 39 s) and longest (L, NX: 6 m 49 s and HX: 21 m 2 s) breath-hold times.
Neurovascular unit proteins.
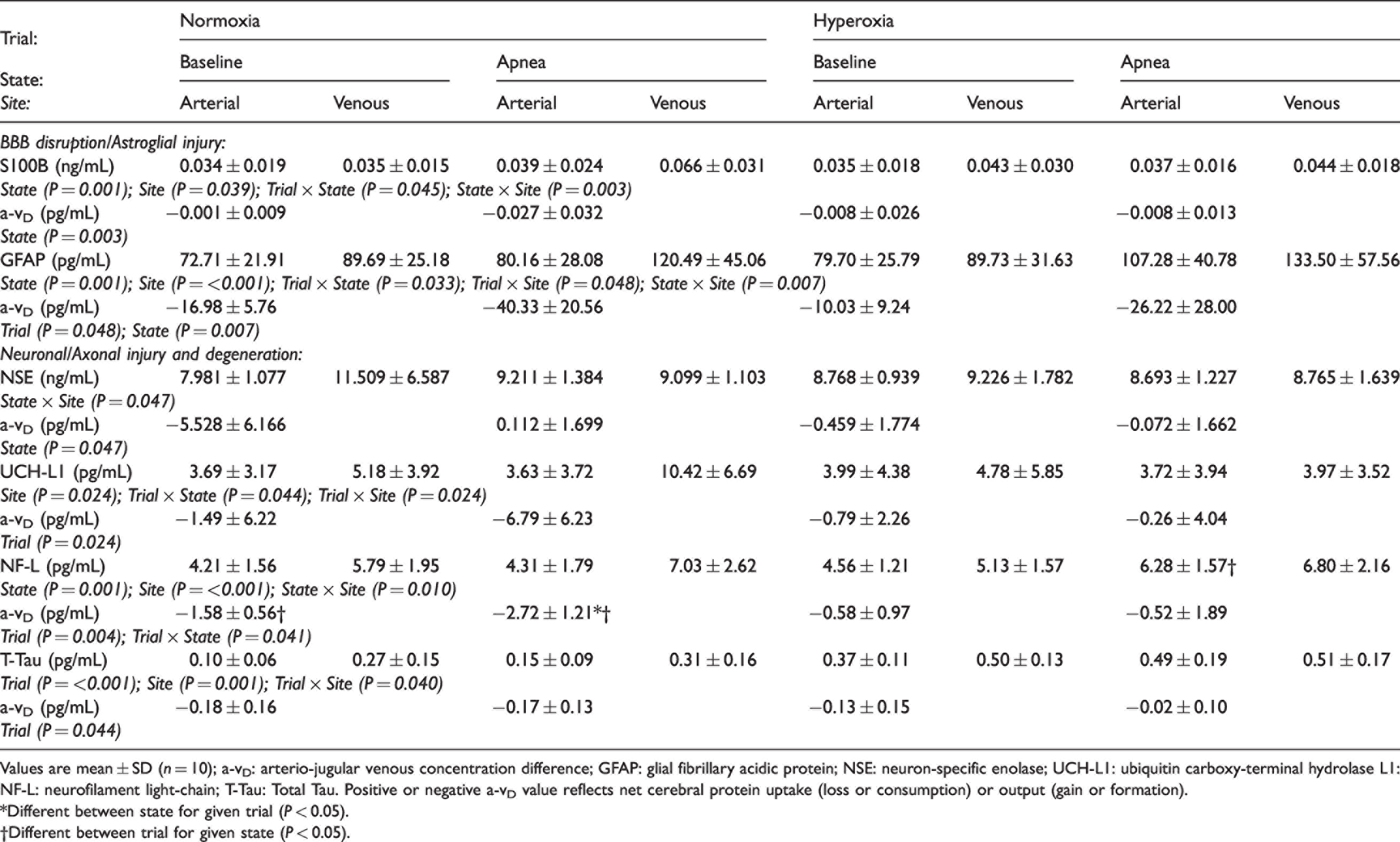
Values are mean ± SD (n = 10); a-vD: arterio-jugular venous concentration difference; GFAP: glial fibrillary acidic protein; NSE: neuron-specific enolase; UCH-L1: ubiquitin carboxy-terminal hydrolase L1: NF-L: neurofilament light-chain; T-Tau: Total Tau. Positive or negative a-vD value reflects net cerebral protein uptake (loss or consumption) or output (gain or formation).
