
Abstract
Vascular theories of migraine and cluster headache have dominated for many years the pathobiological concept of these disorders. This view is supported by observations that trigeminal activation induces a vascular response and that several vasodilating molecules trigger acute attacks of migraine and cluster headache in susceptible individuals. Over the past 30 years, this rationale has been questioned as it became clear that the actions of some of these molecules, in particular, calcitonin gene-related peptide and pituitary adenylate cyclase-activating peptide, extend far beyond the vasoactive effects, as they possess the ability to modulate nociceptive neuronal activity in several key regions of the trigeminovascular system. These findings have shifted our understanding of these disorders to a primarily neuronal origin with the vascular manifestations being the consequence rather than the origin of trigeminal activation. Nevertheless, the neurovascular component, or coupling, seems to be far more complex than initially thought, being involved in several accompanying features. The review will discuss in detail the anatomical basis and the functional role of the neurovascular mechanisms relevant to migraine and cluster headache.
Introduction
Migraine and cluster headache are two of the most debilitating neurological conditions that patients can suffer. Migraine affects 15–18% of the global population,1,2 is ranked the 6th most disabling disorder globally, and is considered the most disabling neurological disorder. 3 It is characterized by attacks of moderate to severe unilateral throbbing head pain, with increased sensitivity to movement, light, sounds, smells, and foods. 4 Patients can also experience autonomic, affective or cognitive symptoms, all of which may occur before (premonitory 5 ), during or after (postdrome 6 ) the headache. Additionally, in approximately one-third of migraine patients, their attacks are associated by neurological deficits which include cortical perturbations, collectively termed migraine aura. 7 Cluster headache is rare compared to migraine, with a prevalence in the population of about 0.1%8,9; but similar to other debilitating neurological disorders, such as multiple sclerosis. It consists of severe unilateral head pain that occurs in association with cranial autonomic features, and attacks are noted for their excruciating pain 4 – described by some as worse pain than childbirth and limb fracture. 10 It is also characterized by the circadian rhythmicity of the short-lived attacks, and the fact that cluster headache very often recurs in bouts, with periods of complete remission.
The headache phenotype of these primary headaches can be very different, but they share important pathophysiology; it is believed that the head pain is mediated by activation of neuronal pathways within the trigeminovascular system. Based on research findings of the last 50–75 years, both migraine and cluster headache are now commonly thought of as neurovascular disorders. They both certainly involve the brain; imaging studies during attacks clearly characterize activation in brain regions specific to these headaches.11–16 However, the involvement of the trigeminovascular system, its specific innervation of the dural vasculature, and the very nature of several symptoms, including autonomic symptoms and migraine aura, suggest the vasculature is still very important, even if it is not responsible for the initiation of attacks. The objective of this review is to describe the neurovascular mechanisms involved in migraine and cluster headache, and their potential contributions to specific symptoms, based on preclinical and clinical studies.
Anatomical connections of the trigeminovascular pain pathway
Migraine and cluster headache present as very different primary headache disorders, but they share much in relation to their pathophysiology, particularly with respect to headache itself. We know that all somatosensory information from craniovascular structures, including nociceptive, is relayed through the trigeminal nucleus caudalis and its cervical extension. Thus, all pain related to the head, but particularly intracranially mediated pain, is relayed through the same mechanisms, be it migraine or cluster headache. While both are thought of as disorders of the brain, and imaging studies demonstrate that brain activation is intrinsic to these disorders, a brief description of this anatomy and physiology (which has been described in detail elsewhere)17–19 illustrates the importance of the cranial vasculature through its innervation by central structures.
Peripheral connections of the trigeminovascular pain pathway
The pain associated with migraine headache is generally in frontal, temporal, parietal, occipital, and higher cervical regions, while the pain in cluster headache predominates peri-orbitally, in and around the eye, or temporally. However, both are thought to be mediated by activation of the trigeminovascular system (Figure 1), likely via slightly different innervations. While the brain is largely insensate, there is a rich plexus of nociceptive nerve fibers (non-myelinated C-fibers and thinly myelinated Aδ-fibers) originating in the trigeminal ganglion (TG) that innervate intracranial structures, including the pial, arachnoid and dural blood vessels, and large cerebral arteries.20–22 This is mainly through the ophthalmic (V1) division of the trigeminal nerve, but there is also innervation through maxillary (V2) and mandibular (V3) divisions. The dura mater is also innervated by nerves from upper cervical dorsal root ganglia.
