
Abstract
Intracerebral hemorrhage (ICH) is a subtype of stroke with high mortality and disability but no specific or effective treatment. In the last two decades, much has been learned about the pathologic mechanisms of ICH. It is now known that after ICH onset, immune and inflammatory responses contribute to blood–brain barrier disruption, edema development, and cell death processes, jointly resulting in secondary brain injury. However, the translation of potential therapies from preclinical to clinical success has been disappointing. With the development of new laboratory technology, recent progress has been made in the understanding of ICH pathomechanisms, and promising therapeutic targets have been identified. This review provides an update of recent progress on ICH and describes the prospects for further preclinical studies in this field. Our goal is to discuss new therapeutic targets and directions for the treatment of ICH and promote the effective transformation from preclinical to clinical trials.
Introduction
The epidemiology of intracerebral hemorrhage
Intracerebral hemorrhage (ICH), the second most common subtype of stroke, occurs when a weakened arterial vasculature ruptures, causing blood to leak into the adjacent brain parenchyma. ICH is associated with high mortality and disability. Indeed, mortality has been reported to be 59% at one year and up to 70% at five years, with more than 80% of survivors experiencing permanent disabilities. 1 ICH most commonly occurs in the basal ganglia, followed by cortex, brainstem, cerebellum, and other deep brain tissues. The prevalence of ICH is rising with the increasing age of the population; with increases in risk factors such as hypertension, obesity, diabetes, and alcohol consumption; and with the widespread use of anticoagulant drugs.
Although no specific treatment has yet been found for ICH, our understanding of its underlying pathologic mechanisms has progressed rapidly in the last two decades.2,3 ICH-induced brain injury consists of primary and secondary brain injury. Primary brain injury occurs a few hours after ICH onset during hematoma formation, resulting in mass effects and increased intracranial pressure, even herniation and death. Secondary brain injury is triggered by the activation of resident cells, infiltration of peripheral immune cells, and secretion of inflammatory factors that aggravate brain edema and cell death.
Thus far, research on sex differences in response to ICH has been inconclusive. Although sex has a strong influence on ICH occurrence and resulting brain damage, it may not affect long-term outcomes. The annual incidence of ICH per 100,000 persons is higher in men than in women (69.6 vs. 62.7). 4 Sex differences in neurological outcomes are emerging with varying results. Marini et al. 5 reported a higher risk of hematoma expansion and mortality in men than in women, whereas other studies have reported no sex-related differences in long-term outcomes after ICH.6–8 In mouse models, inflammatory responses after stroke differ between the sexes. In preclinical ischemic stroke studies, young male mice exhibit higher expression of inflammatory genes in the cortex than female mice. These genes are associated with T cell function, adhesion molecules, cell signaling, cell death, inflammatory cytokines as well as major histocompatibility complex and costimulatory signals. In preclinical studies of hemorrhagic stroke, male mice exhibit significantly more severe brain edema than do female mice at three days and slower recovery of neurological function. 9 Sex differences in short-term outcomes may be mainly attributable to the effects of circulating estrogen and progesterone.10,11
Age is another factor associated with ICH occurrence and ICH-induced brain damage. The incidence of ICH increases with advanced age. In 2010, the incidence of ICH per 100,000 was 5.9 in the 35–54 age group, 37.2 in the 55–74 age group, and 176.3 in the 75–94 age group. 12 Sex and age have a strong correlation with early ICH outcomes, but there is little to no evidence that sex can influence the effect of age on 90-day outcomes after ICH. 6 Recent inpatient database studies based on prospective research have shown that age is a key predictor for poor functional outcomes after ICH. 13 Similarly, ICH-induced brain injury is aggravated in senescence-accelerated prone mice. Activated microglia were abundantly distributed around the hematoma in old senescence-accelerated prone mice. 14 Moreover, the expression of inducible nitric oxide synthase (iNOS) was higher in the elderly mice than in young mice. 14 Another study showed that compared to young rats, older rats had stronger microglial activation and a greater perihematomal induction of heat shock proteins 27 and 32 but a weaker astrocytic reaction to the hematoma. 15 Additionally, it has been reported that the age of mice is correlated to the expression, timing, or levels of many genes that reflect the activation of immune cells and inflammation after ICH, such as interleukin (IL)-6, IL-1β, iNOS, and toll-like receptors (TLRs). 16
ICH occurrence as well as subsequent damage is also influenced by genetics. ICH family aggregation has been observed and the heritability of ICH risk has been estimated at 44%. 17 Two variants of the gene encoding apolipoprotein E (APOE) are thought to increase the risk of ICH. MicroRNAs (miRNAs) also contribute to brain inflammation. In a rat model of ICH, downregulation of miRNA-144 inhibited inflammation and brain edema and improved neurologic function. 18 However, the effect of genetics on ICH occurrence and/or outcomes has not been well studied at the population level. Overall, very little research has focused on the genetics of ICH-associated inflammation in humans, and additional studies are warranted.
Emerging evidence shows that the immune cascade plays a critical role in the progression of ICH-induced brain injury and repair.2,3,16 Upon the onset of ICH, blood components enter the brain. They activate innate and adaptive immune responses, causing the release of a variety of cytokines, chemokines, free radicals, as well as other potentially toxic chemicals. Together, these molecules ultimately lead to blood–brain barrier (BBB) disruption, brain edema, as well as cell death.19–22 Previous reviews have highlighted the preclinical and clinical translation of inflammation.2,3,19–22 The fact that the inflammatory process contributes to acute and chronic brain injury after ICH suggests that regulation during both phases is essential for promoting brain repair. This review aims to summarize updates in preclinical and clinical studies of inflammation and discuss how modulating inflammatory responses in the early and delayed phases of ICH, especially those of microglia/macrophages, astrocytes, leukocytes, mast cells (Figure 1), and hemoglobin (Hb)/iron, might translate into ICH therapy.