*Different between state for given trial (P < 0.05).
†Different between trial for given state (P < 0.05).
Discussion
Local sampling of blood across the cerebral circulation during maximal apnea has provided novel insight into the persistence of functional-structural destabilization of the BBB in world-class BH divers transiently exposed to physiological extremes of arterial O2-CO2 stress. The study of clinically relevant protein biomarkers and the HX trial designed to distinguish between hypoxemic-hypercapnic vasoactive stimuli, highlights two novel mechanistic observations. First, apnea increased CBF and biomarker exchange, culminating in net cerebral output of NVU proteins reflecting increased leakage across the BBB and increased neuronal-gliovascular reactivity; this was a common finding in NX and HX. Second, output was more exaggerated in NX and linked to more marked elevations in ICP and MAP. Collectively, these findings identify the hemodynamic stress to which the apneic brain is transiently exposed, highlighting the key contribution of hypoxemia to BBB disruption.
Cerebral bioenergetics
Despite severe asphyxia, cerebral substrate delivery remained generally well preserved due to a marked compensatory elevation in CBF subsequent to hypercapnia/hypoxemia-induced pial artery dilation and severe arterial hypertension.5,23 However, the hemodynamic consequences underlying substrate conservation may prove maladaptive, especially in the setting of (impaired) cerebral autoregulation 24 and IH. In support, emerging evidence indicates that repeated, prolonged apnea may promote minor BBB disruption and neuronal parenchymal damage, albeit indirectly based on elevated blood protein biomarkers confined exclusively to the systemic (arterial) circulation25–28 and in the absence of supporting neuroimaging data (see Limitations).
NVU integrity
Release of the astrocytic protein S100B to the systemic circulation correlates directly with the extent and temporal sequence of BBB opening confirmed by contrast-enhanced MRI, 29 thus its appearance in blood is considered an established molecular reporter of increased BBB permeability. 15 Herein, we chose to measure cerebral gradients of S100B in combination with a more comprehensive panel of additional protein biomarkers using ultrasensitive Simoa assays, to extend our prior findings 23 and better distinguish potential BBB opening from increased neuronal-gliovascular reactivity.
As reported for BH divers 23 and (non-diver) controls, 30 we confirm that even at baseline before apnea, the human brain continuously releases S100B (v > a), implying that glial synthesis and subsequent extravasation to the periphery may reflect the ‘normal’ physiological exchange status. This may represent ‘fluctuations’ in BBB integrity since it does not constitute a static membrane with occasional ‘leaks’ that may serve to clear excess protein or metabolites. 31 While the same interpretation could equally be applied to the ‘spillage’ of remaining proteins in light of the persistent net output observed, we cannot exclude the possibility that mild BBB disruption was a historical, lingering feature in divers, even before the apneic challenge(s) began. However, that apnea further compounded biomarker output [v > a for all proteins with molecular weights ranging from 10.7 kDa (S100B) to 78.9 kDa (T-Tau) with the exception of NSE (below)] provided convincing molecular evidence for a rapid increase in BBB permeability that was generally more pronounced in NX.
