
Abstract
Cellular RNAs are pervasively tagged with diverse chemical moieties, collectively called epitranscriptomic modifications. The methylation of adenosine at N6 position generates N6-methyladenosine (m6A), which is the most abundant and reversible epitranscriptomic modification in mammals. The m6A signaling is mediated by a dedicated set of proteins comprised of writers, erasers, and readers. Contrary to the activation–repression binary view of gene regulation, emerging evidence suggests that the m6A methylation controls multiple aspects of mRNA metabolism, such as splicing, export, stability, translation, and degradation, culminating in the fine-tuning of gene expression. Brain shows the highest abundance of m6A methylation in the body, which is developmentally altered. Within the brain, m6A methylation is biased toward neuronal transcripts and sensitive to neuronal activity. In a healthy brain, m6A maintains several developmental and physiological processes such as neurogenesis, axonal growth, synaptic plasticity, circadian rhythm, cognitive function, and stress response. The m6A imbalance contributes to the pathogenesis of acute and chronic CNS insults, brain cancer, and neuropsychiatric disorders. This review discussed the molecular mechanisms of m6A regulation and its implication in the developmental, physiological, and pathological processes of the brain.
Introduction
RNA is not merely a passive carrier of genetic information as it is replete with dynamic and reversible chemical modifications that form an additional layer of post-transcriptional gene regulation. To date, 172 modifications are cataloged for different classes of cellular RNAs, which collectively constitute the “Epitranscriptome”. 1 Of these, 72 are variants of methyl group modifications attached at different positions within RNA bases. Some of the well-studied epitranscriptomic modifications include N6-methyladenosine (m6A), pseudouridine (ψ), 5-methylcytidine (m5C), N1-methyladenosine (m1A), ribose 2'-O-methylation (Nm), and N4-acetylcytidine (ac4C) (Figure 1). 2 Among these, m6A is the most abundant modification mRNAs undergo and accounts for ∼80% of all RNA base methylations. 3 In mammals, 0.1–0.4% of total adenines in the transcriptome are m6A methylated at any given time. 4 , 5
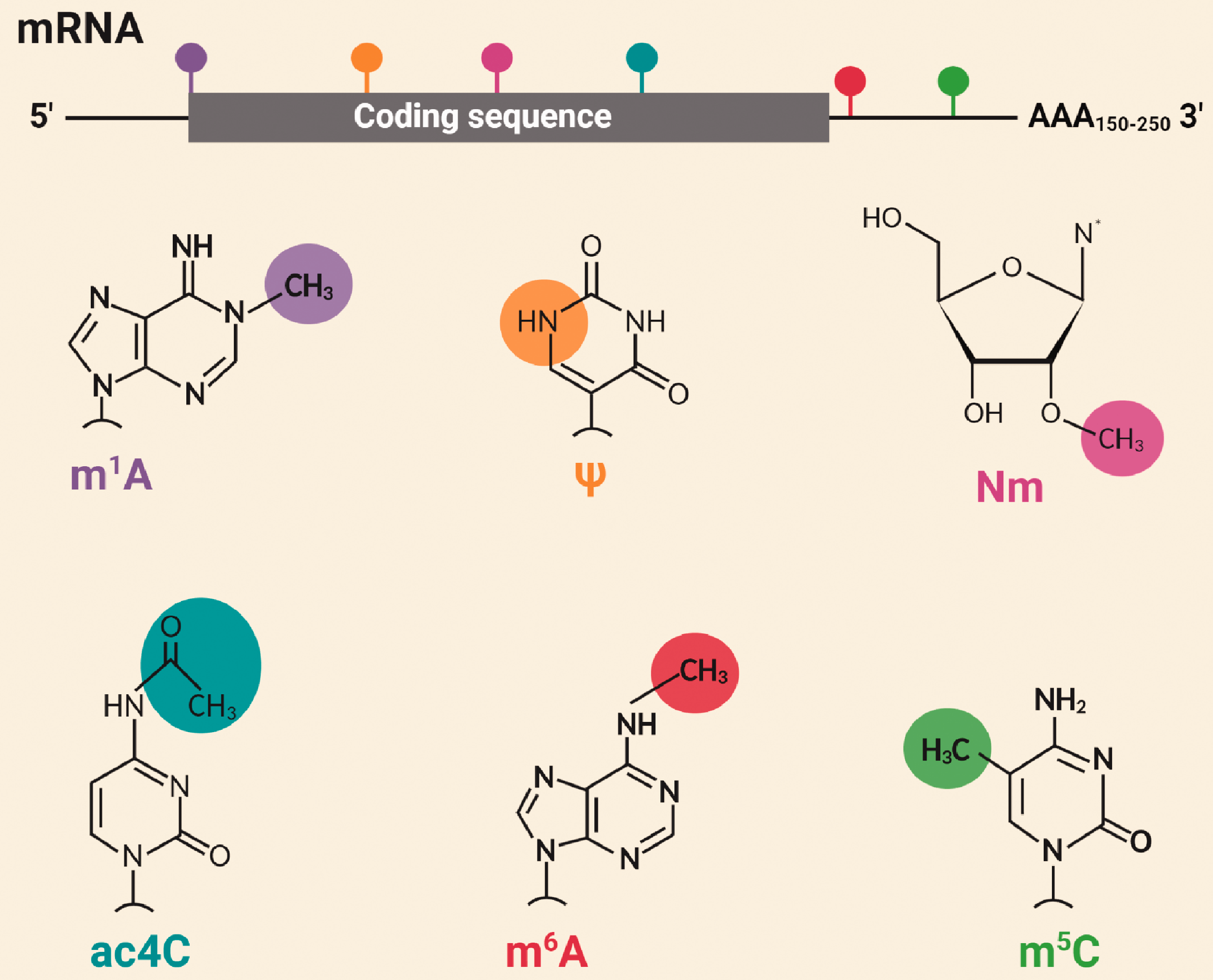
Major epitranscriptomic modifications. Mature RNAs can undergo 172 types of chemical modifications; 72 of those are variants of methyl group modifications. The most studied epitranscriptomic modifications are N6-methyladenosine (m6A), pseudouridine (ψ), 5-methylcytidine (m5C), N1-methyladenosine (m1A), ribose 2'-O-methylation (Nm), and N4-acetylcytidine (ac4C).
Although RNA m6A methylation was discovered in the early 1970s, its prevalence and distribution was not assayed due to lack of high throughput techniques. 3 Two parallel studies used m6A antibody-based next-generation sequencing and found that m6A is widespread in the transcriptome, tagging approximately 7000 mRNAs in mammals. 6 , 7 Furthermore, m6A occurs in the consensus RRACH (R is a purine base, A is adenine that can be modified to m6A, and H is a non-guanine base) motif enriched near stop codons and in the 3'-UTRs of mRNAs, indicating its regulatory potential. 6 , 7
Intriguingly, not all RRACH consensus sites are methylated. The fraction of the methylated sites appears to be highly variable, ranging from 6% to 80%. 8 The m6A methylation machinery, comprising of a writer complex, erasers, and readers that deposit, remove, and recognize m6A, is well-characterized in yeast, plants, flies, and mammals. 9 The m6A methylation controls multiple post-transcriptional mRNA processing steps, including splicing, export, stability, translation, and degradation. 9 Additionally, m6A methylation mediates partition of the mRNA into diverse subcellular compartments such as P-bodies and stress granules. 10
In mammals, the brain has the highest m6A of all organs, which increases from the embryonic stage to adulthood. 6 In the adult brain, m6A is regulated in an activity-dependent manner. For example, neuronal depolarization significantly increases m6A levels in plasticity-related genes. 11 Within neurons, m6A machinery is localized in axons and dendrites and controls the local gene expression. 12 Interestingly, m6A methylation displays distinct profiles between different brain regions. For example, m6A is enriched in cell cycle-related transcripts in the cerebellum, whereas in synaptic plasticity-associated transcripts in the cerebral cortex. 13 With these striking features, m6A signaling is implicated in multiple physiological and pathological processes of the brain.
This review delineates the m6A machinery, emerging techniques to study m6A, molecular mechanisms of m6A-mediated mRNA processing, and its role in brain development (Table 1) and physiology (Table 2). The review further highlights the involvement of m6A epitranscriptomic regulation in acute CNS injuries, chronic neurodegeneration, brain cancer, and neuropsychiatric diseases (Table 3).
Role of m6A methylation in brain development.