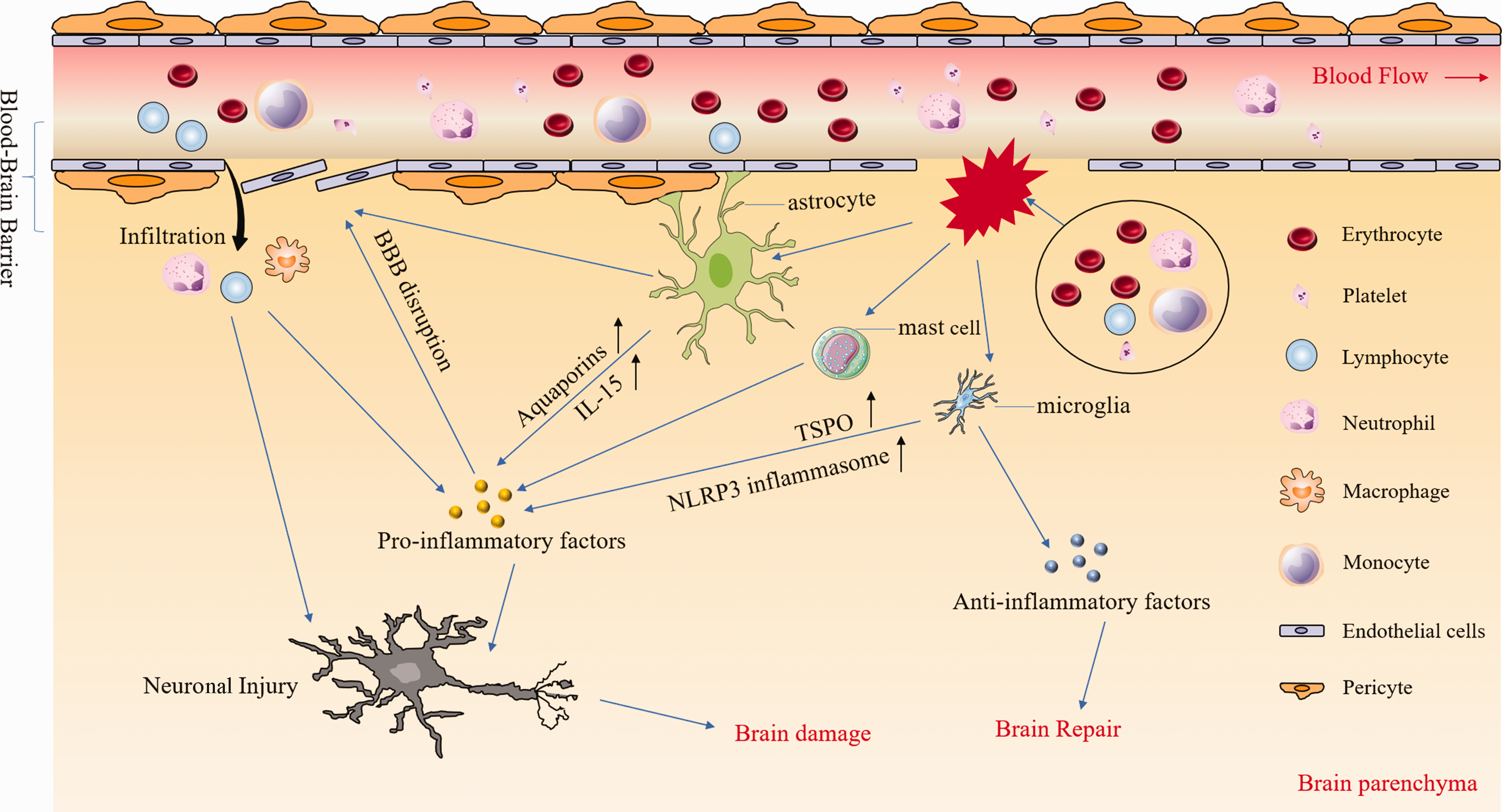
Inflammatory responses after intracerebral hemorrhage (ICH). After ICH, multiple blood components (e.g. erythrocytes, leukocytes, platelets) are released into the brain parenchyma where they can activate microglia, astrocytes, and mast cells by distinct pathways. After activation, microglia upregulate the expression of NLRP3 and TSPO and produce large amounts of pro-inflammatory factors. However, they also produce anti-inflammatory factors such as TGF-β which contributes to brain repair. Astrocytes contribute to the composition of the blood–brain barrier (BBB) and are destroyed directly after ICH, leading to BBB disruption. In addition, astrocytes secrete various pro-inflammatory factors that aggravate neuronal injury and brain damage. Pro-inflammatory factors from diverse cell types not only exacerbate BBB disruption and leukocyte infiltration, but also kill neurons directly. NLRP3: the nucleotide-binding oligomerization domain-like receptor containing pyrin domain 3; TSPO: translocator protein.
Search strategy and inclusion/exclusion criteria
Literature search was conducted on major scientific databases including but not limited to PubMed, Web of Science, and the Internet Stroke Center from October 2010 to June 2019 with different combinations of the following words: spontaneous or intracranial or intracerebral or cerebral or brain or putaminal or intraparenchymal or basal ganglia or thalamic or h(a)emorrhage or h(a)emorrhagic or stroke and inflammation or immune response or immunity. Only English articles were chosen. Articles were shortlisted based on screening of the title and abstract. Then the chosen articles that described neuroprotective effects or improved outcomes in the ICH model were analyzed. References of relevant articles were also screened for eligible studies. Exclusions include case reports, negative results, and studies that examined mechanisms of ICH but did not evaluate inflammation or a putative therapy. Not all the referenced papers are cited due to the stated limit on reference numbers.
Microglia/macrophages
As resident immune cells in the brain, microglia are thought to act as “the first responders” in the early stage of ICH 21 and other acute brain injuries. In normal brains, they are ramified, quiescent, and constitute approximately 5–20% of total neuroglial cells. After the onset of ICH, microglia are activated within 1 h 23 and persist for up to four weeks.2,24 Monocyte-derived macrophages infiltrate into the perihematoma and hematoma region after microglia are activated. Activated, polarized microglia and macrophages contribute critically to acute neuroinflammation and hematoma resolution after ICH. 21 However, when fully activated, phagocytic microglia are difficult to distinguish from macrophages. Recently, we developed a flow cytometry protocol that identifies and sorts microglia and macrophages, 25 offering us the opportunity to study microglia and macrophages separately. Nevertheless, given that activated microglia and macrophages share many similarities including signature genes, surface markers, anti/inflammatory responses, free radical release, trophic factor production, as well as phagocytic function, in this review we do not differentiate these two cell types or discuss them separately in most scenarios.
