
Abstract
This study investigated trans-cerebral internal jugular venous-arterial bicarbonate ([HCO3−]) and carbon dioxide tension (PCO2) exchange utilizing two separate interventions to induce acidosis: 1) acute respiratory acidosis via elevations in arterial PCO2 (PaCO2) (n = 39); and 2) metabolic acidosis via incremental cycling exercise to exhaustion (n = 24). During respiratory acidosis, arterial [HCO3−] increased by 0.15 ± 0.05 mmol ⋅ l−1 per mmHg elevation in PaCO2 across a wide physiological range (35 to 60 mmHg PaCO2; P < 0.001). The narrowing of the venous-arterial [HCO3−] and PCO2 differences with respiratory acidosis were both related to the hypercapnia-induced elevations in cerebral blood flow (CBF) (both P < 0.001; subset n = 27); thus, trans-cerebral [HCO3−] exchange (CBF × venous-arterial [HCO3−] difference) was reduced indicating a shift from net release toward net uptake of [HCO3−] (P = 0.004). Arterial [HCO3−] was reduced by −0.48 ± 0.15 mmol ⋅ l−1 per nmol ⋅ l−1 increase in arterial [H+] with exercise-induced acidosis (P < 0.001). There was no relationship between the venous-arterial [HCO3−] difference and arterial [H+] with exercise-induced acidosis or CBF; therefore, trans-cerebral [HCO3−] exchange was unaltered throughout exercise when indexed against arterial [H+] or pH (P = 0.933 and P = 0.896, respectively). These results indicate that increases and decreases in systemic [HCO3−] – during acute respiratory/exercise-induced metabolic acidosis, respectively – differentially affect cerebrovascular acid-base balance (via trans-cerebral [HCO3−] exchange).
Introduction
The regulation of extracellular pH – which directly affects cells via local changes in intravascular or extravascular/interstitial conditions – is affected by rapid chemical buffering reactions, involving phosphate, glycolysis, and carbon dioxide tension (PCO2). The cerebral vasculature is exceptionally sensitive to changes in arterial PCO2 (PaCO2),1–3 such that cerebral blood flow (CBF) rapidly increases by 6–8% per mmHg increase in PaCO2 (reviewed in: Hoiland et al.
4
) This CBF responsiveness to PaCO2/[H+] acts to stabilize CO2 gradients and thus regulate pH across the blood-brain barrier (BBB).4–6 As CO2 travels rapidly across the BBB – indicated by the swift changes in CBF (<15–30 seconds) following stepwise changes in PaCO27–10–perivascular/interstitial fluid (ISF) and intracellular brain tissue pH are tightly related to PaCO2. The Fick principle explains that elevated PaCO2 will be related to reductions in the trans-cerebral venous-arterial PCO2 difference as: 1) CBF varies directly with PaCO2,1,3,11 and 2) the cerebral metabolic production of CO2 (
Exercise-induced metabolic acidosis results when the rate of ATP hydrolysis (H+ release) eventually exceeds the maximal buffering capacity; the excess CO2 is then removed via hyperventilation21,22 and these PaCO2 changes in part explain the CBF kinetics response with exercise.23–25 With exercise-induced acidosis, arterial [HCO3−] is markedly reduced due to excessive buffering of [H+]21,22 – however, whether this change in arterial [HCO3−] is related to alterations in trans-cerebral [HCO3−] and PCO2 exchange as well as in vivo buffering capacity has not been experimentally addressed in humans. The cerebral metabolic rate of oxygen (e.g., CMRO2 = CBF × arterial-venous oxygen content difference) increases linearly by up to 30% at maximal exercise intensities to support substrate utilization in the face of reductions in CBF.26–33 At maximal exercise, there is a higher relative contribution of cerebral anaerobic glycolysis and lactate oxidation.34–36 There is evidence to support that maximal exercise-induced acidosis facilitates trans-cerebral lactate exchange and related increases in lactate oxidation mediated via pH-sensitive transcellular [H+] gradients37–39 and increases in systemic lactate availability.28,32,40–42 Additionally, metabolic acidosis achieved with exhaustive exercise reportedly increases BBB permeability with direct relevance for transcellular CO2 transport.38,43 Overall, cerebrovascular acid-base regulation is likely interrelated with cerebral substrate prioritization during exercise via pH-sensitive utilization of lactate.44,45
The Henderson-Hasselbalch relationship (equation (2)) explains that acute elevations in arterial [HCO3−] would increase arterial buffering capacity; for example, with increases in arterial [HCO3−] any given change in PaCO2 will result in a lesser change in arterial [H+]/pH.13,46 This relationship would theoretically apply with reductions in arterial [HCO3−] and therefore be reflective of related decreases in arterial buffering capacity. Pre-clinical experiments in anesthetized and artificially ventilated cats show that rapid/transient [HCO3−]/Cl− exchange occurs within 15 seconds between intravascular and extracellular fluid in response to elevated systemic arterial [HCO3−] at maintained PaCO2 47 – these results indicate that extracellular [HCO3−] is partially and transiently altered with changes in arterial [HCO3−], however, such changes in extracellular [HCO3−] are restored (within 1 hour) and these [HCO3−]/Cl- exchange kinetics are appreciably less influential than persistent and freely diffusible CO2 transport.48,49 At rest, cerebrospinal fluid (CSF) PCO2 – typically indexed via internal jugular venous sampling – is 6 to 11 mmHg higher and pH is 0.05 to 0.08 units lower than that of the arterial blood.13,50,51 As such, resting trans-cerebral net exchange of [HCO3−] and PCO2 (e.g., CBF × venous-arterial difference) is a positive value reflective of net release. A narrowing of the venous-arterial difference would indicate a shift toward net uptake and can be achieved, for example by: 1) larger increase in arterial relative to venous value; and 2) reduction in venous with a corresponding increase in arterial values. No study to date has investigated whether alterations in the trans-cerebral venous-arterial [HCO3−] and PCO2 differences during acute respiratory/exercise-induced metabolic acidosis are related to CBF and regulatory systemic changes in arterial [HCO3−] in vivo in humans.