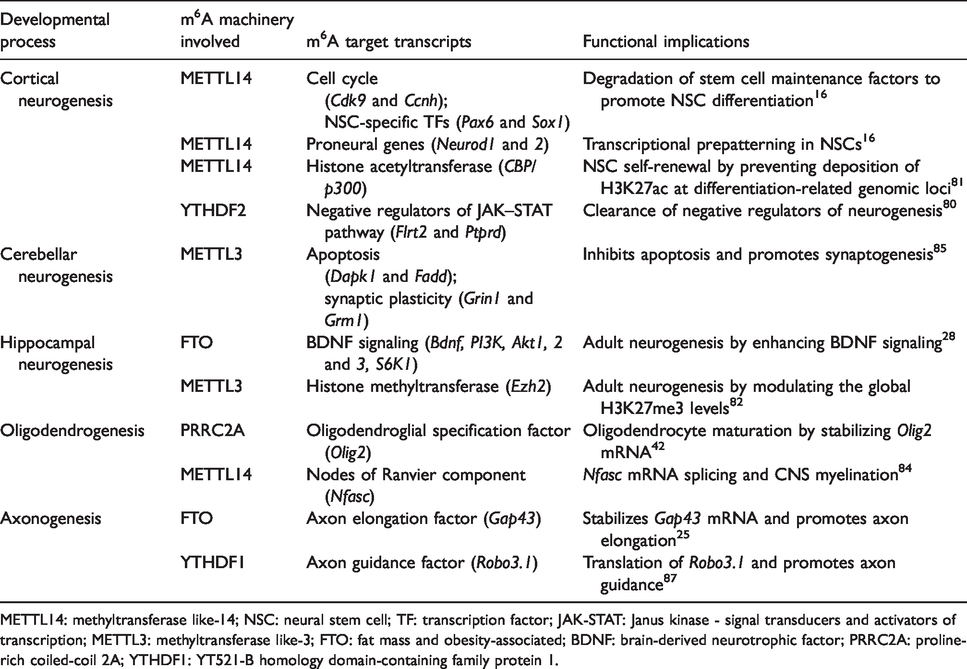
METTL14: methyltransferase like-14; NSC: neural stem cell; TF: transcription factor; JAK-STAT: Janus kinase - signal transducers and activators of transcription; METTL3: methyltransferase like-3; FTO: fat mass and obesity-associated; BDNF: brain-derived neurotrophic factor; PRRC2A: proline-rich coiled-coil 2A; YTHDF1: YT521-B homology domain-containing family protein 1.
The role of m6A methylation in brain physiology.
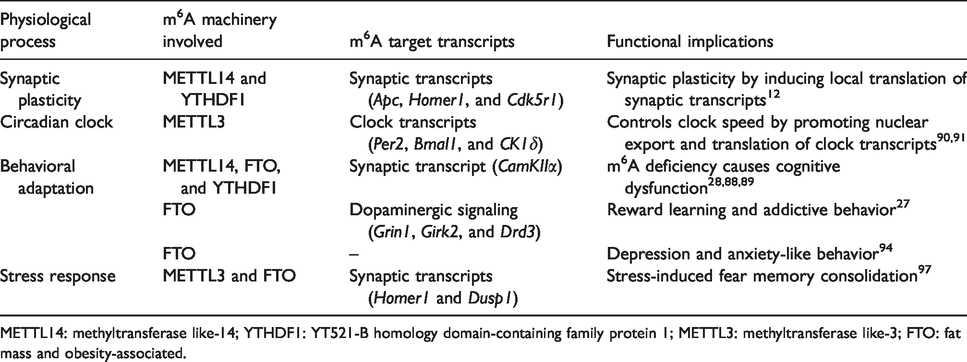
METTL14: methyltransferase like-14; YTHDF1: YT521-B homology domain-containing family protein 1; METTL3: methyltransferase like-3; FTO: fat mass and obesity-associated.
The role of m6A methylation in brain diseases.
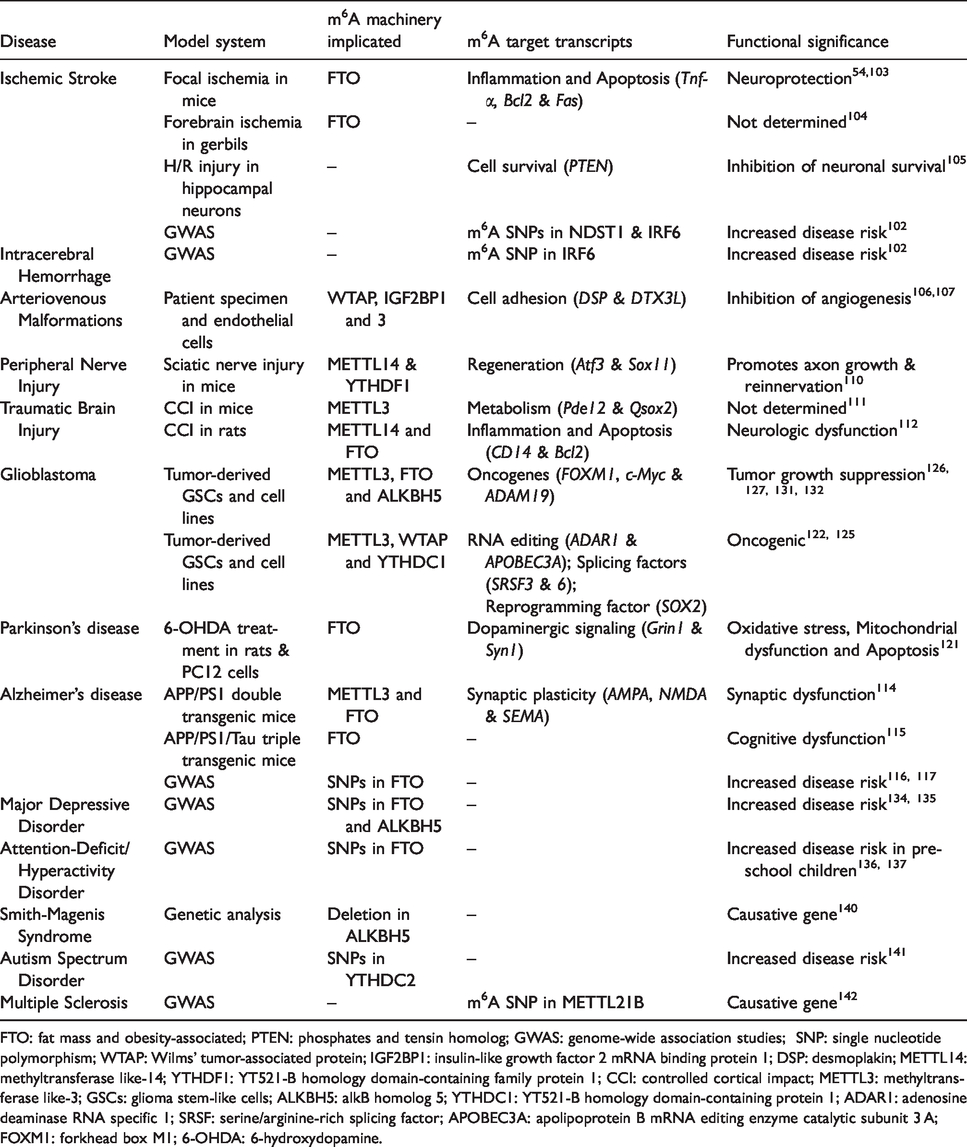
FTO: fat mass and obesity-associated; PTEN: phosphates and tensin homolog; GWAS: genome-wide association studies; SNP: single nucleotide polymorphism; WTAP: Wilms’ tumor-associated protein; IGF2BP1: insulin-like growth factor 2 mRNA binding protein 1; DSP: desmoplakin; METTL14: methyltransferase like-14; YTHDF1: YT521-B homology domain-containing family protein 1; CCI: controlled cortical impact; METTL3: methyltransferase like-3; GSCs: glioma stem-like cells; ALKBH5: alkB homolog 5; YTHDC1: YT521-B homology domain-containing protein 1; ADAR1: adenosine deaminase RNA specific 1; SRSF: serine/arginine-rich splicing factor; APOBEC3A: apolipoprotein B mRNA editing enzyme catalytic subunit 3 A; FOXM1: forkhead box M1; 6-OHDA: 6-hydroxydopamine.
Molecular machinery of m6A methylation
The m6A methylation is controlled by the coordinated activity of the m6A methylase complex (aka m6A writer complex) and m6A demethylases (aka m6A erasers). Furthermore, m6A readers and anti-readers dictate the fate of m6A-modified transcripts (Figure 2).