Activated microglia not only phagocytose the hematoma and cell debris, but also release a variety of proinflammatory cytokines (TNF and IL-1β), anti-inflammatory cytokines (IL-4 and IL-10), chemokines (C–X–C motif chemokine 2), reactive oxygen species (ROS), and nitric oxide, which all aggravate brain injury.21,26 Interestingly, a recent study indicated that microglial depletion with clodronate liposomes increased proinflammatory cytokine levels, induced astrocyte activation, and damaged neurons in addition to BBB integrity. 27 In brief, the overall effect of microglia is detrimental during the acute phase, 28 but beneficial to long-term recovery. 21 Though microglia are activated by multiple blood components, red blood cells induce microglial activation via CD36 29 ; hemoglobin, heme, and fibrinogen induce microglial activation via TLR4 30 ; and thrombin and complement factors induce microglial activation via protease-activated receptors (PARs). 31 Many attempts have been made to target these pathways, but the preclinical efficacy has not translated into clinical success. Thus, it is imperative that we gain a deep understanding of the current state of clinical as well as preclinical studies and seek new and promising cell-specific targets for microglia/macrophages. 32
Clinical studies
Clinical studies have helped investigators to confirm the mechanisms of ICH injury and verify the findings from preclinical studies. Table 1 summarizes recent clinical studies or trials associated with microglia/macrophages. One study of 27 ICH patients revealed that inflammation peaked at two to three days after ICH onset primarily as a result of activated microglia/macrophages and infiltrating leukocytes. 33 This finding was complimentary and consistent with data from preclinical studies. Additionally, in the early stage of ICH, CD163 (a high affinity scavenger receptor for the hemoglobin-haptoglobin complex that is mainly expressed on microglia/macrophages) gradually increased from 6 to 24 h and reached a peak at more than 72 h after ICH onset. Its peak coincides with the maximal expression of heme oxygenase (HO)-1. 33 The CD163/HO-1 pathway regulates inflammation in tissues that surround the hematoma, 33 and Roy-O’Reilly et al. 34 concluded in a study of 51 primary ICH patients with moderate-sized, hypertensive deep hemorrhage that acute soluble CD163 level in serum might be a useful biomarker for predicting hematoma expansion, perihematomal edema expansion, and worse short-term outcomes in patients. Thus, upregulating the CD163 expression to enhance microglia/macrophages phagocytosis may be a therapeutic strategy for ICH treatment. In a similar study of 199 patients with acute ICH, those whose macrophages were deficient in CD36, also a scavenger receptor, had larger hematoma volumes and slower absorption seven days after admission when compared to patients with normal CD36, despite equal hematoma volumes on admission. 35 PPAR-γ agonists have been shown to upregulate CD36 and promote microglia/macrophage-mediated phagocytosis in rodent models of ICH. Pioglitazone, a PPAR-γ agonist, has completed its phase II dose-escalation trial, and has proven to be safe to use for hematoma resolution after ICH (NCT00827892). Minocycline, which inhibited microglial proliferation in pure microglial cultures and reduced the number of microglia and macrophages around the hematoma after ICH in an in vivo model, has also shown efficacy as an adjunctive therapeutic solution. 36 According to a systematic review and meta-analysis of randomized clinical trials for acute stroke, minocycline was associated with improved three-month outcomes in a subgroup of patients with acute ischemic stroke. 37 However, two separate studies using minocycline or placebo to treat ICH patients found no significant difference in clinical outcomes at 90 days.38,39 The negative results may be due to the small sample size (n = 16 or 20) and the selection of the patients. Consequently, the efficacy and safety of minocycline treatment for ICH need further research. Considering the key role of microglia in ICH injuries and recovery, well-designed clinical studies focused on targeting microglia/macrophages are still lacking.
Clinical studies or trials associated with microglia/macrophages.
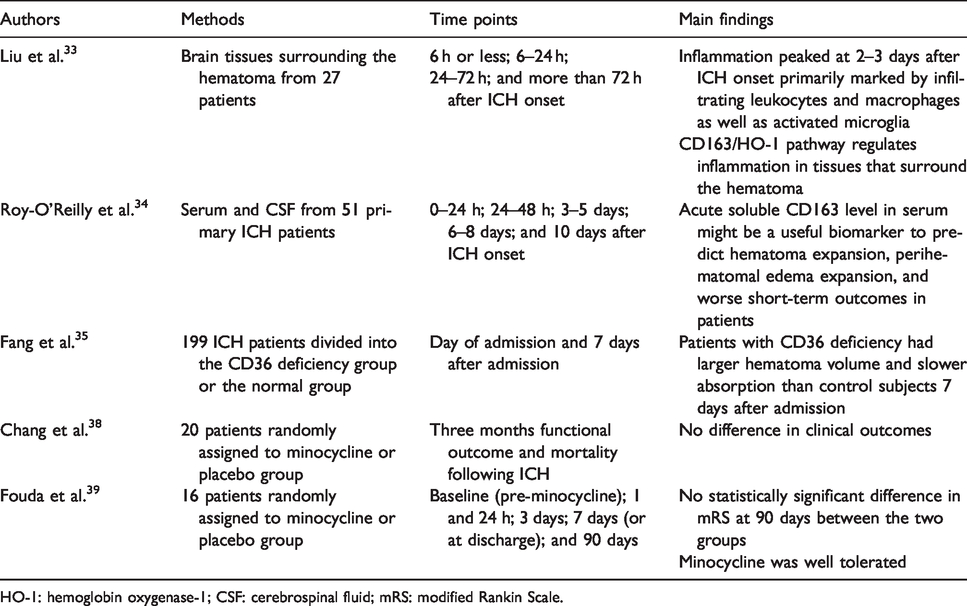
HO-1: hemoglobin oxygenase-1; CSF: cerebrospinal fluid; mRS: modified Rankin Scale.
Preclinical studies
Preclinical studies have provided much new information about the underlying mechanistic role of microglia/macrophages in ICH, clarified the pathologic processes of ICH injury in the early and late stage as well as identified new targets for modulation of microglial/macrophage function that have potential for further study.
The NLRP3 inflammasome
Inflammasomes are newly identified pattern-recognition receptors (PRRs) that lead to the release of proinflammatory cytokines IL-1β and IL-18 by activating caspase-1. 40 In addition, the inflammasome initiates caspase 1-dependent cell death (pyroptosis) and promotes cytokine processing, although the mechanism remains controversial. 41 To date, the NLRP3 inflammasome is the best characterized of the known inflammasomes. It is an intracellular multiprotein complex that contains the skeletal nod-like receptor (NLR), the adaptor protein apoptosis-associated speck-like protein containing a caspase-activating recruitment domain protein (ASC), and the effector procaspase-1. NLRP3 is expressed predominantly in microglia. 42 It can be activated by multiple endogenous and exogenous stimuli, 43 including the binding of pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs) to PRRs. 44 NLRP3 inflammasome plays a key role in a variety of central nervous system (CNS) diseases, such as Parkinson’s disease, 45 subarachnoid hemorrhage, 46 ICH, 47 and autoimmune diseases. 48 In general, the NLRP3 inflammasome requires two separate signals for activation. One is the priming signal which produces pro-IL-1β and pro-IL-18. 50 The second signal activates the NLRP3 inflammasome directly. 49 Once activated, NLRP3 inflammasome cleaves the IL-1β and IL-18 precursors into their biologically active forms. 50 Furthermore, mature IL-1β and IL-18 can recruit peripheral immune cells (neutrophils and macrophages) and amplify the inflammatory response, 51 thus exacerbating brain edema and brain injury. 47 In one study, it was shown that P2X7R small interfering RNA (siRNA) or selective P2X7R inhibitors could suppress the activation of NLRP3 inflammasome and significantly attenuate secondary brain injury after ICH. 52 In addition, endogenous H2S was shown to ameliorate brain edema, microglial accumulation, and neurologic deficits by suppressing P2X7R/NLRP3 inflammasome-regulated neuroinflammation. 53 Activation of dopamine D1 receptor improves neurologic outcome by inhibiting NLRP3-mediated inflammatory responses in a mouse ICH study. 54 Additionally, we recently determined that MCC950, a novel, selective NLRP3 inflammasome inhibitor, reduced brain injury in both autologous blood- and collagenase-induced ICH models. 55 Other groups have verified that andrographolide and low-dose fimasartan ameliorate NLRP3 inflammasome-mediated neuroinflammation and secondary brain injury after ICH.56,57 Thus, NLRP3 inflammasome may represent a potential therapeutic target for ICH treatment that warrants future clinical-pathologic studies and clinical trials.