Indeed, the apnea-induced elevations in venous S100B were comparable to a submaximal (∼30%) disruption of the BBB as previously reported. 32 Furthermore, since biomarker output had partially recovered (after NX) prior to HX indicates that exchange likely reflects transient NVU ‘dysfunction’ and not structural ‘damage’ per se, highlighting active repair mechanisms initiated upon recovery. In further support, peak concentrations of S100B and GFAP were generally below the pathologic thresholds used to diagnose traumatic brain injury and neuronal-gliovascular damage in patients. 33
In contrast, the transition from net output to uptake (a > v) in NSE (comparable ‘mid-range’ molecular weight of 47.3 kDa) during apnea was intriguing and confirmed our prior observations, albeit confined exclusively to NX. 23 Given that no detectable hemolysis was observed (<100 mg of free Hb/L), we are confident that this was an authentic, albeit counter-intuitive observation independent of any artefactual elevation incurred during arterial sampling. 34 Uptake may simply reflect contributions from extracerebral sources (manifest as increased arterial inflow and negative a-vD) including the adrenal glands to foster neuronal repair in response to injury given NSE’s (additional) neurotrophic regulation of neuronal survival, differentiation, and neurite regeneration through activation of phosphatidylinositol-4,5-bisphosphate 3-kinase and mitogen-activated protein kinase signalling pathways.35,36
Hypoxemia
Collectively, these findings highlight the key role of hypoxemia, consistent with prior observations albeit in non-diver healthy controls exposed to severe poikilocapnic (hypocapnic) hypoxia who exhibit hyperperfusion-mediated autoregulatory breakthrough, 37 vasogenic-cytotoxic edematous brain swelling 38 and hemosiderin deposits 39 collectively taken to reflect cerebral capillary ‘stress failure’ subsequent to BBB disruption. 37 It is conceivable that in the setting of hypoxic-hypercapnic cerebral vasodilatation, impaired cerebral autoregulation, previously documented in BH divers 24 and corresponding reduction in arteriolar tone could lead to a more pressure-passive elevation in CBF, further compounded by intrathoracic compression and intermittent surges in intrathoracic/systolic pressure triggered by the IBMs. The resultant elevation in cerebral capillary hydrostatic pressure could cause extracellular (vasogenic) edema subsequent to mechanical disruption of the BBB and potentially elevate ICP, 40 as observed indirectly through the IJVP surrogate. While elevated ICP would be expected to reduce cerebral perfusion pressure (CPP; i.e. CPP = MAP − ICP), MAP continues to increase during the latter stages of apnea due to sympathetically-mediated peripheral vasoconstriction,41,42 allowing ICP to continually rise and maintain CPP to preserve cerebral substrate (O2/glucose) delivery. 43 Indeed, it has been suggested that repeated intermittent exposure to these transient IBM-induced surges in ICP could potentially disrupt barrier integrity. 23
The increased cerebral output of NF-L in the present study was especially pronounced, agreeing with recent reports documenting its systemic appearance in patients with hypoxic brain damage subsequent to out-of-hospital cardiac arrest demonstrating superior prognostic accuracy compared to more established methods such as head computed tomography, somatosensory-evoked potentials, electroencephalogram and bedside clinical tests. 44
Hypertension
The more pronounced general increase in NVU protein output in NX coincided with more marked elevations in MAP and ICP. The latter was inferred from elevated IJVP, an established surrogate for ICP, 22 a likely consequence of greater intrathoracic compression (in the absence of atelectasis defining apnea breakpoint in HX) combined with intermittent surges in intrathoracic/systolic pressure triggered by the IBMs. The more pronounced arterial pressure in NX in the setting of increased barrier permeability and elevated cerebral blood volume would be expected to promote transcapillary fluid filtration and elevate ICP, consistent with the Starling fluid equilibrium applied to cerebral vessels with a leaky BBB. It is also of interest to note that pronounced BBB blood protein leakage and astrogliosis has recently been documented in patients with idiopathic IH. 45