The objective of this study was to investigate acid-base balance via alterations in trans-cerebral internal jugular venous-arterial [HCO3−] and PCO2 exchange utilizing two separate experimental interventions to induce acidosis: Study 1) acute respiratory acidosis via elevations in PaCO2 (range: 35 to 60 mmHg); and Study 2) metabolic acidosis via incremental cycling exercise to exhaustion (range: 7.45 to 7.20). We hypothesized that: 1) acute hypercapnic acidosis would reduce the venous-arterial [HCO3−] and PCO2 differences and this reduction would be related to higher CBF, less trans-cerebral [HCO3−] exchange, and elevated arterial [HCO3−]; and 2) arterial [HCO3−] would progressively decrease with maximal exercise-induced acidosis and – as explained by the non-linear CBF response with incremental exercise – this reduction in systemic [HCO3−] would be unrelated to any change in CBF or trans-cerebral venous-arterial [HCO3−] and PCO2 exchange.
Methods
Ethical approval
All participants provided informed written consent before participating in these studies. The original research studies were approved by the University of British Columbia Clinical Review Ethical Board (CREB: H16-01028, H18-01755, H15-00166, H11-03287) and the Comité d’éthique de la recherche de l’Institut universitaire de cardiologie et de pneumologie de Québec (CER: 21557) and according to the principles established by the Declaration of Helsinki (except for registration in a database). As the current study included a secondary analysis from previous institutional review board approved studies and the use of de-identified data sharing, this study did not require additional institutional review board review.
Participants
These data were obtained during five separate experimental studies previously conducted at the University of British Columbia, Kelowna, British Columbia, Canada and the Université Laval, Québec City, Québec, Canada. Thirty-nine healthy adults completed the CO2 trials (n = 34 males and n = 5 females) and twenty-four healthy adults completed the exercise trials (n = 19 males and n = 5 females). Participants had no history of cerebrovascular, cardiovascular, or respiratory disease and were not taking any prescription medication at their time of participation. Participants refrained from alcohol and caffeine consumption as well as vigorous exercise or activity for at least 12 hours prior to testing.
Experimental overview
The experimental questions addressed in this study involved post-hoc data analysis; therefore, few of the arterial and venous blood gas data have been reported separately in various context for previously published works from these studies; e.g.,.36,52,53 Importantly, the current experimental questions involved new data analyses that have not been previously reported.
Study 1: Respiratory acidosis protocols
Participants completed one of the following protocols to elicit progressive stepwise steady-state elevations in PaCO2 via dynamic end-tidal forcing: 1) +3, +6, +9 mmHg PaCO2 (n = 11 males); 2) +4.5 and +9 mmHg PaCO2 (n = 12 males); 3) +8 mmHg PaCO2 (n = 5 females; n = 7 males); or 4) +10 and +20 mmHg PaCO2 (n = 4 males). The alterations in PaCO2 were calculated from the resting eupneic breathing end-tidal values. All measurements were taken after at least 2–3 minutes of steady-state. Previous investigations from our group have shown that cerebrovascular CO2 reactivity (utilizing the same dynamic end-tidal forcing system) is highly linear in the hypercapnic range up to +20 mmHg.4,54 Additionally, recent retrospective analysis by our research group 109 has shown that following a rapid stepwise change in PETCO2 (via the exact same experimental methods/techniques in the current studies), relevant cerebrovascular and cardiorespiratory variables achieve steady-state within 2-minutes of exposure duration to elevated inspired PCO2. As such, by applying a linear mixed model analysis, there is no statistical or physiological rationale to expect these results to be different whether participants completed the exact same stepwise steady-state elevations in PaCO2 compared to the varied stages utilized in this post-hoc analysis.
Study 2: Exercise protocols
Participants completed supine incremental cycling exercise to exhaustion at various relative (0, 20, 40, 60, 80, 100% maximal workload; n = 12 males) and fixed exercise intensities (0, 50, 75, 100 watts; n = 5 females and 0, 75, 100, 125 watts; n = 7 males). Each workload was 3–5 minutes in duration and steady-state blood samples were drawn within the last 20 s of each exercise stage.