23
Activation of these structures, particularly the dura mater, with mechanical distension or electrical stimulation results in pain very similar to these headaches, as well as other associated symptoms, including nausea and photophobia.20–22 Interestingly, the site of stimulation caused pain to be localized to specific regions of the head. Stimulation of the superior sagittal sinus produced pain in the peri-orbital region (similar to cluster headache); stimulation of the middle meningeal artery produced pain in the parietal or temporal region (similar to migraine and cluster); and stimulation at the floor of the posterior fossa, sigmoid, transverse, and occipital sinuses produced pain in the occipital region (similar to migraine). Furthermore, stimulation away from the dural vessels was much less pain-producing. This suggests that the dural vasculature is especially important in mediating activation of this nociceptive relay, because it specifically receives innervation by the trigeminal nerve.
Anatomy of the neurovascular pathways that modulate trigeminal nociceptive neurotransmission. Schematic representation of ascending neurovascular pathways of the trigeminovascular system, and descending neuronal pathways that modulate trigeminovascular nociceptive transmission in the medullary and upper cervical dorsal horn, which are involved in aspects of migraine and cluster headache (see text for full description). Ins: insula; M1/M2: primary and secondary motor cortex; S1/S2: primary and secondary somatosensory cortex: V1/V2: primary and secondary visual cortex; PtA: parietal cortex; Au: auditory association cortex; Ect: entorhinal cortex; RS: retrosplenial cortex; PAG: periaqueductal gray; LC: locus coeruleus; RVM: rostral ventromedial medulla; TNC: trigeminal nucleus caudalis; TG: trigeminal ganglion; DRG: dorsal root ganglion; SPG: sphenopalatine ganglion; SuS: superior salivatory nucleus.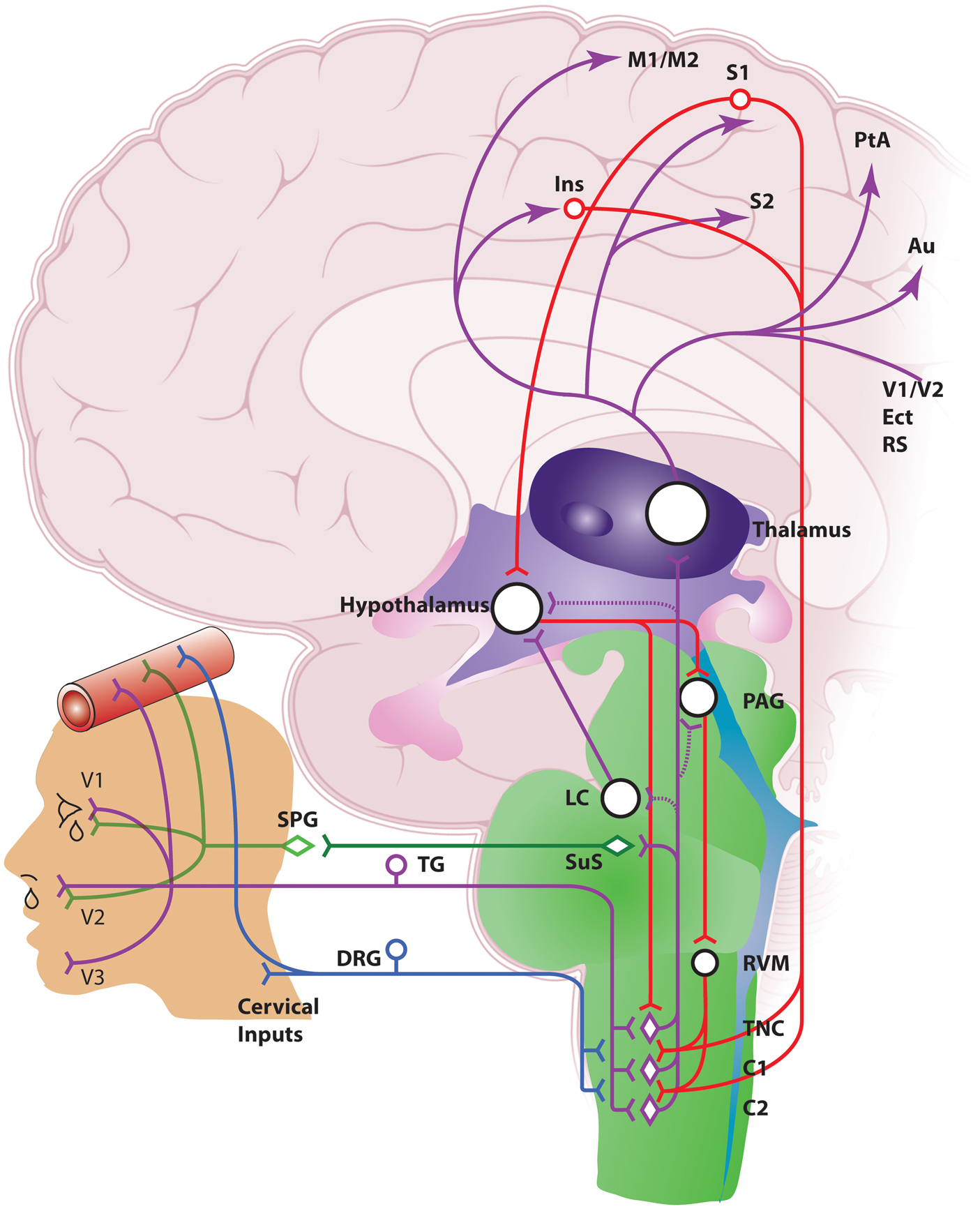
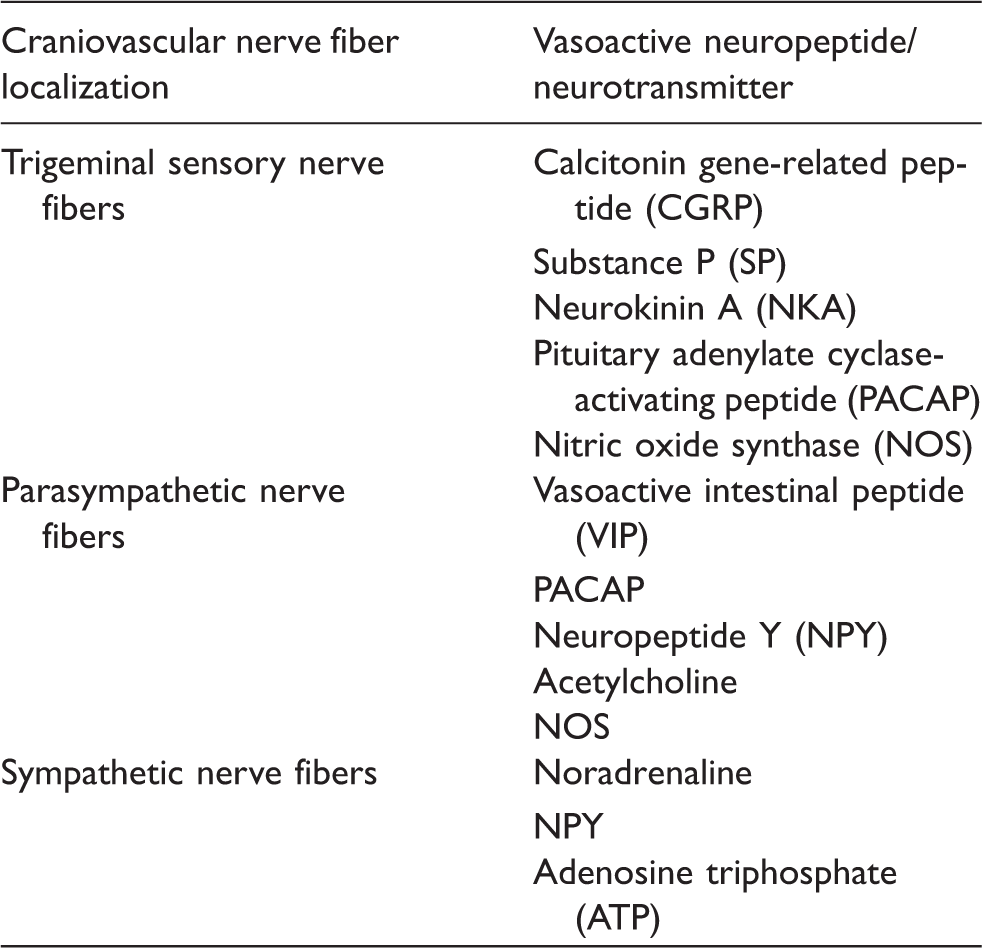
Summary of craniovascular nerve fibers and their vasoactive neuropeptides involved in migraine and cluster headache.