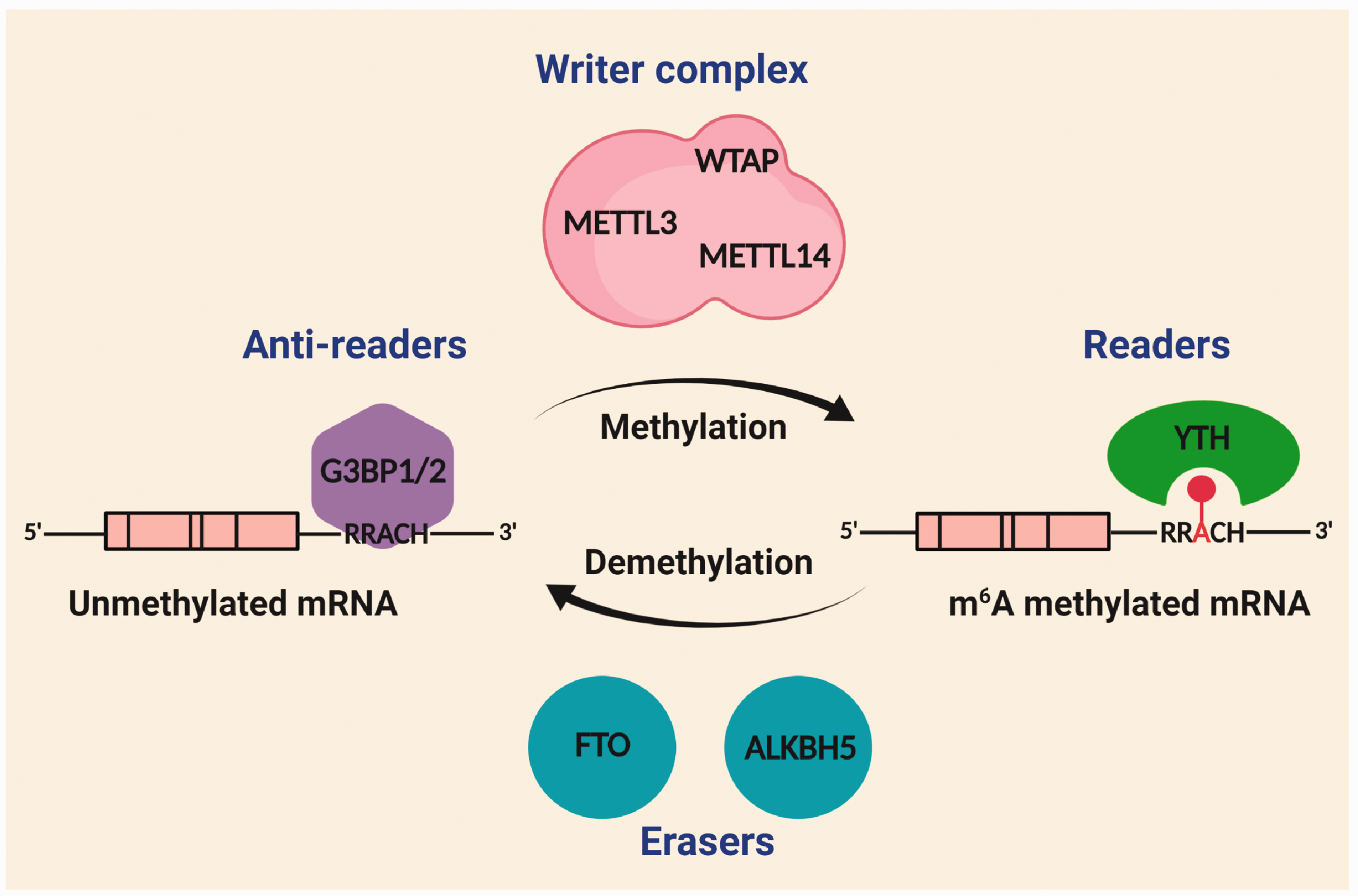
Molecular machinery of m6A methylation. The m6A modification predominantly occurs at consensus RRACH (R is a purine base, A is m6A-modified adenine, and H is a non-guanine base) motif enriched in the 3'-UTR of mRNAs. The m6A is deposited by a multi-subunit methylase complex (aka writer complex) primarily composed of a catalytic subunit METTL3 and a regulatory subunit METTL14. The m6A methylation is reversible and the methylated RNAs are demethylated by FTO and ALKBH5, with distinct erasure mechanisms. The m6A readers bind the methyl group through a characteristic YTH domain to propagate the m6A signaling. Additionally, the m6A methylation is repelled by m6A anti-readers G3BP1/2 that preferentially bind to unmethylated m6A motifs in the RNA.
m6A writer complex
The m6A writer complex contains a catalytic subunit methyltransferase (METTL)-like 3 (METTL3) and two regulatory subunits, METTL14 and Wilms’ tumor-associated protein (WTAP). 14 METTL14 mediates RNA substrate binding and WTAP confines the methylase complex to the nucleus. 15 All three subunits (METTL3, METTL14, and WATP) are indispensable as their individual deletion is embryonically lethal in mice.16–18 The methylase complex contains several other subunits like vir-like m6A methyltransferase associated (VIRMA), zinc finger CCCH-type containing 13, RNA binding motif protein 15/15B, and Cbl proto-oncogene like 1. 19 Specific functions of these subunits are not yet evaluated in detail. The mechanism of transcript-specific and site-specific m6A deposition is also not well understood. However, de novo m6A methylation of mRNAs is influenced by the interaction between transcription factors and methylase complex, elongation rate of RNA polymerase, and epigenetic modifications such as Histone H3 trimethylation at lysine 36. 20 Furthermore, SUMOylation of METTL3 inhibits the in vivo m6A methyltransferase activity. 20 In brain, m6A writer complex is predominantly localized in the neuronal nuclei. Both METTL3 and METTL14 were also detected in the synaptic fractions suggesting the possibility of dynamic m6A methylation in the extra-somatic regions. 12 , 21
m6A erasers
The m6A methylation is reversible. In eukaryotes, m6A is removed by two demethylases, fat mass and obesity-associated (FTO), and alkB homolog 5 (ALKBH5). 22 , 23 Unlike the m6A writer complex, FTO shuttles between nucleus and cytoplasm by interacting with exportin 2. 24 In the neurons, FTO is also detected in the extra somatic regions, including axons and dendritic shafts, suggesting the dynamic erasure of m6A and also certain other epitranscriptomic marks. 12 , 25 , 26 FTO-dependent m6A demethylation is implicated in dopaminergic neurotransmission, adult neurogenesis, and axon elongation.25,27,28 Due to these wide-ranging roles, FTO deficiency results in postnatal growth retardation, microcephaly, and functional deficits in both humans and mice. 29 , 30 FTO catalyzes the removal of m6A in steps by oxidizing the methylated base and then fragmenting it. 31 In contrast, ALKBH5 catalyzes the direct removal of the methyl group in a single step reaction, but with significantly lower catalytic efficiency. 23 ALKBH5 is ubiquitously expressed with the highest expression in the lungs followed by the testes. 32 Unlike FTO, ALKBH5 strictly localizes to nuclear speckles, precluding the dynamic demethylation of cytoplasmic mRNAs. 12 ALKBH5 deficiency in mice results in a highly specific phenotype of compromised spermatogenesis. 23 Although the transcript selectivity for these demethylases is not well understood, a recent study proposed that the interaction of m6A-methylated RNAs with FTO or ALKBH5 depends on the sequence context. 31 Additionally, an RNA-binding protein (RBP), splicing factor proline- and glutamine-rich, confers transcript selectivity by recruiting FTO to promote proximal m6A demethylation. 33
m6A readers and anti-readers
The downstream effects of m6A-methylated RNAs are mediated by specific RBPs called m6A readers that influence the post-transcriptional RNA processing. Readers recognize m6A by direct binding to m6A, by increased accessibility to RNA-binding motifs and by repulsion of RBPs. 9 The m6A readers bind to m6A-methylated RNAs directly via a well-conserved YT521-B homology (YTH) domain. 34 In mammals, there are five YTH proteins, including YTH domain-containing family proteins (YTHDF) 1, 2, and 3 and YTH domain-containing proteins (YTHDC) 1 and 2. YTHDC1 recognizes m6A in nuclear transcripts, while others recognize m6A in cytosolic transcripts. YTH proteins are not site-specific and they bind to m6A motifs at a similar ratio. 35 , 36 YTH proteins regulate the splicing, export, translation, and degradation of the m6A-modified mRNAs. 20 m6A in the short RNA helices destabilizes and unfolds the RNA and thus increases the binding of RBPs. 37 This leads to increased binding of splicing regulatory proteins heterogeneous nuclear ribonucleoprotein (HNRNP) C and G near the m6A sites leading to alternative splicing of the target transcripts. 38 The m6A methylation in the primary microRNAs (Pri-miRNAs) also increases the binding of HNRNPA2B1 leading to Pri-miRNA processing. 39
Insulin-like growth factor 2 mRNA-binding proteins (IGF2BPs) and certain neuronal-specific RBPs like Fragile X mental retardation protein (FMRP) and proline-rich coiled-coil 2 A (PRRC2A) are also potential m6A readers.40–42 FMRP reads m6A and promotes the nuclear export of methylated mRNAs during neural differentiation.