Blood sampling
Approximately 1.0 ml of radial arterial and internal jugular venous blood were drawn at the same time into pre-heparinized syringes (SafePICO, Radiometer, Copenhagen, Denmark) and analyzed immediately using a commercial blood gas analyzer (ABL90 FLEX, Radiometer) at each experimental stage (n = 39). This analysis included measurements of PaCO2 and oxygen tension (PaO2), arterial oxygen saturation (SaO2), arterial oxygen content (CaO2), base excess, [H+], hemoglobin concentration ([Hb]), and pH. Data collected at the Université Laval were analyzed within 10−15 minutes for n = 12 (ABL800 FLEX, ABL825, Radiometer).
Arterial oxygen content (CaO2) was calculated as:
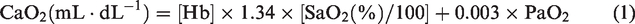
Acid-base balance data analysis
Blood gas analyzers do not typically have the capacity to directly measure [HCO3−]; instead, it is calculated from measured PaCO2 and pH, by rearranging the Henderson-Hasselbalch equation (equations (2) and (3)).
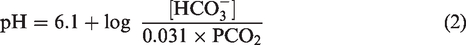
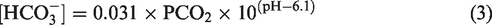
The following calculations were used to quantify an index of in vivo buffering capacity termed “extracellular pH defense”.58,59 All in vivo buffering capacity calculations were analyzed with the average venous-arterial values of [HCO3−], [La], and pH as an estimate of cerebral tissue values. These results were consistent when expressed as arterial or internal jugular venous buffering capacity.
Exercise:

The reduction in [HCO3−] per unit pH is reportedly attributable to hyperventilation and HCO3− buffering.
58

This index includes lactate ([La]) and is considered total pH defense by hyperventilation, as well as HCO3− and non-bicarbonate buffers. 58
Respiratory acidosis:

Equation (4) above has been adapted as [HCO3−] increases with respiratory acidosis.
Cardiorespiratory
Detailed cardiorespiratory experimental methods and results have been previously reported elsewhere; e.g., literature.36,52,53 Briefly, the partial pressures of end-tidal CO2 and O2 (i.e., PETCO2 and PETO2, respectively) were controlled during acute respiratory acidosis using a custom-designed dynamic end-tidal forcing system to effectively regulate end-tidal gases across wide ranges of PETCO2 and PETO2,60,61 independent of ventilation (
Cerebrovascular
Detailed cerebrovascular experimental methods and results have been previously reported elsewhere; e.g., literature.36,52,53 Briefly, extra-cranial blood velocity and diameter of the internal carotid artery (ICA) and vertebral artery (VA) were measured using a 10-MHz multifrequency linear array Duplex ultrasound (Terason t3000; Teratech, Burlington, MA, USA). Pulse-wave mode was used to measure beat-to-beat peak blood velocity and arterial diameter was instantaneously measured via B-mode imaging; data were analyzed using custom edge-detection and wall tracking software (BloodFlow Analysis, version 5.1). The vessel location was decided on an individual basis to allow for reliable image acquisition, with the same location and consistent insonation angle (60°) repeated within participants and between trials.
Blood flow was calculated as:

Total cerebral blood flow (CBF) was calculated as:
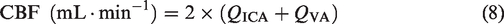
Net [HCO3−] exchange was calculated as:
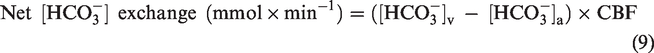
Net PCO2 exchange was calculated as:
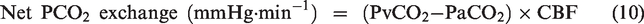
Statistical analyses
All data are presented in-text as mean ± SD and as individual values in figures. Statistical analyses were performed using SPSS software (IBM statistics, Version 23.0) and Prism (GraphPad Software, Version 9.1.0). Normality was assessed using Shapiro-Wilk tests and by visual inspection of Q-Q plots. Statistical significance was set at P < 0.05. Relationships between select variables were analyzed using linear regression. A linear mixed-model analysis with fixed effects of either arterial [H+], pH, or PaCO2 was used to compare PCO2, [HCO3−], [La], in vivo buffering capacity, trans-cerebral exchange, and CBF separately for the respiratory acidosis and exercise interventions. Subjects were included as a random effect.