Central and ascending connections of the trigeminovascular pain pathway
Central projections of dural-nociceptive primary afferents enter the caudal medulla of the brainstem, via the trigeminal tract, terminating predominantly in the superficial laminae, I–II, as well as deeper laminae V–VI of the spinal trigeminal nucleus caudalis (TNC; Sp5C), and also the upper cervical spinal cord (C1–C2).31–37 There is convergence of these dural-nociceptive trigeminal neurons that also receive inputs from facial skin and muscle, including the greater occipital nerve.38–40 This suggests that the trigeminal nucleus extends beyond its caudalis boundary to the dorsal horn of the upper cervical region in a functional continuum that includes the cervical extension – together described as the trigeminocervical complex (TCC). The convergence of primary afferent inputs from vascular-intracranial and extracranial structures relayed through the TCC32–34 most likely explains the distribution of pain perception in migraine and cluster headache. 41 From here ascending connections are made to other areas of the brainstem and diencephalon, including nuclei of the rostral ventromedial medulla (RVM), locus coeruleus (LC), periaqueductal gray (PAG), hypothalamic, and thalamic nuclei, and cortical regions involved in the processing of somatosensory information, to determine the ultimate perception of these inputs (reviewed elsewhere,17–19 and summarized in Figure 1).
Physiological modulation of trigeminal neurovascular pain pathways
In experimental cranial pain studies, capsaicin injection – which is a potent vasodilator largely by causing local release of CGRP – into the forehead (ophthalmic trigeminal division) is pain-producing and causes vasodilation of the internal carotid artery. It also causes regional cerebral blood flow increases in several brain regions associated with non-specific pain processing, but not in the brainstem or hypothalamus.42,43 In addition, capsaicin injection into the chin (mandibular trigeminal division) or shin is equally pain-producing, but failed to produce vasodilation. Imaging during migraine highlights specific activation in the midbrain, including dorsal rostral pons close to the PAG and raphe nuclei during the premonitory and headache phase,13–15,44 and posterolateral hypothalamic activation during the premonitory phase, 11 as its signature. Cluster headache, similar to other trigeminal autonomic cephalalgias (TACs), is associated with mainly hypothalamic grey matter activation.12,45–47 Together, these data tell us that activation in these brain regions is specific to headache sub-types, rather than a consequence of trigeminally mediated pain. This demonstrates that this brain activation reflects not just migraine or cluster headache symptoms, but also an important role in their underlying pathophysiology. It also highlights the importance of neurovascular coupling specifically in the ophthalmic trigeminal division, where head pain in these primary headaches is localized, rather than in other trigeminal or non-trigeminal regions. Finally, these data suggest that craniovascular outcomes, if they occur during these headaches, are likely the outcome of neurovascular mechanisms linked to brain activation, rather than the cranial vasculature being the driver of pain and brain activation itself.