43
Recently, stress granule proteins such as GTPase-activating protein (SH3 domain) binding protein 1 and 2 (G3BP1 and G3BP2) were named as anti-readers as they bind to unmethylated m6A motif CA
Techniques to study m6A methylation
As methyl group does not alter the base pairing of adenosine and m6A is chemically non-modifiable, it cannot be detected by standard hybridization and sequencing technologies. 6 Although dot blot and immuno-northern blot using m6A antibodies allow faster and easier estimation of global m6A levels, these techniques are less sensitive and semi-quantitative. 44 The m6A levels can be quantitated with high sensitivity by liquid chromatography/mass spectrometry, but this technique will not provide the sequence context and localization information, 45 whereas, m6A immunoprecipitation followed by next-generation sequencing (MeRIP-seq) allows analysis of m6A methylation with 50–200 nucleotide resolution. 6 Furthermore, m6A individual-nucleotide resolution cross-linking and immunoprecipitation enables identification of specific m6A residues at single-nucleotide resolution. 46 However, m6A antibodies cross-react with m6Am; but the abundance of m6Am is <5% of the total methylated RNA residues, 47 whereas, Site-specific Cleavage and Radioactive labeling followed by Ligation-assisted Extraction and Thin-layer chromatography (SCARLET) allows the stoichiometric quantification (the fraction of RNAs that are m6A modified at a given site) within the total RNA pool. However, this is a tedious technique and hence suitable only to validate the proposed m6A changes at a given site. 8
More recently, RNA digestion via m6A-sensitive RNase MazF-sequencing (MAZTER-seq) and deamination adjacent to RNA modification targets-sequencing (DART-seq) allowed single-nucleotide resolution and stoichiometric quantification of transcriptome-wide m6A methylation simultaneously. In MAZTER-seq, upstream of unmethylated ACA motifs, but not the methylated m6ACA motifs, will be selectively cleaved with a bacterial RNase (MazF) and the ACA readthrough will be correlated with m6A abundance. 48 A caveat of this method is the requirement of the ACA motif in single-stranded RNAs, and hence only 16–25% of m6A sites in the mammalian system can be captured. 48 DART-seq employs an engineered fusion protein in which the N-terminus of the m6A-binding domain YTH fused to the editing domain of cytosine deaminase, apolipoprotein B mRNA editing enzyme, catalytic polypeptide 1. The YTH domain directs the editing machinery to the m6A site facilitating the conversion of proximal C-to-U, and the editing events adjacent to the m6A sites can be evaluated by RNA sequencing. Notably, this technique requires only 10 ng total RNA. 49
Recent studies employed clustered regularly interspaced short palindromic repeats (CRISPR)-based site-specific epitranscriptomic editing to dissect the functional contribution of a specific m6A site in a modified transcript. Rauch et al. tethered m6A readers YTHDF1 and YTHDF2 to catalytically inactive cas13 protein (dCas13) to specifically target and degrade isomerase B mRNA using a complimentary guide RNA. 50 Liu et al. fused dCas9 protein to methyltransferase domains of METTL3 and METTL14 and demethylases ALKBH5 and FTO. 51 Using this engineered m6A writer, they promoted site-specific methylation of heat shock protein 70 (HSP70) mRNA leading to its increased translation upon heat shock stress, recapitulating the context-specific m6A regulation. 51 More recently, Li et al. used an engineered ALKBH5-dCas13 fusion protein to decrease the proliferation of cancer cells by site-specific demethylation, and consequent destabilization of the oncogenic transcripts Egfr and Myc. 52 These novel tools are indispensable for dissecting the precise roles of m6A at particular regions within an RNA (Figure 3).
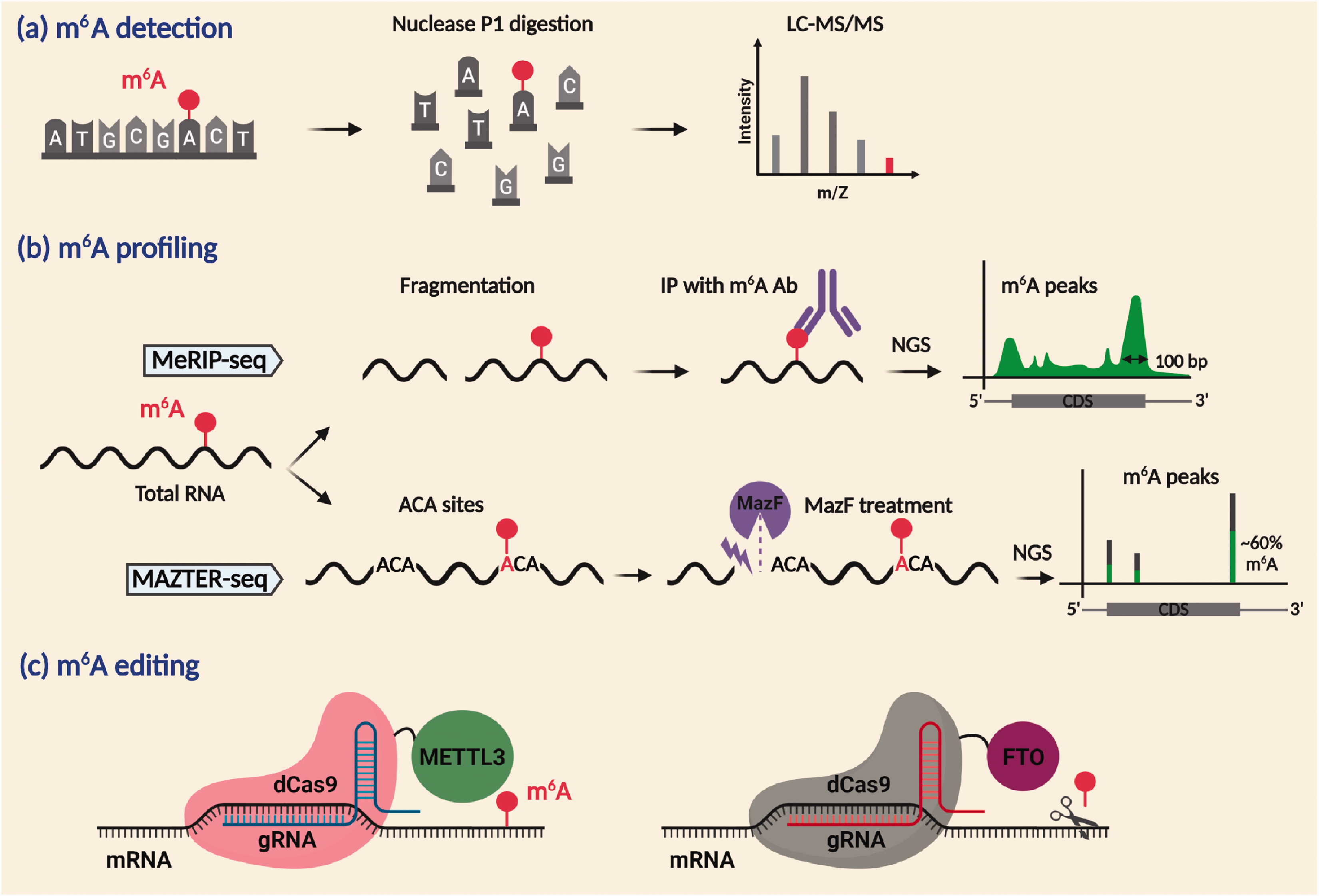
Techniques to study m6A methylation. LC-MS/MS can quantitate the m6A modification (a). MeRIP-seq can identify the transcriptome-wide m6A profiles. MAZTER-seq can show the m6A sites along with the stoichiometry of methylation with single-nucleotide resolution (b). CRISPR/Cas9 genomic editing using METTL3 or FTO fused to inactive dCas9 targeted near the vicinity of m6A site by sequence-specific gRNA can modulate site-specific m6A methylation (c).
Cell-type-specific distribution of m6A in the brain
Elucidating cell-specific methylation patterns is crucial for understanding the cell–cell communication under physiological and pathological conditions. MeRIP profiling in the brain revealed a higher prevalence of m6A in neuronal than glial transcripts. 13 Moreover, m6A effector proteins are more widely expressed in the neurons than in the glial cells, endorsing this bias. 53 The m6A-modified transcripts and their interactomes are localized in axons and dendritic shafts, indicating their role in activity-dependent gene regulation.12,25 Nevertheless, the presence of m6A in the non-neuronal cells cannot be overlooked. Astrocytic transcripts, such as Sparcl1 and Slc1a2 involved in synapse formation, as well as a microglial transcript CX3CR1 involved in the complement cascade, are highly m6A modified. 12 The brain microvascular pericytes from the spontaneously hypertensive rats showed ∼7500 m6A peaks in 5000 mRNAs related to inflammatory response, proximal tubule development, mitogen-activated protein kinase (MAPK) cascade, and cAMP response element binding, indicating the significance of m6A methylation in non-neuronal functions. 54 The m6A is also highly prevalent in oligodendrocyte-specific transcripts such as Olig2. 42 Taken together, m6A signaling occurs predominantly in the neurons, but can be seen to some extent in other CNS cell types.