Results
Study 1: Respiratory acidosis
Total CBF increased by 4 ± 1 ml ⋅ 100 g−1 ⋅ min−1 per mmHg elevation in PaCO2 across a wide range from 35 to 60 mmHg PaCO2 (P < 0.001; Figure 1(a)); this hypercapnia-mediated increase in CBF was related to narrowing of the venous-arterial PCO2 difference (P < 0.001; Figure 1(b)) such that trans-cerebral PCO2 exchange was unaltered with stepwise increases in PaCO2 (P = 0.057; Figure 1(c)). Arterial [HCO3−] increased by 0.15 ± 0.05 mmol ⋅ l−1 per mmHg elevation in PaCO2 (P < 0.001; Figure 1(d)). There was a relationship between respiratory acidosis and narrowing of the venous-arterial [HCO3−] and PCO2 differences; e.g., −0.16 ± 0.11 mmol ⋅ l−1 and −0.52 ± 0.22 mmHg reduction in venous-arterial [HCO3−] and PCO2 differences per nmol ⋅ l−1 increase in arterial [H+], respectively (both P < 0.001; Figure 1(e) and (f)). The venous-arterial [HCO3−] and PCO2 differences were each related to hypercapnia-induced increases in CBF (both P < 0.001; Figure 1(g)). As such, [HCO3−] exchange was reduced by -0.05 ± 0.08 mmol ⋅ min−1 per nmol ⋅ l−1 increase in arterial [H+]; i.e., less venous [HCO3−] efflux/release with respiratory acidosis (P = 0.004; Figure 1(h)).
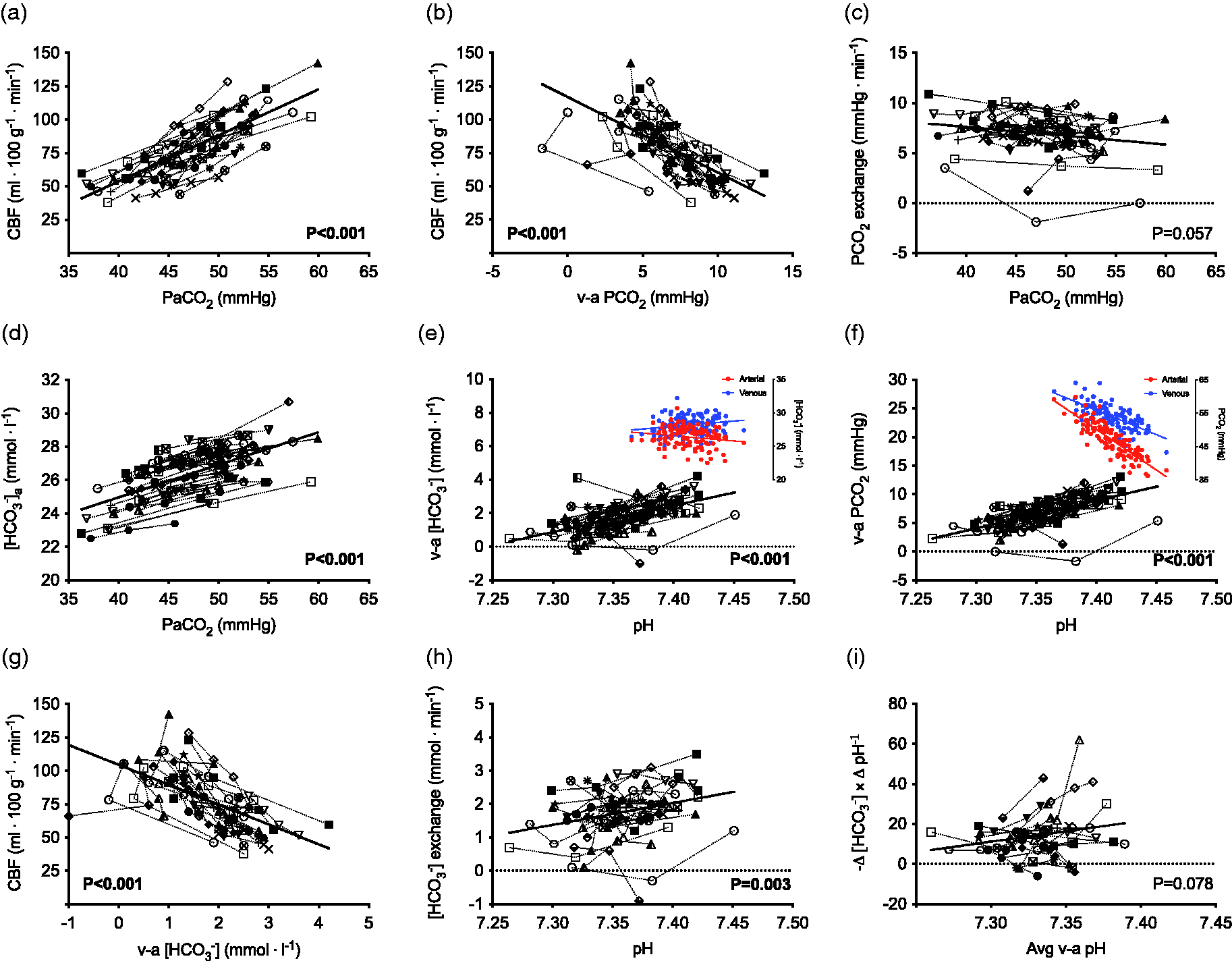
Venous-arterial [HCO3−] and PCO2 exchange with hypercapnic acidosis in humans. Total cerebral blood flow (CBF) increases with stepwise elevations in arterial PCO2 (PaCO2) (a); this hypercapnia-mediated increase in CBF is related to narrowing of the venous-arterial PCO2 difference (b) such that – explained via the Fick principle – trans-cerebral PCO2 exchange is unaltered with respiratory acidosis (c). Arterial [HCO3−] increases with progressive elevations in PaCO2 (d); this response may theoretically contribute to localized changes in CBF with severe respiratory acidosis – via increases in extravascular [HCO3−] – and thus, regulatory changes in [HCO3−] may help explain the maximal cerebrovascular vasodilatory reserve to PaCO2. There is a relationship between reductions in arterial pH (e.g., respiratory acidosis) and narrowing of the venous-arterial [HCO3−] (e) and PCO2 (f) differences. The reduction in venous-arterial [HCO3−] difference is achieved by a decrease in venous [HCO3−] and increase in arterial [HCO3−]; whereas, PaCO2 increases to a larger extent relative to PvCO2 with respiratory acidosis (illustrated by the coloured inlay figures). This narrowing is reflective of less [HCO3−] exchange (h) in part attributable to increases in [HCO3−] and CO2 ‘wash-out’ due to higher CBF with hypercapnia (g); i.e., the venous-arterial [HCO3−] and PCO2 