TSPO
The translocator protein (18 kDa, TSPO), formerly known as peripheral benzodiazepine receptor (PBR), is a five-transmembrane unit localized primarily in the outer mitochondrial membrane. It is the rate-limiting protein for the synthesis of neurosteroid. 58 TSPO is widely expressed in many tissues, such as adrenal glands and gonads. 59 In the CNS, microglia express TSPO sporadically under normal conditions, but during pathologic states, microglial expression increases sharply, with smaller increases in reactive astrocytes and other neuronal cell types.58,60 TSPO is also associated with increased production of ROS, imbalanced calcium homeostasis, and disordered ATP production. 58
TSPO-PET imaging is considered to be an effective biomarker of neuroinflammation and treatment response in many CNS diseases. 61 In a mouse model of multiple sclerosis (MS), TSPO was prominently upregulated in activated microglia and macrophages. Treatment with etifoxine, a high-affinity TSPO ligand, 58 delayed this increase, decreased the activation of microglia, reduced infiltration of T cells, and eventually protected the brain and promoted recovery. 60 Similarly, the expression of TSPO is elevated after ICH, mostly in Iba-1+ microglia or macrophages, but not in GFAP+ or NeuN+ cells. 62 In addition, the TSPO ligand etifoxine significantly reduced neurodeficits and perihematomal brain edema in both collagenase (type IV-S)- and autologous blood-induced mouse models of ICH by decreasing microglial activation and the production of proinflammatory factors, thereby improving BBB integrity. 63 Interestingly, the protective effects of etifoxine were abolished by microglial depletion. 63 Therefore, TSPO may not only represent a potential therapeutic target for ICH, but may also be useful as a biomarker for monitoring disease activity.
TGF-β1
Transforming growth factor (TGF)-β1 is a member of the TGF-β superfamily of cytokines, which are involved in microglial development, immune homeostasis, and apoptosis. 64 In the CNS, TGF-β1 is expressed primarily by microglia/macrophages. Some authors have reported that inhibiting TGF-β receptor 1 or TGF-β1 itself lessens TGF-β1-induced inflammation and apoptosis in a mild traumatic brain injury model. 65 However, the mainstream view is that TGF-β1 is a neuroprotective factor. For example, increasing the TGF-β1 level was shown to reduce senile plaque formation in brain parenchyma in a mouse model of Alzheimer’s disease. 66 Additionally, in a mouse model of acute ischemic stroke, the upregulation of microglial Zinc finger E-box binding homeobox 1 (ZEB1) inhibited the production of astrocytic CXCL1 through the TGF-β1-dependent signaling pathway. 67 TGF-β1 might be a key regulatory factor of microglial-astrocyte interaction. However, because the pathomechanisms of hemorrhagic and ischemic stroke differ, brain inflammatory reactions may also differ.
After ICH, multiple proinflammatory factors increase in the first three days but rapidly decrease thereafter. In contrast, TGF-β1 increases from day 1 to day 10 and remains elevated at day 14. 68 The perihematomal area transitions from a pro-inflammatory to an anti-inflammatory state within the first two weeks after ICH, similar to the microglial phenotype transition. TGF-β is also required to promote the proliferation and differentiation of regulatory T cells (Tregs), which regulate microglial polarization to the M2-like phenotype via the IL-10/GSK3-β/PTEN axis and reduce inflammation after ICH. In primary cultured microglia exposed to thrombin, TGF-β1 causes a reduction in CCL2, TNF, and IL-6 gene expressions. 68 TGF-β1 also participates in brain tissue repair by macrophages through the Smad2 and Smad3 signaling pathways. What’s even more remarkable is that the improved outcome promoted by TGF-β1 persists. In clinical evaluation, patients whose TGF-β1 plasma levels increased during the first few days after ICH had better long-term outcomes even after adjusting for ICH severity. 68 TGF-β1 is a potential mediator of the microglial phenotype switch toward a neuronal repair status and critical to long-lasting recovery after ICH.
Astrocytes
Astrocytes are the most abundant glial cells in the CNS, generally 10 times more numerous than neurons. They play an important role in the maintenance of normal brain function and are considered to be active elements of the brain circuitry. 69 They provide neurotrophic and structural support to neurons and help to maintain homeostasis in the extracellular environment. 69 Astroglial activation, through reactive astrogliosis, is a common response to CNS events such as neurotrauma, stroke, and neurodegeneration.70,71 Reactive astrogliosis involves a series of continuous changes, including alterations in gene and protein expression, cellular proliferation and migration, cellular hypertrophy, and glial scar formation.69,71 Moreover, the resulting glial scar impedes axonal regeneration and the rehabilitation of neuronal connectivity. 72 Interestingly, inhibition of glial scar formation does not always attenuate brain injury in experimental models. 73
In the early stage of ICH, astrocyte activation is robust in the perihematomal area. 74 The number of active astrocytes was shown to increase on day 1 and remain elevated until day 7. Astrocytes secrete various types of cytokines and chemokines that may contribute to microglial differentiation and ICH pathology. 21 Reactive astrogliosis and microglia can interact to induce activation of matrix metalloproteinases (MMPs) and Nrf2-HO-1, which have important roles after ICH. Astrocyte-derived HO-1 preserves neuronal viability and BBB integrity and accelerates the secretion of anti-inflammatory factors after ICH. 75 Moreover, microglia-derived HO-1 may mediate the proinflammatory process that aggravates brain injury early after ICH. 23 Therefore, the therapeutic target of HO-1 needs to be considered comprehensively. Astrocyte-derived IL-33 may also have bidirectional effects by increasing the production of both proinflammatory 76 and anti-inflammatory cytokines. 77 Although astrocytes have received far less attention than microglia, preclinical studies have opened up this field and suggest that some astrocyte targets deserve more in-depth research. Nevertheless, the functional significance of astrocytic responses after ICH remains unclear, as few clinical studies have been carried out.