Mechanisms of post-transcriptional regulation by m6A methylation
The molecular effects of epigenetic modifications are often singular as they either activate or repress the gene expression, whereas epitranscriptomic modifications like m6A methylation influences a multitude of downstream functions, including splicing, export, stability, translation, and degradation of RNAs (Figure 4). The functional outcome of m6A RNA methylation is determined by the m6A readers, which mobilize specific cellular machinery to the target RNAs. Furthermore, m6A regulation is dynamic as the fate of a target RNA is determined by a specific reader depending on the context. For example, heat shock stress increases m6A in the 5'-UTR region of Hsp70 mRNA to promote cap-independent translation. 55 In contrast, hypoxic stress increases m6A in mRNA targets such as Glut1, c-Myc, and Jun to enhance their stability without affecting the translation efficiency. 56
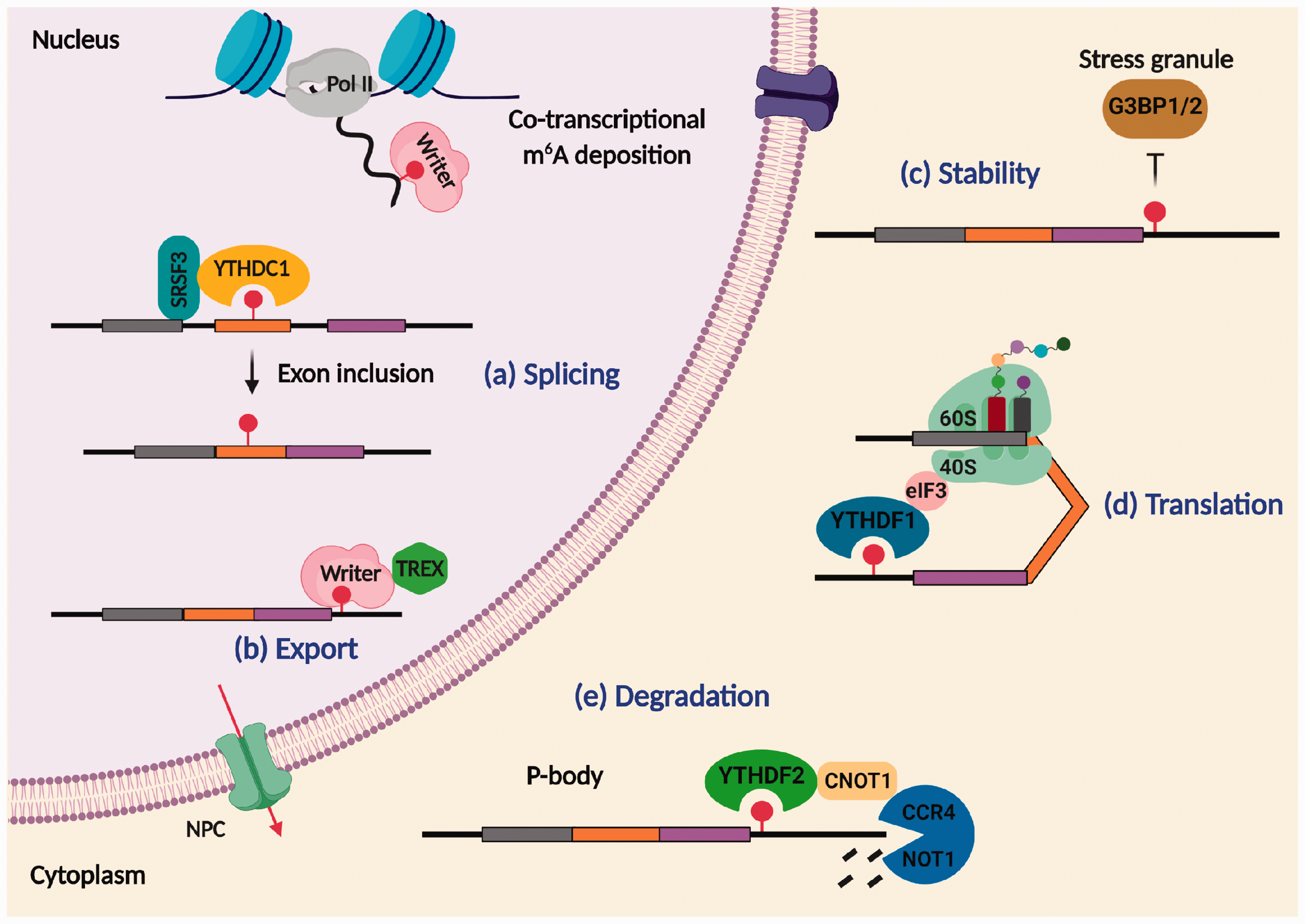
Post-transcriptional regulation by m6A methylation. The m6A methylation occurs co-transcriptionally in the nascent pre-mRNAs. The fate of methylated transcripts is dictated by m6A readers that bind and recruit diverse cellular machinery. In the nucleus, YTHDC1 binds to exonic m6A and recruits splicing regulatory factor SRSF3 to promote exon inclusion and thereby controls alternate splicing (a). The m6A writer facilitates the nuclear export of spliced mRNAs by interacting with export machinery TREX complex to guide them through the nuclear pore complex to the cytoplasm (b). In the cytoplasm, the presence of m6A repels the binding of stress granule proteins G3BP1/2 that regulates the stability of target RNA (c). Binding of YTHDF1 to methylated RNA recruits translation machinery leading to RNA translation (d). In contrast, YTHDF2 recruits mRNA decay enzymes in the P-bodies leading to degradation of the methylated transcripts (e).
Splicing
In METTL3 knockdown cells, m6A abundance correlated with differentially spliced exons indicating its role in alternate splicing. 7 Colocalization of m6A writer complex with pre-mRNA splicing factors in nuclear speckles further supports this notion. 57 A closer examination of exonic m6A peaks from the MeRIP data revealed that m6A occurs in the vicinity of 5' and 3' splice sites. Alternate splicing caused by differential splice site selection is primarily controlled by the binding of serine/arginine-rich splicing factors (SRSFs) to cis-regulatory sequences proximal to splice sites to cause exon inclusion or exclusion. 58 In adipocytes, exonic m6A was found to enhance the recruitment of SRSF2 to an RNA promoting exon inclusion. 59 The nuclear m6A reader YTHDC1 was shown to promote exon inclusion by engaging SRSF3 while inhibiting SRSF10 binding to splicing enhancer elements. 60 Thus, m6A controls alternate splicing by modulating the access of splicing regulatory factors to target RNA.
Export
ALKBH5-deficient cells showed a dramatic increase in cytoplasmic mRNA fraction and overexpression of ALKBH5 normalized it, indicating the role of m6A methylation in modulating the mRNA export. 23 Nuclear mRNA export is driven by the multi-subunit protein complex transcription-export (TREX), which interacts with spliced mRNAs to guide them through the nuclear pore complex to the cytoplasm. 61 The m6A methyltransferase was shown to recruit TREX to m6A-modified RNAs for efficient export. Interestingly, depletion of the m6A methyltransferase subunits VIRMA and WTAP inhibits this process. 62 The m6A reader YTHDC1 plays an essential role in facilitating RNA export as its knockdown increases the nuclear retention of m6A-modified transcripts. 63 Another m6A reader FMRP also mediates the nuclear export of m6A-containing mRNAs, which was blocked in FMRP-deficient mouse. 64 FMRP was also shown to regulate neural differentiation by promoting the nuclear export of Notch- and Hedgehog signaling-related mRNAs. 43 Mechanistically, FMRP associates with exportin 1 to mediate the export of m6A-modified RNAs. 43 Thus, m6A modulates the nuclear export of selective transcripts by recruiting export factors in a stimulus-dependent manner.