differences were each related to increases in CBF (both P < 0.001; subset n=27). There was no relationship between the average venous-arterial in vivo buffering capacity (−Δ [HCO3−] × Δ pH−1) with stepwise respiratory acidosis when indexed against average venous-arterial pH (i). Data are individual values across stepwise progressive increases in PaCO2 for n = 27 (a, b, c, g, h, i) and n=39 (d, e, f) participants via dynamic end-tidal forcing.
Study 2: Exercise-induced metabolic acidosis
Total CBF was related to PaCO2 throughout progressive cycling exercise to exhaustion corresponding to 1 ± 1 ml ⋅ 100 g−1 ⋅ min−1 per mmHg change in PaCO2 (P = 0.004; Figure 2(a)). There was no relationship between CBF and the venous-arterial PCO2 difference (P = 0.177; Figure 2(b)); therefore, trans-cerebral PCO2 exchange was unrelated to PaCO2 during exercise (P = 0.155; Figure 2(c)). Arterial [HCO3−] was reduced by −0.48 ± 0.15 mmol ⋅ l−1 per nmol ⋅ l−1 increase in arterial [H+] with exercise-induced acidosis at maximal cycling exercise (P < 0.001; Figure 2(d)). There was no relationship between the venous-arterial [HCO3−] difference and arterial [H+] with exercise-induced acidosis (P = 0.801; Figure 2(e)). There was, however, a relationship between widening of the venous-arterial PCO2 difference by 0.12 ± 0.20 mmHg per nmol ⋅ l−1 increase in arterial [H+] during incremental cycling exercise to exhaustion (P = 0.006; Figure 2(f)); i.e., increased PCO2 uptake reflective of acidosis. The exercise-induced CBF response was unrelated to the venous-arterial [HCO3−] difference (P = 0.682; Figure 2(g)); as such, there were no changes in trans-cerebral [HCO3−] exchange during exercise when indexed against arterial [H+] or pH (P = 0.933 and P = 0.896, respectively; Figure 2(h)).
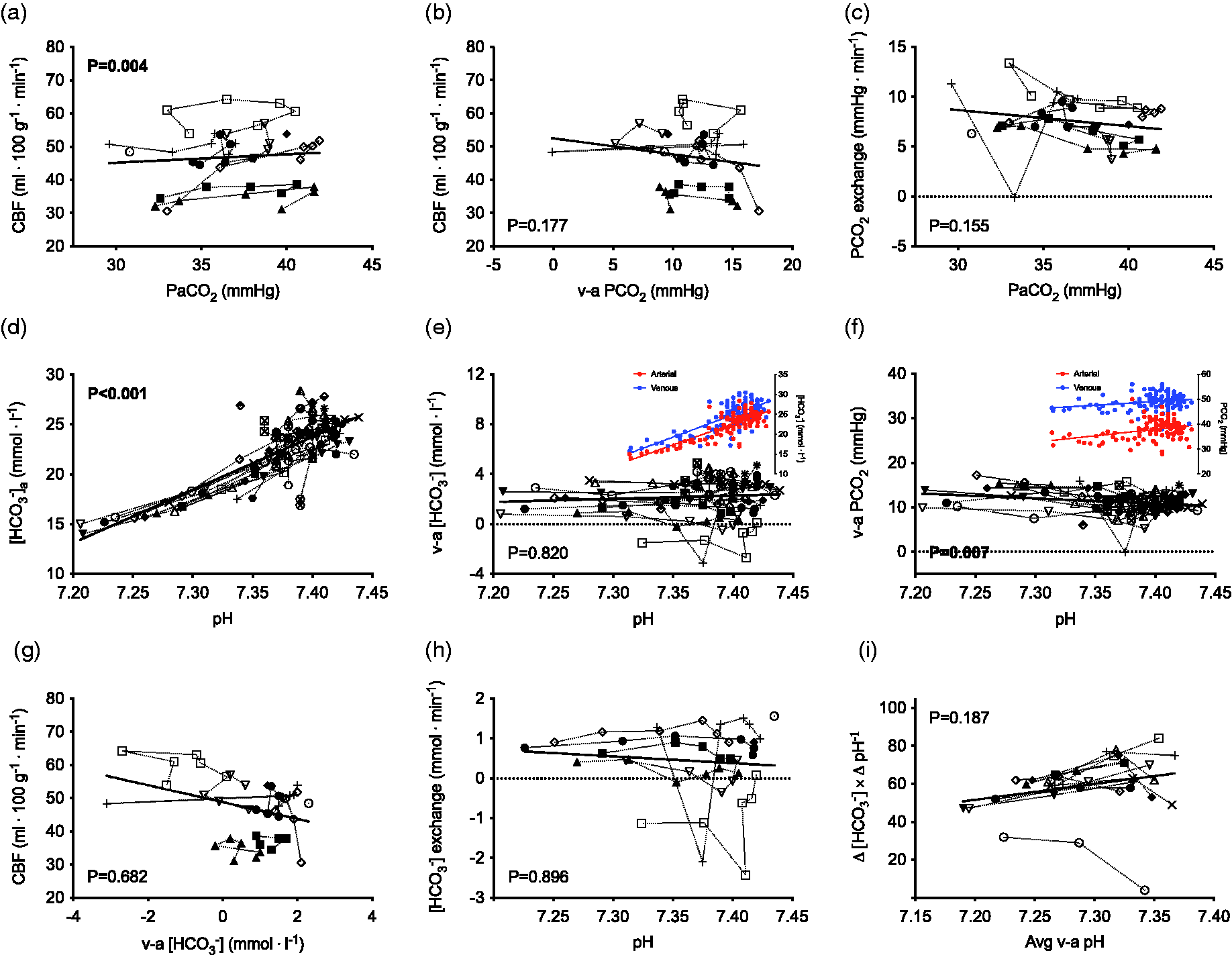
Venous-arterial [HCO3−] and PCO2 exchange with progressive submaximal to maximal cycling exercise. Total cerebral blood flow (CBF) is positively related to arterial PCO2 (PaCO2) throughout progressive cycling exercise to exhaustion (a); however, there is no relationship between CBF and the venous-arterial PCO2 difference (b), therefore, trans-cerebral PCO2 exchange is unrelated to PaCO2 during exercise (c). Arterial [HCO3−] is reduced with exercise-induced acidosis at maximal exercise (d). There is no relationship between the venous-arterial [HCO3−] difference and arterial pH (e) – as indicated by the equivalent reduction in arterial and venous [HCO3−] – however, there is widening of the venous-arterial PCO2 difference with exercise-induced acidosis (f) as indicated by a larger