Preclinical studies
Aquaporins
Aquaporins (AQPs) are a family of membrane transporters associated with water permeabilization. They have a central role in water transmembrane transport and intracellular and extracellular homeostasis. 78 In the CNS, AQPs are distributed mainly on astrocytes and contribute to the development of brain edema after stroke, neurotrauma, and other disorders. 78 AQP4 is the most numerous isoform in the brain 79 and is primarily localized to astrocyte perivascular endfeet. It is closely associated with the occurrence of brain edema.
After ICH, brain edema peaks within three to four days and may persist for more than a week. 80 This timeline is consistent with changes in AQP4 expression, suggesting that AQP4 is involved in the early development of brain edema after ICH. In fact, the expression of AQP4 on astrocyte endfeet facilitates water entry and drainage in brain tissue. Two studies have demonstrated that AQP4 gene deletion in mice destroys BBB integrity, aggravates brain edema, and exacerbates neurologic deficits.81,82 Conversely, other studies have reported that downregulation of AQP4 and AQP9 expression by recombinant hirudin, a specific thrombin inhibitor, or by adipocytokine apelin-13, reduces brain edema and improves neurologic function after ICH. 83 Interestingly, a study of two independent cohorts of ICH patients concluded that AQP4 gene mutations might affect susceptibility to primary ICH and influence the age of onset. 84 Therefore, it can be inferred that AQP4 is an important component of the BBB and that modulation of AQP4 expression rather than complete elimination should be tested as a potential treatment for edema after ICH.
IL-15
IL-15 is an important proinflammatory and immunomodulatory factor for the maintenance and function of multiple cell types. 85 In the CNS, astrocytes are the main source of IL-15 after injuries, with minor contributions from microglia and neurons. 86 In MS, astrocyte-derived IL-15 regulates the activation of CD8+ T lymphocytes that migrate within brain tissue, promotes the killing activity of CD8+ T lymphocytes, and aggravates brain damage. 87 We recently found that astrocyte-derived IL-15 regulates the migration and activity of natural and acquired immune cells to brain tissue, and influences the killing activity of CD8+ T and natural killer cells through trans-presentation, eventually exacerbating ICH-induced brain damage.
In our initial exploration of the effects of astrocyte-derived IL-15 on ICH (unpublished research), we found that IL-15 expression was significantly upregulated in astrocytes and that GFAP-IL-15tg mice, which overexpress IL-15, exhibit significantly larger lesion sizes and greater neurologic deficit than wild-type mice after ICH. In addition, GFAP-IL-15tg mice show significant upregulation of proinflammatory factors (TNF-α, IFN-γ, etc.) after ICH. These unpublished results suggest that astrocytes and astrocyte-derived IL-15 may play a vital role in modulating inflammation, cellular immunity, and brain damage after ICH. Therefore, IL-15 could represent a potential target for treating ICH and be of great significance for improving the CNS immune response after ICH.
Leukocytes
Under physiologic conditions, leukocytes in the blood rarely enter the brain parenchyma and do not remain for long periods unless they are activated by antigen in the CNS. At the early stage of ICH, local release of cytokines, chemokines, prostaglandins, proteases, ferrous iron, and other immunoactive molecules by infiltrating leukocytes is an important pathophysiologic hallmark of ICH. Peripheral leukocyte counts have been reported to be an important predictor of neurologic deterioration in primary ICH. In the periphery, acute leukocytosis is a well-established response to ICH and associated with an elevated risk of early neurologic deterioration. 88
Clinical studies
As we reviewed previously, clinical studies have provided evidence that indicates peripheral neutrophil infiltration is toxic in ICH.2,3 Five recently published clinical studies have shown that a lower admission lymphocyte count and higher neutrophil-to-lymphocyte ratio are independently related to worse long-term outcome in ICH patients.89–93 Another study indicated that a higher peripheral monocyte count on admission was independently associated with 30-day case fatality. 94 On the other hand, according to a study of 1302 ICH patients, higher peripheral leukocyte count on admission was associated with a reduced risk of hematoma expansion. In particular, a higher neutrophil count reduced the risk of hematoma expansion, whereas a higher monocyte count correlated with greater risk. 95 One reason for these differing results may be that neutrophils promote thrombus formation and thrombin generation to produce a procoagulant state that reduces the risk of hematoma expansion, but they also release various inflammatory factors that aggravate the brain injury. Another possible explanation is that higher neutrophil count is associated with higher baseline hematoma volume. If the hematoma volume peaks, neutrophils will not cause continued expansion. The therapeutic implication is that leukocytes may be a practicable inflammatory biomarker after ICH and that fine modulation of leukocytes rather than broad, nonselective anti-inflammatory treatment could be an option for neuroprotection. The S1PR1 modulators, such as fingolimod and RP101075, which deplete circulating lymphocytes by inhibiting their migration from lymph nodes and preventing their recirculation, ultimately improve neurologic function and attenuate brain edema in mice after ICH.96,97 In an open-label, phase 2 clinical study, administration of oral fingolimod attenuated neurologic deficits and promoted recovery in 23 patients with small-to-moderate-sized deep primary ICH. 98 Additionally, a clinical trial of a novel S1PR modulator (Siponimod) is currently recruiting ICH patients to study efficacy, safety, and tolerability (NCT03338998).
Preclinical studies
Neutrophils
Neutrophils are the first leukocyte subtype to infiltrate into the CNS in the early phase of ICH. Infiltrating neutrophils can be observed around the hematoma within 4 h, 99 with peak levels at approximately three days in a rat ICH model. 20 Once they are at the site, they release various inflammatory factors, such as ROS, MMP-9, TNF, and iNOS, and they induce neuronal death by excitotoxicity or oxygen–glucose deprivation. Moreover, neutrophils undergo apoptosis within two days after influx into the perihematoma, further stimulating microglial activation and consequent inflammatory responses.20,23 However, neutrophils could benefit the ICH-compromised brain by secreting iron-binding protein lactoferrin and hemoglobin-binding haptoglobin. 100 A recent preclinical study that used a mouse autologous blood-induced ICH model showed that immunoregulatory cytokine IL-27 accelerated neutrophil maturation in the bone marrow, reducing the secretion of pro-inflammatory factors, such as iNOS, MMP-9, and NOX2. It also increased the secretion of beneficial molecules, such as lactoferrin and haptoglobin, thereby enhancing hematoma clearance, reducing brain edema, and improving neurologic outcomes. 101 Exogenous administration of IL-27 or lactoferrin improved iron removal and reduced both brain edema and neurologic deficits,100,101 consequently modulating neutrophil function may warrant research as a potential therapeutic target for ICH.