Stability
The mRNA abundance of approximately one-tenth of the neural transcriptome is determined by the mRNA stability. 65 The mRNA stabilization involves the competitive binding of RBPs to target mRNAs to guard them against degradation. It was shown that RBPs specifically bind to the m6A methylated transcripts to control their stability. For example, IGF2BP1, 2 and 3 directly bind to ∼80% of m6A methylated transcripts and recruit mRNA stabilizer human antigen R to promote mRNA stability. 41 Additionally, certain neuronal-enriched m6A readers, including FMRP and PRRC2A, compete with YTHDF2 for binding to methylated transcripts, and thus prevent their degradation.42,66
Translation
The m6A methylation was thought to reduce the half-life of mRNAs. 67 However, the interaction of the m6A reader YTHDF1 with the translation initiation factor 3 (eIF3) promotes the translation of the m6A-modified transcripts, and the YTHDF1 knockdown curtailed this process. 68 Furthermore, YTHDF3 binds to YTHDF1 to promote the translation of the m6A-modified transcripts. 69 The translation regulatory mechanisms of m6A are site-specific within the mRNA. YTHDF1-directed translation regulation is mainly observed for m6A sites in the 3'-UTR region of the mRNAs, 69 whereas, m6A sites present in the 5'-UTR region directly interacts with eIF3 to cause cap-independent translation initiation. 70 Surprisingly, ∼13% of endogenous circular RNAs are also m6A methylated and the m6A reader YTHDF3 recruits the translation initiation factors eIF4G2 and eIF3A, leading to their translation. 71
Degradation
The m6A reader YTHDF2 recruits RNA decay machinery, including CCR4-NOT deadenylase complex and RNase P/MRP to the m6A-methylated RNAs, and thus regulates their decay in the P-bodies.72–74 YTHDF2 knockdown was shown to increase the half-life of m6A modified transcripts by ∼30%. 72
Transcription
The m6A methylation of RNAs is also implicated in transcription regulation. The lncRNA X-inactive specific transcript (XIST) has the highest number of m6A sites than any other RNA, and depletion of m6A by METTL3 knockdown impairs XIST-mediated transcriptional repression of X-linked genes. 36 The m6A sites are highly prevalent in RNAs within R-loop structures that regulate transcription termination. 75 , 76 METTL3 depletion leads to fewer R-loops near transcription end sites indicating that m6A is crucial for efficient transcription termination, 75 whereas, YTHDF2 binds to m6A-modified R-loops to destabilize and prevent their accumulation. 76 In stem cells, METTL3 deposits m6A on chromosome-associated regulatory RNAs and YTHDC1 enable their degradation, resulting in chromatin closure and transcription inactivation. 77
Role of m6A methylation in brain development
Neurogenesis
Epigenetic modifications are known to regulate the proliferation and differentiation of neural stem cells (NSCs). 78 Several studies showed that m6A methylation also controls neurogenesis. During development, METTL3 expression peaks during the early stage, whereas FTO expression peaks during the later stage of neurogenesis. 16 , 28 Loss of METTL3 or METTL14 prolongs the cell cycle progression of NSCs, and thereby delays the production of upper-layer neurons in the postnatal mouse cortex. 16 Deletion of METTL14 and YTHDF2 leads to enlarged ventricles and decreased cortical thickness, respectively. 16 , 79 Furthermore, in the E13.5 stage mouse forebrain, several transcripts related to cell cycle (Cdk9, Ccnh, and Cdkn1C) and NSC-specific transcription factors (Pax6, Sox1, and Emx2) were shown to be m6A tagged and their stability increased in METTL14-deficient NSCs. 16 Neurod1 and Neurod2 transcripts were also m6A tagged in NSCs, indicating the role of m6A methylation in transcriptional prepatterning. 16 Furthermore, YTHDF2-deficient NSCs showed decreased proliferation and differentiation, and neurons derived from YTHDF2-deficient NSCs have shorter neurites and are vulnerable to oxidative stress. 79 Moreover, transcripts related to the JAK–STAT pathway that negatively regulate the stem cell proliferation like Flrt2 and Ptprd are hypermethylated and stabilized in YTHDF2-deficient NSCs. 79 In METTL14-deficient NSCs, a significantly increased abundance of histone H3 at lysine 27 acetylation (H3K27ac), lysine 27 trimethylation (H3K27me3), and at lysine 4 trimethylation (H3K4me3) was observed. 80 Particularly, loss of m6A increased gene-silencing H3K27me3 mark near promoters of proliferation-related genes like Egr2 and Egr3, and gene-activating H3K27ac mark near promoters of differentiation-related genes like Kif26a, Gas7, and Pdgf1b. 80 Modulating these changes using inhibitors of histone acetyltransferase CBP/p300 and histone methyltransferase Ezh2 promoted proliferation of METTL14-deficient NSCs. 80 This indicates that m6A induces histone alterations to regulate the NSC gene expression. Mechanistically, failure to clear m6A-tagged CBP/p300 impairs neurogenesis in METTL14-deficient embryonic NSCs. 80 METTL3 knockout decreased m6A in the Ezh2 transcript responsible for global H3K27me3, reduced the NSC proliferation and differentiation, which was rescued by Ezh2 overexpression. 81 Altogether, these studies indicate the cross-talk between RNA modifications and histone modifications during neurogenesis.
The m6A erasers also play a role in neurogenesis. Although depletion of FTO or ALKBH5 did not affect the proliferation of embryonic NSCs, 80 loss of FTO elevated m6A in the brain-derived neurotrophic factor (BDNF) signaling pathway-related transcripts Bdnf, PI3K, Akt1, Akt2, Akt3, and S6K1, which were consequently degraded, leading to reduced proliferation of adult NSCs. 28 FTO deficiency also reduced mature BDNF protein levels, and thereby decreased the activation of the prosurvival MAPK signaling pathway leading to impaired adult neurogenesis. 82
Gliogenesis
The m6A signaling in the context of glial lineage progression is less studied except for the oligodendrogenesis. PRRC2A was observed to compete with YTHDF2 to bind and stabilize m6A methylated oligodendroglial lineage determination factor Olig2 mRNA, and hence PRRC2A-deficient mice fail to develop mature oligodendrocytes and display hypomyelination. 42 Notably, FTO demethylates and prevents PRRC2A-dependent stabilization of Olig2, and hence FTO-overexpressing mice phenocopied PRRC2A-deficient mice. 42 Moreover, ablation of the m6A writer METTL14 in oligodendrocyte lineage cells led to hypomethylation of several transcription factors (Hey1, Klf19, and Zeb2), histone modifiers (Hdac3, Kdm2b, and Prdm2), and growth factors (Igf-1, Fgf, and Bmp) that promote oligodendrocyte maturation and hence reduced the number of mature oligodendrocytes and caused hypomyelination. 83 METTL14 deficiency also resulted in decreased oligodendrocytes-specific neurofascin (Nfasc) splice isoform, subsequently producing an aberrant node of Ranvier morphology. 83 Furthermore, METTL14- and YTHDF2-deficient NSCs failed to differentiate to astrocytes. 16 , 79 Collectively, these studies show that m6A promotes the maturation of glial cells.
Cerebellar development
Interestingly, the cerebellum shows two times higher m6A methylation than the cerebral cortex. 13 , 84 Several apoptotic (Dapk1, Fadd, and Ngfr) and synaptic transcripts (Grin1, Atp2b3, Grm1, and Lrp8) were shown to be hypomethylated in the cerebellum of METTL3-deficient mice, leading to severe cerebellar granule cell apoptosis and hypoplasia. 84 Apart from constitutively methylated mRNAs, a unique set of stage-specific m6A-methylated mRNAs were observed in the cerebellum during postnatal development. 85 This indicates that m6A methylation is essential for cerebellar development.
Axonal growth
The m6A methylation regulates the translation of mRNAs involved in axon guidance and elongation during development. RNAs in the axons of dorsal root ganglion (DRG) and dorsal spinal cord (DSC) show a high abundance of m6A methylation and m6A regulatory proteins FTO and YTHDF1.25,86 In the axons of DRG neurons, FTO demethylates Gap43 to promote its translation leading to axon elongation, 25 whereas, YTHDF1 promotes translation of methylated axon guidance-related mRNA Robo3.1 in the DSC. 86
The role of m6A methylation in brain physiology
Synaptic plasticity
The m6A methylation is highly prevalent in synaptic transcripts (∼76% of the post-synaptic and ∼30% of pre-synaptic mRNAs are methylated in mice). 13 A recent study showed that METTL14, FTO, and YTHDF1, 2, and 3 are localized in the dendritic processes of cortical and hippocampal neurons and > 1200 transcripts related to synapse organization, assembly, maturation, and modulation of synaptic transmission are hypermethylated. 12 Furthermore, FTO-deficient mice displayed increased methylation of synaptic transmission-related mRNAs in the midbrain. 27 The loss of the m6A reader YTHDF1 in hippocampal neurons caused structural and functional deficits in excitatory synapses, including reduced spine head width, lower levels of post-synaptic density-95, and curtailed excitatory post-synaptic currents. 12 , 87 METTL14 deletion in striatal neurons decreased the m6A methylation in synaptic plasticity-related transcripts that correlate with impaired neuronal excitability. 88 Together, these observations highlight the crucial role of m6A methylation in synaptic plasticity.
Circadian clock
Inhibition of m6A methylation of clock transcripts like Per1, Per3, Tef, Dbp, Nfil3, Bhlhe41, and Nr1d with 3-deazaadenosine prolonged the circadian clock oscillation in mice. 89 Furthermore, METTL3 inhibition caused the nuclear accumulation of unmethylated clock transcripts Per2 and Bmal1. 89 The CK1δ kinase, which is responsible for the degradation of clock proteins, was observed to be heavily m6A-methylated, and targeted ablation of the m6A motif in the CK1δ gene increased its translation in the brain, and those mice showed longer rhythmic locomotor activity. 90 Thus, m6A methylation might play a significant role as the circadian pacesetter.