relative reduction in PaCO2 versus PvCO2 (illustrated by the coloured inlay figures). The exercise-induced CBF response was unrelated to the venous-arterial [HCO3−] difference (g); as such, there were no changes in trans-cerebral [HCO3−] exchange during exercise when indexed against arterial pH (h). There are no relationships between the average venous-arterial in vivo buffering capacity (Δ [HCO3−] × Δ pH−1 or −Δ [La] × Δ pH−1) during incremental cycling exercise to exhaustion (i). These data are reflective of the linear relationship between pH, reductions in [HCO3−], and increases in [La] with exercise-induced acidosis and indicate an unaltered in vivo buffering capacity at maximal cycling exercise. Data are individual values during supine incremental cycling exercise to exhaustion for n = 12 (a, b, c, g, h, i) and n=24 (d, e, f). Exercise stages included various relative (0, 20, 40, 60, 80, 100% maximal workload; n=12 males) and fixed exercise intensities (0, 50, 75, 100 watts; n = 5 females and 0, 75, 100, 125 watts; n=7 males). The in vivo buffering capacity was calculated at 60, 80, 100% maximal workload (I).
In vivo buffering capacity during acute respiratory acidosis and exercise
Linear mixed-model analysis revealed no significant relationship between in vivo buffering capacity (−Δ [HCO3−] × Δ pH−1) and the average venous-arterial pH during stepwise respiratory acidosis (P = 0.078; Figure 1(i)). Additionally, there were no relationships between in vivo buffering capacity (Δ [HCO3−] × Δ pH−1 or -Δ [La] × Δ pH−1) and the average venous-arterial pH during incremental cycling exercise to exhaustion (P = 0.187 and P = 0.392, respectively; Figure 2(i)).
Discussion
The results of this study indicate that trans-cerebral [HCO3−] and PCO2 exchange are differentially regulated in response to acute respiratory and exercise-induced acidosis. This finding is supported by: 1) acute hypercapnic acidosis elevated arterial [HCO3−]; however, trans-cerebral [HCO3−] exchange was reduced (i.e., indicating a shift from net release toward net uptake of [HCO3−]) and this was reflective in narrower venous-arterial [HCO3−] and PCO2 differences with higher CBF (Figure 1); and 2) arterial [HCO3−] was progressively reduced with maximal exercise-induced acidosis and this was unrelated to venous-arterial [HCO3−] and PCO2 exchange (Figure 2). Additionally, these results show there are no appreciable changes in the in vivo buffering capacity across a wide range of respiratory acidosis (up to +20 mmHg PaCO2) or incremental cycling exercise to exhaustion in humans (Figures 1(i) and 2(i)).