In addition to protein diffusion across the BBB interface, elevated ICP would be expected to influence venous syphoning and glymphatic protein and small molecular weight clearance from the brain. 46 Direct compression of the intrathoracic vessels (e.g. vena cava) attributable to high lung volumes typical of BH divers, in combination with IBM-induced surges in intrathoracic pressure/ICP, could also increase back-pressure and vascular resistance, thereby reducing the pressure passive gradient for cerebral venous drainage. 43 However, while we cannot entirely exclude interstitial and/or glymphatic mechanism(s) of clearance, blood biomarkers were seen to increase immediately at apnea breakpoint, arguing for prompt passage across a permeated BBB. Net molecular drainage from the parenchymal to the meningeal compartment via the interstitial fluid is a process that likely exceeds 47 the more rapid extravasation readily detectable within a single a-v transit, further supporting acute ‘trans-BBB’ leakage/exchange as the more plausible mechanism. An important issue relates to the fate of the continuous output of NVU proteins. Previous findings have shown that at least for S100B, renal filtration is sufficient to maintain a constant level of this protein in spite of persistent spillage from the brain.32,48
NVU protein turnover
Furthermore, while the apnea-induced systemic (arterial) elevations in NVU proteins may appear subtle, local exchange kinetics reveal that the ‘rates’ of cerebral output were impressively high and far exceeded systemic accumulation, providing novel insight into the kinetics of protein ‘turnover’. In support, the arterial concentration of S100B (for example) increased by 0.005 pg/mL in NX and assuming dilution within the extracellular space (∼12 L), 6 the total amount of S100B accumulated would have equated to ∼60 pg/mL or ∼12 pg/min over the course of apnea. This implies that the global (i.e. non mass-normalized) cerebral output (∼17 pg/min) was almost 50% higher than the observed rate of systemic accumulation linked to renal disposal.
Indeed, the brain would have contributed to ∼29% of the S100B content in the blood during every minute of apnea. Simple subtraction of the systemic rate of accumulation from cerebral output highlights that the turnover rate of S100B is high, estimated in the order of ∼6 pg/min, assuming steady state kinetics and that the brain is the primary source of S100B during apnea notwithstanding (unlikely albeit unquantified) contributions from extracranial sources including adipose and skeletal tissue. 15 Furthermore, while beyond the scope of this study, we likely underestimated ‘peak’ rates of protein flux/turnover given delayed extravasation into the local circulation following initial injury to the NVU.15,25 Equally, it was intriguing to note the more marked elevations in the arterial concentrations of GFAP, NF-L and T-Tau during HX, confirming the brain as the primary source contributing to systemic accumulation.
Clinical perspectives
While it is conceivable that the cardiovascular system and cerebral microcirculation of BH divers may already have been remodelled given recurrent exposure to apneic stress, available evidence suggests that, at least during resting conditions, blood pressure and sympathetic nervous activity, 49 spleen, 50 cardiac structure and function, 43 and cerebrovascular function51,52 are all normal, that is, comparable between BH divers and healthy controls. Although apnea divers typically exhibit larger lung volumes, 53 there is no CT evidence of long-term damage to the pulmonary vasculature and parenchyma (unpublished findings, Z. Dujic). However, longitudinal studies are needed to better address to what extent recurrent intermittent exposure to such dramatic extremes of hypertension-hypoxemic-hypercapnic stress impact integrated pulmonary/cardio-cerebrovascular health over the long-term. 4
Our findings advance a plausible mechanism for the transient loss of motor control or ‘samba’ acutely experienced by BH divers during training/competition, 54 since focal motor seizures occur immediately following osmotically-induced BBB disruption in patients 55 and in experimental models of inflammation associated with neurovascular damage. 56 Furthermore, NVU destabilization precedes multiple infectious, inflammatory and degenerative central nervous system (CNS) pathologies 57 known to be exacerbated by hypoxia that has recently been linked to human tau seeding and propagation. 58 These observations may contribute to longer-term neurological complications with emergent evidence for persistent short-term memory impairments 59 and cerebral ischemic lesions related to neurological decompression sickness26,60 among BH divers.