Brainstem modulation of trigeminovascular pain pathways
While activation in these specific brain regions may not directly cause craniovascular outcomes, somatosensory trigeminovascular neurons are under the control of pain-modulatory circuits in the brainstem and hypothalamus, which ultimately affects how the nociceptive signal is processed and perceived. There are many examples now from preclinical studies which demonstrate that brainstem and hypothalamic nuclei specifically activate and modulate innocuous and noxious somatosensory signals coming from intra- and extracranial neurovascular structures. Imaging studies in migraine focused original preclinical studies on brainstem structures, such as the PAG and RVM. The RVM and PAG are a cluster of neurons in the medullary and midbrain region of the brainstem. They are part of the descending endogenous pain processing pathway, involved in modulating somatosensory inputs to the medullary and spinal dorsal horn48–50 (reviewed in detail elsewhere17,51). Electrical stimulation or chemical manipulation of the ventrolateral PAG (vlPAG) in animal models causes transient differential modulation of nociceptive-evoked activation of dural-responsive trigeminovascular neurons, as well as basal trigeminal tone.52–54 These responses are mediated by manipulation of GABAergic,53,55 P/Q-type voltage-gated calcium channels, 55 5-HT1B/1D receptors, 56 and CB1 receptor pharmacologies. 57 Similarly, injection of bupivacaine directly into the RVM inhibits durovascular-evoked cutaneous peri-orbital hypersensitivity. 58 Finally, the nucleus raphe magnus (NRM), within the RVM, and thought to provide the primary source of serotonergic projections to medullary and spinal neurons,59,60 also provides control over trigeminovascular nociceptive transmission. Conditioning electrical stimuli applied to the NRM inhibits nociceptive dural-evoked trigeminocervical neuronal responses. 61 These inhibitory responses, which would normally gate the nociceptive response, are suppressed by the cortical neurovascular phenomenon cortical spreading depression (CSD). 61
The contribution of these brainstem mechanisms to the pathophysiology of migraine is strongly debated. The implication being that if dysfunction in these nuclei affects trigeminovascular nociceptive processing, 17 they should equally affect pain processing at the spinal level. However, manipulation of descending mechanisms mediated by the vlPAG tends to only alter dural-evoked Aδ-fiber trigeminovascular nociceptive responses, as well as basal trigeminal tone, but it does not alter either noxious or innocuous cutaneous facial, particularly C-fiber, responses.54,62–65 Whereas previous studies clearly indicate that the PAG-RVM pathway provides descending control of only noxious cutaneous C-fiber responses at the spinal level,66,67 with innocuous inputs and spinal tone unaffected. This may indicate that the nociceptive trigeminovascular projections, particularly to migraine-related intracranial structures in the ophthalmic division, are mediated predominantly by Aδ-fibers, rather than C-fibers. Therefore, descending modulation from the brainstem may be mediated by a separate population of neurons to those that project to the spinal cord and modulate spinal nociceptive processing.
Hypothalamic modulation of trigeminovascular pain pathways
Migraine and cluster headache are also associated with activation and changes that occur within hypothalamic nuclei. The hypothalamus has direct and indirect reciprocal connections with many structures involved in medullary trigeminovascular and spinal pain processing,68–71 and it is involved in a number of crucial physiological functions including controlling circadian rhythm, sleep-wake cycle, feeding, thirst, arousal and urination, as well as autonomic and endocrine regulation.71,72 These mechanisms and connections implicate it in the origins of both migraine and cluster headache aetiology.
The posterior (PH) and paraventricular (PVN) hypothalamic regions are both activated by dural vascular stimulation,73,74 and are regions potentially activated during migraine and cluster headache. Unfortunately, imaging studies in patients lack the spatial resolution to define particular nuclei. However, in preclinical studies, where precise manipulation of specific brain nuclei is possible, local chemical manipulation in the PH (orexins and somatostatin) and PVN (GABA, 5-HT1B/1D and PACAP) modulates the transmission of durovascular-nociceptive trigeminal neurons.70,75,76 Orexin peptides are related to feeding, sleep and arousal and thus may provide a causal link between the origins of triggering migraine and/or cluster headache, and control of trigeminal neurovascular mechanisms. In addition, octreotide, a somatostatin analogue agonist, is effective in the treatment of cluster headache, but not migraine.77,78 This suggests that hypothalamic neurotransmitter systems are likely important in mediating cluster headache mechanisms, and these are not universal to all primary headache disorders, and trigeminally mediated pain. The PVN has descending projections to the superior salivatory (SuS) nucleus, which likely has a role in cranial autonomic symptoms. Furthermore, the response to GABA was additionally suppressed in stressed rats. This suggests the powerful modulatory role of the PVN in processing dural trigeminovascular nociceptive neurotransmission, and links it to many aspects in the pathophysiology of triggering and symptoms in migraine and cluster headache. This demonstrates the importance of these specific descending mechanisms to the pathophysiologies of different primary headache disorders. Finally, the A11 hypothalamic nucleus, located along the rostrocaudal axis of the periventricular grey of the caudal hypothalamus, 79 is believed to provide the sole source of direct dopaminergic inhibitory projections to the spinal cord dorsal horn.79–81 Electrical stimulation or lesioning of this nucleus differentially causes inhibition or facilitation, respectively, of noxious dural and cutaneous facial-evoked inputs of trigeminovascular neurons. 82 The inhibitory responses are mediated through D2 dopamine receptors, whereas the facilitatory response appears to be mediated by both D2 dopamine and 5-HT1B/1D receptors.82,83 It appears the A11 nucleus is involved in the control of somatosensory trigeminovascular neurotransmission, particularly of inputs that arise from the dural vasculature.