Behavioral adaptation
By modulating neural plasticity, m6A methylation can influence adaptive behavior. Perturbation of m6A methylation was shown to alter cognition, addiction, anxiety, and depression-like behavior in mice. 2 Furthermore, behavioral training by fear conditioning also significantly altered the m6A methylome in the mouse brain.11,91 Ablation of FTO or METTL14 or YTHDF1 was shown to impair cognition in mice.28,87,88 The m6A methylation is also thought to be critical for reward mechanisms. Cocaine-induced locomotor and reward-stimulatory actions were observed to be impaired in FTO-deficient mice 27 and certain FTO variants strongly correlate with increased alcohol consumption in humans. 92 FTO deficiency also suppressed depression- and anxiety-like behavior in mice which was observed to be due to decreased inflammation caused by gut microbial dysbiosis. 93
Stress response
In non-neuronal cells, various environmental stressors, such as heat shock, hypoxia, oxidative stress, and UV irradiation, alter the m6A epitranscriptome. 55 , 56 , 94 , 95 Acute restraint stress in mice caused m6A hypomethylation in the prefrontal cortex and hypermethylation in the amygdala. 96 All the enzymes of m6A machinery harbor glucocorticoid response elements in the 10 kb upstream region of the transcription start site, indicating their putative stress-responsive regulation. 96 Furthermore, stress-induced m6A methylation suppressed translation of synaptic plasticity-related and morphogenesis-related transcripts like Camk2n1, Dusp1, and Homer1. 96 Importantly, neuronal-specific deletion of METTL3 or FTO improved cued/contextual fear memory, mimicking stress. 96 Two recent studies showed increased m6A levels with a concomitant loss of FTO expression in the prefrontal cortex and hippocampus following fear-conditioning.11,91 Conversely, FTO inhibition in the hippocampus or prefrontal cortex also enhanced the fear memory. 11 , 91 Collectively, these studies hint the role of m6A signaling in stress-related disorders.
The role of m6A methylation in brain diseases
Stroke
Secondary brain damage following stroke is synergistically mediated by several pathologic events, including excitotoxicity, edema, apoptosis, mitochondrial dysfunction, oxidative stress, endoplasmic reticulum stress and inflammation. 97 Recent studies showed that epigenetic changes modulate post-stroke dysfunction. 98 , 99 Many studies indicated that epitranscriptomic mechanisms also promote stroke and its consequences. The Mannheim–Heidelberg stroke study showed a genetic association between a FTO variant and transient ischemic attack. 100 Furthermore, 4000 m6A-single nucleotide polymorphism (SNPs), including in IRF6 and NDST1 genes, were shown to be associated with ischemic stroke. 101 Using a rodent model, we and others reported that ischemic stroke down-regulates the cerebral FTO levels, leading to increased m6A methylation. 53 , 102 We observed that several transcripts that mediate apoptosis (Fas, Trib3, and Bcl2a1c) and inflammation (Tnf, IL-6, and Sele) were hypermethylated in the ischemic brain. 53 Additionally, stroke upregulated the expression of the m6A reader YTHDF1, which might bind to m6A-methylated inflammatory and apoptotic transcripts to increase their translation. 53 Furthermore, FTO overexpression was shown to protect the cortical neurons from apoptosis following oxygen–glucose deprivation by stabilizing anti-apoptotic transcript Bcl2. 102 Forebrain ischemia in gerbils was also shown to decrease the hippocampal FTO protein expression. 103 When hippocampal neurons were subjected to hypoxia, m6A levels were elevated in the phosphates and tensin homolog transcript that induced cell death, and over-imposing hypothermia normalized this and enhanced the neuronal survival. 104
A recent study showed reduced expression of the m6A writer complex subunit WTAP in the brains of arteriovenous malformation (AVM) patients suffering from congenital vascular dysfunction. 105 Ablation of WTAP decreased the m6A levels and expression of the cell adhesion molecule, desmoplakin (DSP), leading to the reduced angiogenic potential of cultured endothelial cells. 105 An inverse correlation was also observed between METTL3 expression and the nidus size of cerebral AVMs. 106 Notch pathway regulator DTX3L was implicated in AVM formation, and in METTL3-deficient endothelial cells, DTX3L mRNA was observed to be hypomethylated and silenced, indicating that m6A is crucial for its stability. 106 Furthermore, m6A SNP in IRF6 transcript correlated with intracerebral hemorrhage in humans. 101 Collectively, these studies indicate the role of m6A methylation in stroke incidence and post-stroke brain damage.
Peripheral nerve injury
Injury to a peripheral nerve leads to discontinuity of the axon, loss of myelin fibers, and eventual death of the neurons. 107 Epigenetic modifications, including histone acetylation and DNA hydroxymethylation, were shown to promote axon regeneration after peripheral nerve injury by increasing the transcription of regeneration-associated genes (RAGs). 108 A recent study reported that m6A RNA methylation increases in many RAGs (Atf3, Sox11, Gadd45a, and Tet3) as well as several ribosomal subunits (Rps14, Rps20, Rps23, Rps28, Rps29, Eif1a, and Eif3b) in the DRG following sciatic nerve injury in mice. 109 Furthermore, depletion of either METTL14 or YTHDF1 decreased the translation of RAGs in the injured DRGs and significantly reduced the axon growth and reinnervation leading to curtailed functional recovery. 109
Traumatic brain injury
Several studies showed the functional significance of epigenetic modifications in brain damage after traumatic brain injury (TBI). 98 TBI in adult mice decreases METTL3 expression and global m6A abundance in the hippocampus within 6–24 h post-injury. 110 Many of the differentially m6A-methylated transcripts in the post-TBI hippocampus were observed to control neuronal metabolic processes. 110 TBI in rats was also shown to decrease the mRNA expression of METTL14 and FTO in the cerebral cortex and many of the m6A-modified transcripts such as CD14 (inflammation), Dll4 (Notch signaling), and Bcl2 (apoptosis) are known promoters of post-TBI pathology. 111 More importantly, FTO inhibition exacerbated the neurologic dysfunction after TBI but did not affect the cognitive function. 111
Alzheimer’s disease
Epigenetic dysregulation is thought to drive the protein aggregation and neuronal loss in Alzheimer’s disease (AD). 112 The hippocampus of the double transgenic AD mice showed induction of METTL3 and suppression of FTO, leading to altered global m6A methylation of RNAs involved in dendrite development, synaptic growth, pre- and post-synaptic assembly, glutamate receptor signaling, axon guidance, and long-term potentiation. 113 This implies aberrant m6A methylation as a promotor of AD pathogenesis. Expression of the m6A demethylase FTO was shown to be increased in the brains of triple transgenic AD mice, and its knockdown decreased the phosphorylated tau without affecting Aβ accumulation in the neurons. 114 This effect was further shown to be mediated by methylation of the tuberous sclerosis complex 1, which is an upstream inhibitor of the mammalian target of rapamycin kinase that regulates tau. Interestingly, FTO-deficient AD mice showed significantly better cognitive function compared to wild-type mice. 114 In Caucasians, the presence of FTO AA-genotype was found to increase the risk of AD by 1.6-fold. 115 Furthermore, three SNPs in the FTO gene leading to decreased FTO expression was shown to be associated with AD risk in Caucasians and Caribbean Hispanics. 116 Overall, FTO modulation seems to be a potential strategy to alleviate the AD-associated deficits.