Respiratory versus metabolic acidosis – influence of [HCO3−] exchange
The two experimental interventions of acute respiratory acidosis and exercise-induced metabolic acidosis each provoke equivalent reductions in pH with a markedly disparate arterial [HCO3−] response. For example, the reductions in arterial [HCO3−] during exercise are reflective of a 3-fold larger rate of change in [HCO3−] versus the small increases in [HCO3−] observed with respiratory acidosis (e.g., approx. -0.48 vs. +0.15 mmol ⋅ l−1 [HCO3−] per nmol ⋅ l−1 increase in [H+]). Additionally, it is noteworthy to consider how alterations in CMRO2 with exercise and respiratory acidosis will affect
Increases in PaCO2 facilitate rapid passive diffusion of CO2 across the BBB via reduced CSF driving pressure leading to increases in intra- and extracellular [H+], thus provoking acidosis through acid-base re-equilibration67,68 in accordance with Le Chatelier’s Principle (rightward shift in equation (11)).
69
Overall, CO2 transport is the sum of the diffusion of 1) dissolved CO2; and 2) CO2 bound as HCO3− (i.e., “facilitated CO2 diffusion”) – this process involves a flux of H+ equivalent to that of HCO3− as per equation (11).70–72
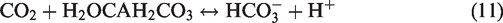
Acute respiratory acidosis increases arterial [HCO3−] via continuous conversion of CO2 – rapidly catalyzed by carbonic anhydrase (CA) – shifting the equilibrium relationship toward the [HCO3−] buffer reaction. This is reflective of rapid increases in extracellular [HCO3−] due to intracellular [HCO3−] exchange with Cl- to buffer the [H+] produced via CO2 hydration. 73 Importantly, however, the ratio of arterial [HCO3−] to PaCO2 is lower such that pH is reduced (as per equation (2)).
At rest, the relative contributions of dissolved CO2, chemically bound to hemoglobin and protein carbamate, and HCO3− to total CO2 exchange are approximately 5%, 10% and 85%, respectively. During severe exercise, HCO3− facilitated CO2 diffusion is reduced to approximately 2/3 of total CO2 exchange (as the contribution of dissolved CO2 increases sevenfold with exercise-induced acidosis).71,74 With maximal exercise-induced acidosis, arterial [HCO3−] is markedly reduced due to compensatory buffering of [H+] leading to production of CO2 and H2O (leftward shift in equation (11)); the excess CO2 is then removed via hyperventilation.21,22
Acute elevations in cerebral blood flow stabilize [HCO3−] and PCO2 gradients
Acute respiratory acidosis provokes three key regulatory responses: 1) arterial [HCO3−] increases (via inter-conversion of CO2) in response to elevated PaCO2 (Figure 1(d)); 2) there is rapid/transient exchange of HCO3− and Cl− across the BBB and between brain tissue and extracellular fluid;47,73 and 3) total CBF increases thus reducing the difference between arterial and extravascular PCO2, thereby limiting the rise in intracellular tissue PCO2 (Figure 1(b)). The increase in CBF in response to PaCO2 is restricted by the maximal vasodilatory response to hypercapnia in vivo in humans; e.g., +15–20 mmHg PaCO2 equates to 150% increase in CBF. 54 Elevations in arterial [HCO3−] may theoretically contribute to localized changes in CBF with severe respiratory acidosis – via increases in extravascular [HCO3−] – and thus, regulatory changes in [HCO3−] may help explain the maximal cerebrovascular vasodilatory reserve to PaCO2. The internal jugular venous-arterial [HCO3−] difference is reduced with acute severe hypercapnic acidosis (Figure 1(e)) – a response likely explained by increased extravascular HCO3− ‘wash-out’ as the reduction in trans-cerebral [HCO3−] difference is related to the hypercapnic CBF response (P < 0.001; Figure 1(g)). Arvidsson and colleagues (1981) showed that intravenous infusion of NaHCO3 in hypercapnic dogs causes a 50–70% reduction from the PaCO2-induced higher CBF. Additionally, cerebrospinal fluid [HCO3−] ([HCO3−]CSF) was appreciably higher following NaHCO3; these data indicate that acutely elevated PaCO2 may facilitate the transport of exogenous HCO3− across the BBB (via higher CBF) resulting in cerebral vasoconstriction due to increased extravascular pH. 75 The present results support that the CBF response to acute respiratory acidosis contributes to tight regulation of cerebrovascular pH (via narrowing of the trans-cerebral [HCO3−] and PCO2 differences) in vivo in humans.