Lymphocytes
Lymphocytes constitute one of the most abundant leukocyte cell populations for immune surveillance and homeostasis maintenance in the peripheral system. They rarely enter the brain parenchyma under normal physiologic conditions. At the onset of ICH, resident glial cells and migrant lymphocytes produce cytokines and chemokines, which, coupled with cell death products, further activate intrinsic and migrant lymphocytes and guide the subsequent homing of more lymphocytes from the periphery. 102 Lymphocytes contribute to the formation of edema and secondary brain injury after ICH. In the collagenase-induced ICH mouse model, cytometric analysis detected the presence of CD4+ T cells in the perihematomal tissue as early as 6 h after ICH. CD4+ T cells are the predominant lymphocyte population in both collagenase and blood injection models of ICH, whereas CD8+ T cells constitute the smallest infiltrating leukocyte population. 103 Foxp3p-Tregs can sustain immune homeostasis and self-tolerance by restricting the activation and release of cytokines from a wide range of inflammatory cells. 104 In other words, upregulating the numbers or percentages of Tregs significantly ameliorates ICH-induced inflammatory and perihematomal edema, decreases cell death, and improves both short- and long-term outcomes.105,106 Based on their various neuroprotective characteristics, modulation of Tregs may be considered as a promising therapeutic strategy for ICH immunotherapy.
Mast cells
Mast cells (MCs), a type of granulocyte derived from hematopoietic lineage, are effector cells of the innate immune system. 107 , 108 They are normal residents in several brain structures and leptomeninges from the time of birth. They can also move through the BBB under both physiologic and pathologic conditions. Upon activation, MCs rapidly produce multiple mediators, such as histamine, heparin, neutral proteases, cytokines, and chemokines, 109 and are involved in almost all major CNS diseases such as MS, 110 ischemic stroke,111,112 Alzheimer’s disease, 113 and Parkinson’s disease. 114
Perivascular MCs are some of the earliest participants in MS. They aggravate the existing condition by releasing histamine, TNF-α, and other cytokines via rapid degranulation, disruption of the BBB as well as inducing an early influx of leukocytes. 115 MCs can also promote BBB breakdown and neutrophil infiltration after ischemic stroke, as shown in an in vivo model of MC deficiency. 112 Similarly, in an autologous whole blood-induced rat model of ICH, stabilization of MCs reduced brain swelling and hematoma volume growth, decreased mortality, and improved neurologic function scores. 116 In another in vivo study, hydrogen inhalation diminished phosphorylation of Lyn kinase and release of tryptase (markers of MC activation), decreased accumulation and degranulation of MCs, attenuated BBB disruption, and improved neurobehavioral function. 117 In a collagenase induced-ICH model, intravenous immunoglobulin attenuated post-ICH MC activation via the FcγRIIB/SHIP1 pathway. 118 MC deactivation led to a decrease in brain inflammation and brain edema and improvement in neurologic function and BBB integrity. Moreover, MC stabilization or deficiency reduced the hemorrhage formation and mortality after tPA administration in a rat model of ischemic stroke. 119 MC stabilization even reversed the harmful effects of tPA administration after ICH. 120 These results suggest that MCs may serve as new therapeutic target for ICH treatment.
MCs have not garnered much attention concerning CNS injury, but some studies have suggested that they may represent the real first responders to brain injury, even before microglia.121–123 Once activated, MCs can recruit macrophages and interact with brain-resident cells (microglia, astrocytes, and neurons). 124 MC-derived granules activate both microglia and astrocytes, resulting in the release of multiple cytokines and chemokines. For example, MC-derived tryptase induces microglial activation and secretion of pro-inflammatory factors via the protease-activated receptor 2 signaling pathway, 125 and MC-derived histamine stimulates microglial activation and the subsequent production of pro-inflammatory factors.126,127 MC proteases activate astrocytes and cause IL-33 release by activating p38 and ERK1/2 MAPKs and the NF-κB signaling pathway. 128 In turn, the activated microglia and astrocytes further stimulate MCs to release histamine, IL-6, and TNF-α and upregulate PAR2 and TLR2/TLR4 expression.125,129–131 The MC-microglia-astrocyte network amplifies the cascade of inflammatory responses and exacerbates neuronal necrosis and apoptosis after ICH (Figure 2). It is hypothesized that MCs might represent one of the central mediators of hematoma expansion and inflammatory responses. The relationship between MCs and oligodendrocytes is still unclear and should be explored. With the development of genetic and fluorescent labeling techniques, we can detect the dynamic changes of fluorescent-labeled MCs before, during, and after ICH, further delineating the mechanisms of MC-associated secondary brain injury. Targeting MCs might be the most fundamental strategy to inhibit inflammation after ICH.
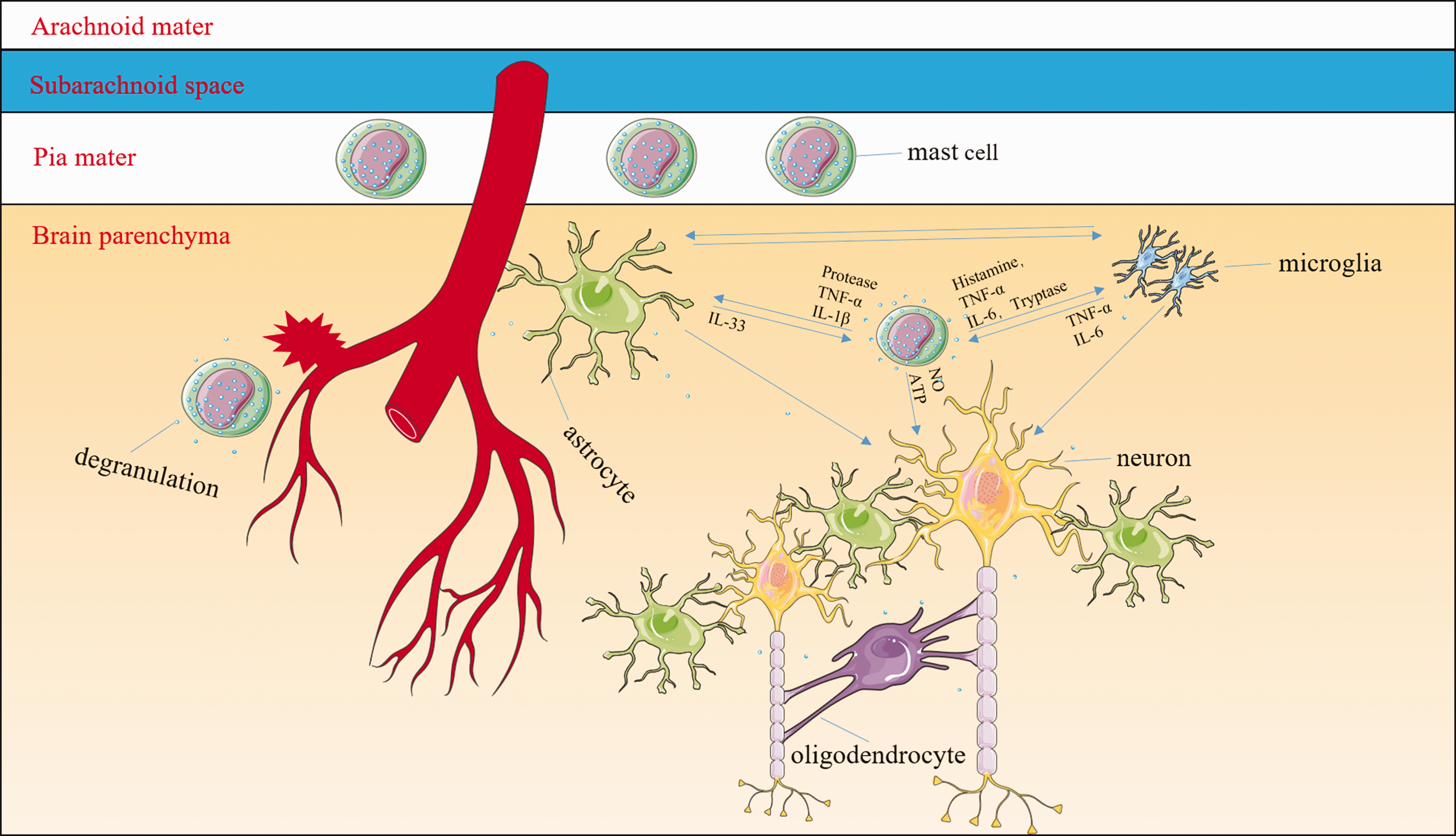