Thus, we need to remain cautious when performing studies of this nature and consider the ethical implications given the general lack of longitudinal research in BH divers. However, it is important to state that these athletes voluntarily perform extended apneas during training and especially during competition. Thus, these studies afford an opportunity to better define the BH cerebral phenotype and provide clinical supervision/support albeit within the confines of laboratory experimentation.
Limitations
Several experimental limitations warrant careful consideration. First, we were unable to conduct complimentary dynamic contrast-enhanced and dynamic susceptibility contrast MRI 61 or CSF to blood protein quotients 62 to (more) directly assess BBB integrity, hence the focus on ‘surrogate’ albeit validated peripheral biomarkers. 15 Second, we specifically chose to limit blood sampling to baseline (i.e. pre-apnea) and apnea breakpoint, the latter coinciding with the most severe hypoxemic-hypercapnic stress and corresponding peak elevations in CBF and (potentially) cerebral output of NVU proteins. While it would have been interesting to take serial measurements to assess the ‘temporal dynamics’ of transcerebral exchange kinetics as hypoxemia-hypercapnic stress ‘evolved’ throughout the BH, we were ethically constrained by blood volume loss given that this constituted a repeated measures design. Thus, we were unable to determine (likely) ‘maximal’ rates of transcerebral protein flux given differences in synthesis/extravasation and corresponding time courses, notwithstanding marked differences in estimated half-lives of the proteins measured (S100B: 2–6 h; 63 UCHL-1: 7–9 h; 64 T-Tau: 10 h; 65 NSE: 30 h; 66 GFAP: 48 h; 63 NFL: 21 d 67 ) Finally, we adopted a mixed sexes design, and although an entirely separate experimental question, we were not adequately powered to determine if males responded differently to their female counterparts.
Conclusions
In conclusion, these findings demonstrate that NVU integrity becomes increasingly compromized during extreme apnea-induced hypoxemic- compared to hyperoxemic-hypercapnic stress, highlighting hypoxia as a key stimulus underlying mild BBB disruption and increased neuronal-gliovascular reactivity. These findings may have broader implications for the management and treatment of models of CNS disease characterized by severe hypoxemia-hypercapnia including identifying more sensitive-specific peripheral biomarkers of NVU health.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: DMB was supported by a Royal Society Wolfson Research Fellowship (#WM170007), Royal Society International Exchanges Award (#IES\R2\192137), Japan Society for the Promotion of Science (#JSPS/OF317) and Higher Education Funding Council for Wales. PNA was funded by a Canada Research Chair (CRC) and Natural Sciences and Engineering Research Council of Canada (NSERC) Discovery grant. ARB was funded through a NSERC postdoctoral fellowship. RLH was funded by a NSERC grant. OFB was funded by the Autonomic Province of Vojvodina, Serbia (#142-451-2541). NM was supported by ANR-Hepatobrain, ANR-Epicyte, ERaNet Neu-Vasc, ANSES Epidemicmac and MUSE-iSite University of Montpellier. OFB, ID, PNA and ZD were funded by a Croatian Science Foundation Grant (#IP-2014-09-1937).
Acknowledgements
We thank BS Stacey and A Iannetelli for assistance with transport/storage of blood samples. Original data arising from this research is available directly from DMB upon reasonable request.
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: DJ is affiliated to the company FloTBI Inc. focused on the technological development of novel biomarkers of brain injury in humans.
Authors’ contributions
DMB, ARB, ID, DBM, PNA and ZD conceived and designed the research. DMB, ARB, RLH, OFB, ID, PNA and ZD obtained funding. DMB, ARB, RLH, OFB, ID, DBM, PNA and ZD performed the experiments; DMB, ARB, RLH, OFB, ID, CH, SL, NM, DJ, DBM, PNA and ZD contributed to data analysis. DMB, ARB, NM, DJ, PNA and ZD interpreted results of the experiments. DMB drafted the manuscript and revisions thereof. DMB, ARB, RLH, OFB, ID, CH, SL, NM, DJ, DBM, PNA and ZD edited and revised the manuscript(s) and approved the final version submitted for publication.