Regulation of cerebrovascular acid-base balance during exercise
Trans-cerebral venous-arterial [HCO3−] exchange was unaffected by maximal exercise-induced acidosis (e.g., pH 7.20; Figure 2(e)) and there was a widening of the venous-arterial PCO2 difference during incremental cycling exercise to exhaustion (Figure 2(f)). Total CBF increases steadily by 10−20% up to intensities of approximately 60–70% of maximal oxygen uptake (
Experimental considerations
A key strength of this study was the invasive direct Kety-Schmidt technique to quantify trans-cerebral venous-arterial exchange of [HCO3−], PCO2, and [H+] in vivo in healthy humans; we recognize that this relies on the assumption that these sampling sites are reflective of trans-cerebral exchange due to practical and ethical experimental constraints. 85 Additionally, the Duplex ultrasound derived indexes of extra-cranial blood flow utilized in the current experiment are assumed to indicate cerebral tissue nutritive flow. Although we verified PaCO2 values with blood gas sampling, without temperature correcting to adjust for higher blood temperature during exercise, we may have underestimated PaCO2 by 1–2 mmHg (due to reduced CO2 solubility at higher temperatures).86,87 Additionally, the influences of exercise88–90 and temperature91–95 may conceivably have a small effect on PCO2, H+, and HCO3− via alterations in carbonic anhydrase activity in the exercise trial96,97 – however, this is unlikely to differentially affect the trans-cerebral venous-arterial exchange values within participants during this trial.
A subset of female participants completed the +8 mmHg PaCO2 respiratory acidosis protocol as well as the fixed intensity submaximal exercise protocol (n = 5 females and n = 7 males). Previous literature is equivocal, with studies reporting that cerebrovascular CO2 reactivity is higher in females,98–100 higher in males,101–103 or not different between sexes.104,105 Whether sex-related differences in cerebrovascular CO2 reactivity exist between females and males in response to a fixed targeted elevation in PaCO2, this is unlikely to affect our results as we saw no change in the in vivo buffering capacity (Δ [HCO3−] × Δ pH−1) when indexed against the average venous-arterial pH during respiratory acidosis. These results indicate that any within-subject change in the sensitivity of CBF to PaCO2 would be reflected in an equivalent pH response (with which we indexed against our dependent variables). Additionally, recent studies have shown no difference in the CBF response to moderate-intensity exercise between females and males106,107 with sex-related differences in CBF regulation only apparent at severe intensity exercise (80–100% maximal workload). 108 As such, we do not expect that the inclusion of 5 female participants in the fixed intensity submaximal exercise protocol (0, 50, 75, 100 watts) would affect the variability of our results. Future investigations are merited to address any sex-related differences in cerebrovascular acid-base regulation in response to respiratory and exercise-induced metabolic acidosis.
Conclusion
Taken together, these results indicate that increases and decreases in systemic arterial [HCO3−] – during acute respiratory/exercise-induced metabolic acidosis, respectively – differentially contribute to cerebrovascular acid-base balance. The previously recognized tight regulation of cerebral interstitial pH during acute hypercapnic acidosis19,20 is likely attributable to: 1) narrower internal jugular venous-arterial [HCO3−] and PCO2 differences; 2) higher CBF; and 3) reductions in trans-cerebral [HCO3−] exchange indicating less venous [HCO3−] efflux/release. These results are supportive of unaltered trans-cerebral [HCO3−] exchange with exercise-induced acidosis – consistent with the unchanged in vivo buffering capacity – which was unrelated to the CBF response throughout progressive cycling exercise to exhaustion.
Footnotes
Data availability statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Funding
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Philip N. Ainslie was funded by the Natural Sciences and Engineering Research Council of Canada (NSERC) and a Canada Research Chair. Hannah G. Caldwell was funded by a NSERC PGS-Doctoral Scholarship. Patrice Brassard was funded by the Foundation of the Institut universitaire de cardiologie et de pneumologie de Québec and a NSERC Discovery Grant. Damian M. Bailey was supported by a Royal Society Wolfson Research Fellowship (#WM170007) and Higher Education Funding Council for Wales (Postdoctoral Fellowship for Benjamin S. Stacey).
Acknowledgements
We would like to extend our thanks to the volunteers who participated in these studies. Additionally, we would like to thank past and present members of the Centre for Heart, Lung and Vascular Health at UBCO for their contribution to this series of studies by our group.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
Study design: RLH, KJS, PB, ARB, DMB, PNA. Data collection: HGC, RLH, KJS, PB, ARB, MMT, CAH, JMJRC, BSS, DMB, AD, MSS, DBM, PNA. Data analysis: HGC, RLH, KJS, PB, ARB. Data interpretation: HGC, RLH, JMJRC, DMB, PNA. Drafted manuscript: HGC. Critically reviewed manuscript: HGC, RLH, KJS, PB, ARB, MMT, CAH, JMJRC, BSS, DMB, AD, MSS, DBM, PNA. Approved final version: HGC, RLH, KJS, PB, ARB, MMT, CAH, JMJRC, BSS, DMB, AD, MSS, DBM, PNA.