Mast cells (MCs) might be some of the central mediators of inflammatory responses. MCs are normal resident cells in the leptomeninges and brain structures surrounding blood vessels. After the onset of intracerebral hemorrhage (ICH), MCs rapidly respond by degranulating and releasing preformed and stored mediators including histamine, serotonin, heparin, TNF-α, and other cytokines. MC-derived granules can activate both microglia and astrocytes as well as exacerbate neuronal necrosis and apoptosis. For example, MC-derived tryptase induces microglial activation and secretion of pro-inflammatory factors via the protease-activated receptor 2 signaling pathway; MC-derived histamine stimulates microglial activation and the subsequent production of pro-inflammatory factors. MC proteases activate astrocytes and cause the release of IL-33 by activating p38, ERK1/2 MAPKs, and the NF-κB signaling pathway. In turn, activated microglia and astrocytes further stimulate MCs to release histamine, IL-6, and TNF-α. The interaction between MCs and oligodendrocytes is still unclear. The MC–microglia–astrocyte network amplifies the cascade of inflammatory responses and aggravates neuronal necrosis and apoptosis after ICH.
Hemoglobin/iron toxicity
After ICH, erythrocytes lysis leads to the release of cytotoxic degradation products such as Hb, heme, and iron. Hb can be phagocytosed by microglia/macrophages and metabolized into ferrous/ferric iron. Iron deposition following uptake is a detrimental mechanism leading to long-term histologic and functional deficits after ICH. The excess ferrous iron initiates Fenton reaction and yields highly toxic hydroxyl radicals. The radicals attack DNA, proteins, and membrane lipids, leading to DNA damage, lipid peroxidation, and neuronal death. 132 Thus, enhancing hematoma clearance with concomitant or subsequent iron chelation might be a therapeutic strategy for ICH treatment.133,134
Clinical studies
Deferoxamine (DFX), a potent iron chelator, has been found to be effective in preclinical studies, and has been tested in several clinical trials (https://clinicaltrials.gov/). In mouse ICH models, administration of DFX decreased iron accumulation and neuronal death, reduced production of ROS, and improved functional outcomes. 133 In rat models of ICH, DFX alleviated iron overload, attenuated brain edema and neurological deficits, and reduced ICH-induced brain injury in both young and aged rats.135,136 DFX also decreased hemin release from hematoma and reduced hemopexin expression in piglets after ICH. 137 In fact, it has been used to treat iron intoxication clinically for over 40 years. Additionally, in a clinical trial with 42 ICH patients, Yu et al. 138 reported that DFX mesylate was able to attenuate brain edema but delayed hematoma absorption. Although consecutive daily infusion of DFX mesylate (low doses) was shown to be feasible and well-tolerated without serious adverse side effects after ICH, 139 no significant differences existed between the experimental group and the placebo group on 90 days after ICH. 140 Data from this recently completed phase II clinical trial (NCT02175225) indicated that further study of the efficacy of DFX mesylate with anticipation of good clinical outcomes at day 90 would be ineffective. 140 However, DFX mesylate might be effective in terms of good clinical outcomes at day 180, and should be further investigated. 140
Tissue plasminogen activator (tPA), well-known as the only FDA-approved drug for thrombotic stroke, has been groundbreakingly applied with surgical aspiration to remove clots in ICH patients in a phase II trial. The safety and efficacy had been proven in that clinical trial. 141 An optimized tPA treatment regimen has also demonstrated its safety and efficacy to promote the clearance of hematomas without triggering neurotoxicity or increasing brain edema in a rat ICH model. 142 However, the recent randomized 500 subject MISTIE III trial did not reach its major endpoint of improved functional outcomes at 365 days after ICH. 143 Similarly, the CLEAR phase-III trial in which tPA was infused through ventricular drains to lyse ventricular hemorrhage failed to improve functional outcome scores, although mortality was reduced. 144 Considering the important role of inflammation in the ICH-induced secondary brain injury, future clinical trials evaluating treatment for ICH are needed to explore the efficacy of minimally invasive surgery plus tPA in combination with inflammatory modulators.
Preclinical studies
Considering that DFX might be ineffective in clinical usage and that the Hb/iron-induced toxicity contributes to post-ICH inflammation, alternative, brain permeable iron chelators are highly desired for preclinical and clinical ICH studies. The brain permeable iron chelator VK-28 has advantages over DFX, which is unstable and has poor cell permeability. VK-28 has been proven to provide neuroprotection with a marked reduction in iron deposition in models of Parkinson’s disease and Alzheimer’s disease. Using a mouse ICH model, we showed that VK-28 decreased iron-deposition and microglial activation around hematoma, decreased reactive oxygen species (ROS) production and cell death, and improved neurologic function. 145 It had better efficacy and less toxicity when compared to DFX. 145 Nevertheless, the efficacy of VK-28 needs to be confirmed in other species and the dose–response and the therapeutic window need to be determined in young and aged animals.