Collectively, these data illustrate the importance of brainstem and hypothalamic nuclei in the pathophysiology of migraine and cluster headache. They are intrinsically involved in the control of trigeminovascular neurotransmission, particularly somatosensory neurovascular inputs coming from intracranial-dural and extracranial-facial cutaneous structures. The net result of this integration of peripheral neurovascular and central pain mechanisms within the TCC is migraine and cluster headache, due to an altered perception of craniovascular inputs. This is also thought to contribute to autonomic, cognitive, endocrine and affective symptoms, which can accompany migraine and cluster headache.
Neuropeptides involved in modulating trigeminovascular pain pathways
Neuropeptides play an essential role in the pathophysiology of migraine and cluster headache. The landmark studies by Goadsby et al.84–97 in the late 1980s and early 1990s have remarkably expanded our understanding of the importance of neuropeptides in the pathobiology of these disorders and have been a success story of translational research as these neuropeptides and their receptors are currently under investigation as potential treatment targets.
In principle, activation of the trigeminovascular system involves activation of the trigeminal nerve with its main neuropeptides/neurotransmitters CGRP, SP, NKA, PACAP, and nitric oxide (NO). Trigeminovascular activation also involves the cranial vascular innervation of the parasympathetic nervous system, with its neurotransmitters being VIP, PACAP, neuropeptide Y (NPY), acetylcholine (ACh) and NO, as well as the sympathetic nervous system, with its neurotransmitters norepinephrine, NPY, and adenosine triphosphate (ATP). Some, but not all of these neurotransmitters are released during migraine and cluster headache, and some of them may even trigger migraine or cluster attacks. The pathophysiological basis for this triggering effect has not been entirely elucidated as their vasoactive effects, as well as their pro-inflammatory properties, have been refuted as an attack-triggering mechanism. As some of these substances may directly modulate nociceptive neuronal activity, it is thought that this modulating effect is likely to be the crucial mechanism behind their role in the pathophysiology of migraine and cluster headache. 19
CGRP
CGRP is a 37 amino-acid neuropeptide that is synthesized from the CALC1 gene as an alternate splicing product to calcitonin. It is localized in Aδ- and C-fibers of the trigeminovascular system and is found within this system in the meningeal afferents, TG, TCC, PAG, hypothalamus, and thalamus.98–106 Beyond the trigeminal system CGRP has been identified in multiple areas of the peripheral and central nervous systems as well as a number of extracranial regions including the blood vessels, heart, kidney, lungs, and intestinal tissues. 107 In general, CGRP is stored in dense-core vesicles within sensory nerve terminals. 108 During neuronal activation, CGRP is released from these vesicles by exocytosis.
Upon its release from sensory nerve fibers, CGRP binds to its receptor which consists of three proteins, the calcitonin receptor-like receptor (CLR), 109 the receptor activity modifying protein 1 (RAMP1) 110 and the receptor component protein (RCP). 111 If dimerization occurs between CLR and RAMP2 or RAMP3, instead of RAMP1, the receptor acts as an adrenomedullin receptor. Dimerization between CLR and RAMP2 creates the AM1 receptor and dimerization between CLR and RAMP3 creates the AM2 receptor. However, even as adrenomedullin receptors, AM1 and AM2 bind CGRP but with a much lower affinity. 112 Within pain pathways relevant for migraine and cluster headache, the CGRP receptor can be found in the periphery in the smooth muscle of meningeal arteries as well as pre- and postsynaptically located in the TG.99,100,113,114 Interestingly, it has not been identified in the meningeal trigeminal afferents 114 explaining the observation that application of CGRP onto the meninges does not induce activation of the trigeminal pathway. 115 Within the central nervous system, the CGRP receptor complex is found in the TCC and the spinal trigeminal tract.