Parkinson’s disease
Epigenetic changes such as reduced DNA methylation at the promoter of SNCA (the gene that transcribes α-synuclein) as well as decreased histone acetylation are implicated in α-synuclein aggregation and Parkinson’s disease (PD) progression. 117 , 118 Interestingly, FTO deficiency increased the m6A methylation and decreased the translation of dopaminergic pathway-related transcripts like Drd3, Girk2, and Grin1. 27 Increased expression of m6A demethylases ALKBH5 and FTO and decreased levels of m6A were observed in the striatum of rats subjected to 6-hydroxydopamine (6-OHDA)-induced PD. 119 FTO inhibition also protected PC12 cells against 6-OHDA-induced cell death. 119
Brain cancer
Several studies showed that the expression of m6A writers, erasers, and readers is significantly perturbed in the glioblastoma multiforme (GBM) patients.120–122 Notably, modulation of m6A machinery in patient-derived glioma stem-like cells (GSCs) altered their self-renewal and tumorigenic properties. 123 , 124 Tumor tissue resected from GBM patients showed high levels of the m6A writer METTL3 that correlated with poor patient survival. 120 However, role of METTL3 in regulating the tumorigenic potential of GSCs remains controversial. Initial studies proposed METTL3 to be a tumor suppressor as METTL3 knockdown in vivo promoted tumor progression and worsened survival. 124 , 125 Furthermore, METTL3 overexpression in a GBM cell line U251 inhibited the migration and proliferation of cells and concomitantly induced apoptosis. 125 In contrast, silencing METTL3 prevented m6A methylation of the glioma reprogramming factor Sox2, leading to apoptosis and radiosensitization of GSCs, and reduced tumor size in mice. 123 Furthermore, METTL3 was shown to regulate many transcripts related to oncogenic signaling and RNA editing, such as adenosine deaminase RNA specific 1 and apolipoprotein B mRNA editing enzyme catalytic subunit 3A that are critical for glioma progression. 126 METTL3 was also shown to drive glioma growth by regulating the alternative splicing machinery and SRSFs. 120 These contradictory suggestions for the role of METTL3 to act both as a tumor suppressor or promoter by different studies might be due to the high heterogeneity of gliomas. Increased levels of the methyltransferase subunit WTAP, as well as m6A erasers, ALKBH5 and FTO, were shown to predict poor prognosis in GBM patients.127–129 Particularly, ALKBH5 demethylates and stabilizes a master transcription factor forkhead box M1 that activates proliferation-inducing genes in GSCs, and its depletion or inhibition prevented migration and invasiveness of GBM cells in mice. 129 , 130 Treatment of GSCs with FTO inhibitors, meclofenamic acid or R-2-hydroxyglutarate, suppressed their growth, stemness, and tumorigenicity. 124 , 131 Expression of m6A readers (YTHDF1, YTHDF2, YTHDF3, and YTHDC2) was also shown to be upregulated in GBMs. 121 YTHDC1 deficiency reduced the sphere formation capacity of GBM cells. 120 Collectively, the above studies suggest that m6A signaling is integral to GBM pathogenesis.
Other CNS disorders
The m6A RNA methylation is also shown to be involved in various other neurodevelopmental and neuropsychiatric conditions. Certain genetic variants of FTO strongly confer risk for major depressive disorder (MDD) and attention‐deficit/hyperactivity disorder.132–135 An SNP in ALKBH5 is associated with various clinical features of MDD, including anxiety, retardation, and cognitive dysfunction. 136 ALKBH5 was further shown to demethylate and stabilize a mood-associated transcript FAAH that contributes to depression-like behavior in mice. 137 ALKBH5 was also found to be a causative gene for a rare neurodevelopmental disorder called Smith–Magenis syndrome. 138 The m6A reader YTHDC2 was reported to be a potential risk locus for the Autism spectrum disorder in the Japanese population. 139 The m6A SNP (G to A mutation) in the METTL21B gene was found to correlate with multiple sclerosis. 140 Overall, key regulators of m6A methylation are implicated in various CNS disorders.
Conclusion and future directions
The brain has evolved multiple RNA regulatory mechanisms to optimize energy expenditure, plasticity, and spatial regulation in polarized structures. Conceptually, these mechanisms involve binding RBPs and noncoding RNAs to the sequence elements present in the RNA, thus recruiting diverse cellular machinery to affect the fate of target RNA. 141 Additionally, RNA acquires chemical modifications to ascertain the fidelity and dynamics of this process. To that end, m6A methylation is an epitranscriptomic mechanism primarily employed by neurons to tightly control gene expression during development and disease (Figure 5). More importantly, the reversible nature of m6A makes it a powerful tool to achieve context- and stimulus-dependent regulation. Among the two well-characterized m6A erasers, FTO is dysregulated in various CNS pathologies ranging from brain tumors to acute and chronic brain damage. The lack of compensatory demethylation by ALKBH5 indicates that each eraser controls distinct targets in the brain.
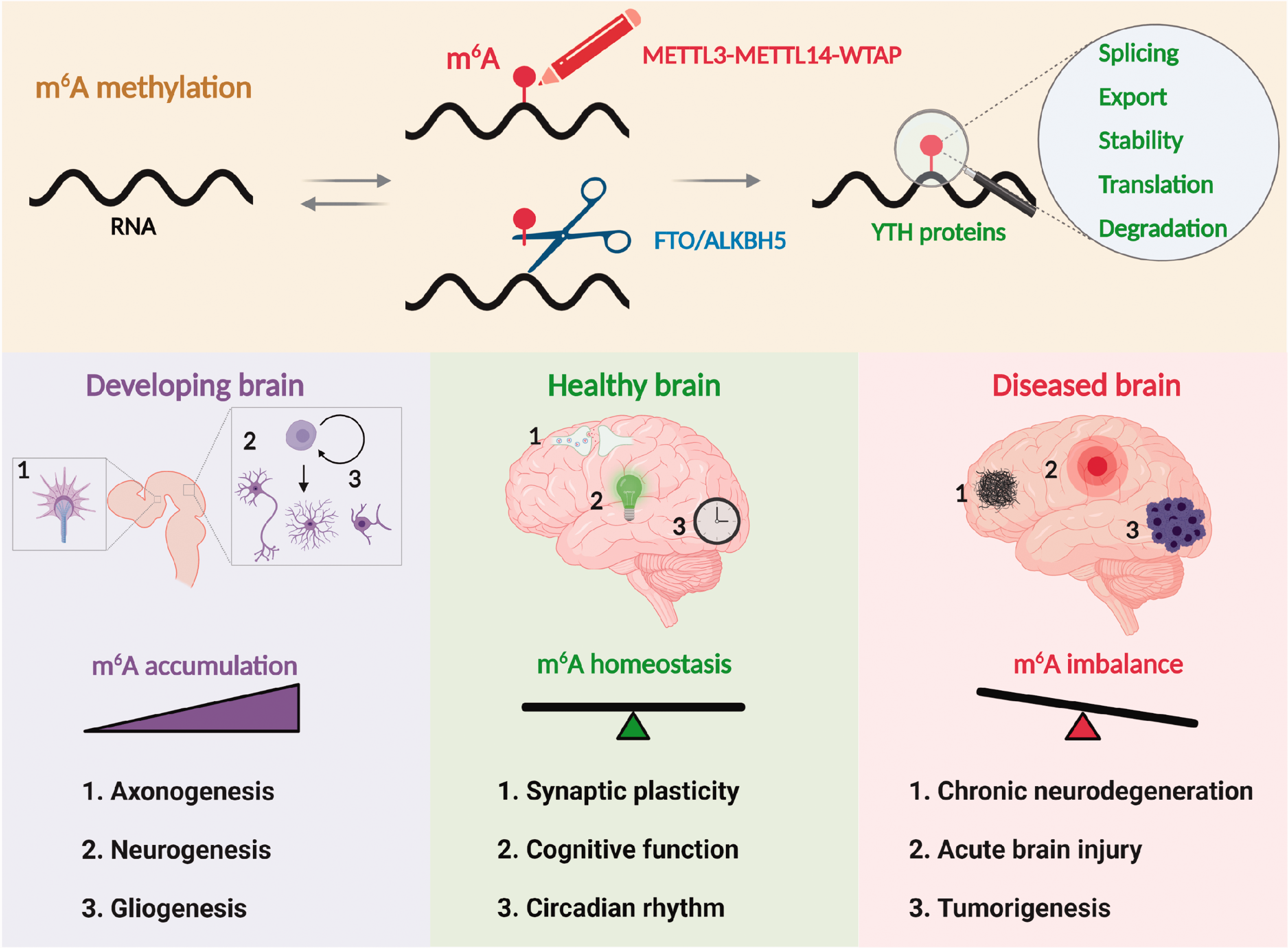
Role of m6A methylation in brain development, physiology, and disease. The m6A, with its multifaceted role in mRNA processing, is considered as the fifth nucleobase in the mammalian brain. In the developing brain, m6A gradually increases to control axonogenesis, neurogenesis, and gliogenesis. In the adult brain, m6A maintains essential physiological processes such as synaptic plasticity, cognitive function, and circadian rhythm. The m6A dysregulation is implicated in many pathological processes, including chronic neurodegeneration, acute brain injury, and tumorigenesis.
Although the m6A dysregulation was observed in various neurological diseases, its functional significance in disease outcomes is not yet completely understood. A challenge is to understand the functional significance of hundreds of transcripts of distinct functional classes that show m6A methylation. However, new studies are emerging to show that dosing (number of m6A consensus motifs and their methylation at a given time) is important to promote a functional change. 142 Techniques like CRISPR-based site-specific m6A editing might allow studying its role in disease outcomes.50–52 Currently, several small-molecule inhibitors of m6A editing enzymes are also available to decipher the roles of writers, erasers, and readers.130,143–148 Finally, several studies showed the cross-talk between m6A and other regulators such as epigenetic modifications and microbiome.80,81,93,149
Footnotes
Acknowledgements
Figures are created using Biorender.com.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was partly supported by the NIH grant RO1 NS109459 and AHA grant 20PRE35120233.