Ferroptosis, an iron-dependent form of nonapoptotic cell death, has been identified in cancer cells and during mouse embryonic development.146,147 Ferroptosis also occurs after ICH and coexists with necrosis and autophagy.148,149 Administration of ferrostatin-1, a lipid ROS inhibitor, prevented neuronal death in brain slice cultures exposed to Hb; intracerebral administration of ferrostatin-1 exhibited marked brain protection with improved functional outcomes after ICH. 148 In another study, Karuppagounder et al. 150 showed that N-acetylcysteine inhibited hemin-induced ferroptosis and improved functional outcomes following a hemorrhagic stroke in mice. Thus, early inhibition of ferroptosis offers a new promising therapeutic strategy for ICH treatment.
Conclusions and perspectives
For the past two decades, we have made great progress in understanding the mechanisms of ICH-induced brain injury through preclinical and clinical studies. Mounting evidence has indicated that many cellular and molecular signal pathways contribute to inflammatory responses and aggravate secondary brain injury. Despite successful preclinical testing as described and discussed above, however, multiple clinical trials have failed. The reasons for failure in these clinical trials include (1) experimental models do not fully mimic the pathomechanisms of human ICH. The success in preclinical studies does not predict the success in clinical trials; (2) humans respond to drugs differently compared to animals. The initiation of treatment was delayed in clinical trials of ICH compared to preclinical studies; (3) toxicity issues related with human subject safety such as unexpected adverse events; (4) some clinical trials have flawed research design, for example, inappropriate endpoints or unclearly defined inclusion and exclusion criteria; and (5) some clinical trials failed due to lack of funding. To overcome these obstacles, we propose: (1) before a drug candidate can begin the clinical trial, it should be tested in different ICH animal models (rodents, rabbits, pigs, and primates) in different labs; (2) explore the translational potential of the clinically used drugs for its new application in ICH treatment; (3) design clinical trials reasonably and scientifically, choose appropriate primary efficacy endpoints, and comply with inclusion and exclusion criteria strictly; and (4) government and funding agencies should use a more rigorous evaluation and selection criteria to support the more meritorious clinical trials.
We must identify new therapeutic targets. All of the novel targets described have shown efficacy in animal models and are promising for clinical trials. Moreover, clinically used drugs have already been developed for some of these targets (Table 2). Agents with multiple targets or multi-agent combinations that act on different signaling pathways are particularly attractive as potential therapeutic strategies.
Summary of potential targets and drugs for ICH treatment.
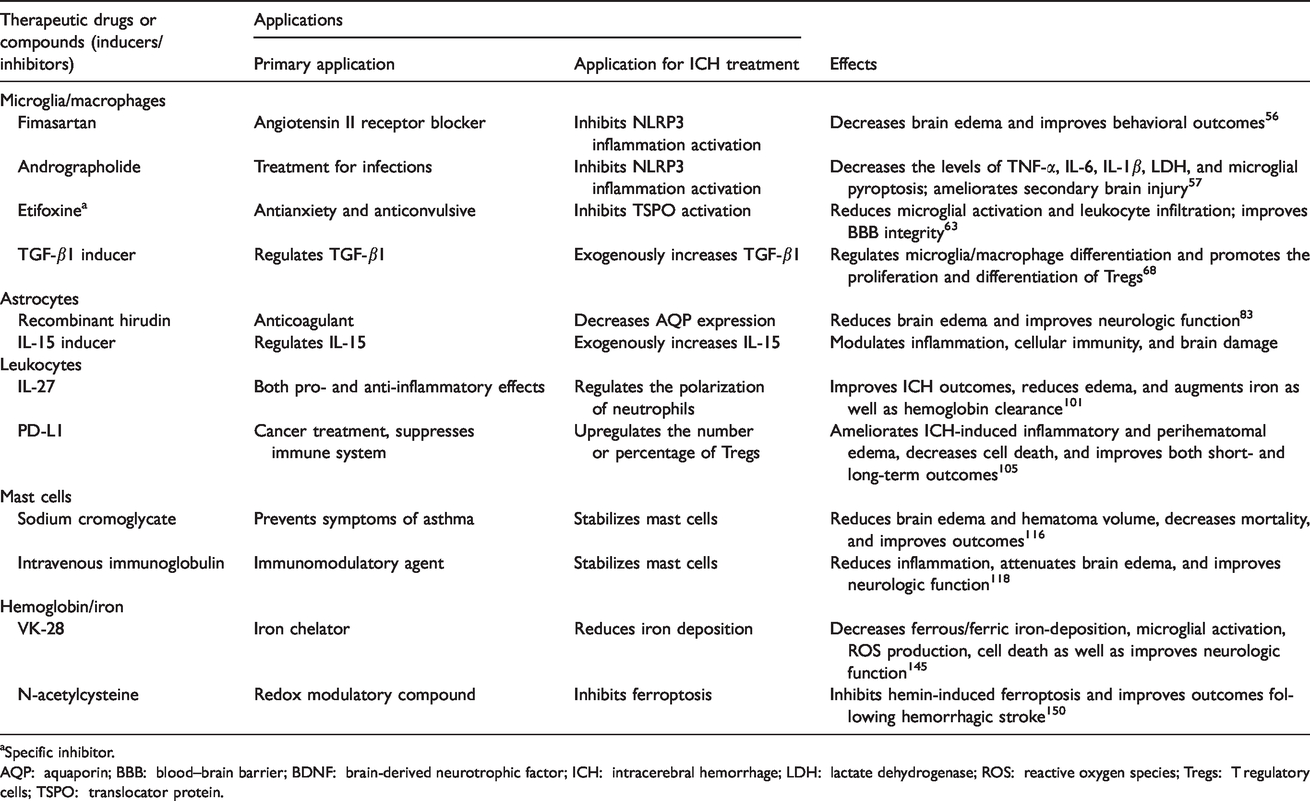
aSpecific inhibitor.
AQP: aquaporin; BBB: blood–brain barrier; BDNF: brain-derived neurotrophic factor; ICH: intracerebral hemorrhage; LDH: lactate dehydrogenase; ROS: reactive oxygen species; Tregs: T regulatory cells; TSPO: translocator protein.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: J. Wan was supported by a Postdoctoral Fellowship Award (18POST33970007); J. Wang was supported by the National Institutes of Health (R01 NS078026, R01 AT007317, the American Heart Association (Grant-in-Aid 17GRNT33660766), and a Stimulating and Advancing ACCM Research (StAAR) grant from the Department of Anesthesiology and Critical Care Medicine, Johns Hopkins University.
Acknowledgements
We also thank Claire Levine, MS, ELS, Anh Le, and Leo Huang for assistance with manuscript preparation.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.