CGRP is the strongest vasodilating neuropeptide known in man and therefore has an important function in the regulation of blood flow. 116 While it plays a prominent role in the regulation of blood flow during pathological conditions, it is most likely not involved in maintaining baseline blood flow under physiological conditions. 107 CGRP-induced vasodilation is mediated by NO-dependent, as well as, NO-independent pathways. In the NO-dependent pathway, CGRP binds to receptors located on the endothelial cells triggering a phosphokinase A (PKA)-mediated cascade that leads to activation of endothelial NO synthase (eNOS) and thereby enhances NO production. NO then diffuses to the adjacent vascular smooth muscles inducing vasodilation. In the NO-independent pathway, CGRP binds directly onto receptors located on the vascular smooth muscle inducing its relaxation and thereby vasodilation. 107 Over the past 25 years, it has become evident that CGRP has a number of functions that go far beyond the regulation of blood flow. CGRP appears to play a role in inflammatory processes as it may reduce the production of pro-inflammatory cytokines117,118 and its plasma concentrations are elevated in several inflammatory conditions.119–121 In primary headaches, the crucial effect of CGRP is its ability to modulate nociceptive neuronal activity in several regions of the nervous system including the TCC, most likely through a facilitation of glutamatergic neurotransmission. 122 In contrast, at least in migraine and cluster headache, the CGRP-induced vasodilation is highly unlikely to play a role in the triggering of single attacks.123,124
The importance of CGRP in migraine and cluster headache was first suggested with the clinical observation that CGRP is released into the extracerebral circulation during trigeminal activation, 85 as well as during spontaneous attacks of migraine and cluster headache.84,125 The elevated CGRP plasma levels during spontaneous migraine attacks normalized after effective treatment with sumatriptan. 87 As CGRP receptor antagonists are effective in acute treatment, and antibodies against the neuropeptide or its receptor are effective in the preventive treatment, of migraine,88,89,91–95,126,127 it may be assumed that CGRP is involved in the triggering as well as the maintenance of a migraine attack and its associated headache.
Preclinical studies have elucidated the mechanisms leading to CGRP release and the functional consequences it may induce. Despite the large body of evidence on the localization of CGRP and its receptors throughout the trigeminovascular system and central pain-processing pathways, it has not been entirely clarified which site or sites of action are relevant for the generation and maintenance of an attack of migraine or cluster headache. Given that it is thought that both headache disorders initiate within the brain, most likely in processes involving the hypothalamus, a central site of action seems likely. This hypothesis was fueled by the observation that CGRP and its receptor are localized in the TCC 114 and that local application of CGRP facilitates neuronal activity in this central relay site. 122 In line with these results, the peptidergic and non-peptidergic CGRP receptor antagonists, CGRP8-37 and olcegepant (BIBN4096BS), respectively, inhibit stimulus-evoked neuronal activity in the TCC induced by stimulation of the meninges, 128 superior sagittal sinus, 122 local glutamate application 122 or by systemic NO infusion. 129 Finally, local application of CGRP or the CGRP receptor antagonist olcegepant on the meninges115,128 or the TG 130 does not modify neuronal activity in the TCC. Furthermore, systemic administration of CGRP had little effect on peripheral primary afferent trigeminal responses over 60 min. 115 Together these data suggest a lack of peripheral neuronal action of CGRP, but rather a central site of action in these headache models. The observation made in the TG is particularly interesting as CGRP as well as its receptor have been identified in the TG although they do not seem to colocalize, indicating that CGRP is present in one group of neurons influencing another group of neurons upon its release. 100
However, the clinical efficacy of antibodies against CGRP or its receptor has cast a shadow on this hypothesis as they do not cross the blood–brain barrier due to their molecular size making a central site of action unlikely. Nevertheless, this observation is still debated as it has not been clarified if the blood–brain barrier remains intact during a spontaneous migraine attack or if it is equally tight throughout the whole brain, as specific regions of the brain, for example the hypothalamus, may be less protected by the blood–brain barrier than other areas.
VIP and PACAP38
VIP and PACAP38 are two pharmacologically similar neuropeptides that play a major role in parasympathetic communication. VIP is a 28 amino-acid neuropeptide initially isolated from porcine small intestine, 131 while PACAP38, initially isolated from ovine hypothalamus, contains 38 amino-acid residues. 132 Following the discovery of PACAP38, a second neuropeptide of the PACAP-family, PACAP27, was identified. Both neuropeptides originate from the same precursor protein prepro-PACAP. However, if PACAP27 plays a significant role in pain processing is not known. VIP and PACAP38 bind to a series of G-protein coupled membrane receptors (GPCRs) which have now been classified as VPAC1-, VPAC2-, and PAC1-receptors. 133 While VIP and PACAP38 show an almost equal affinity to VPAC1 and VPAC2-receptors, PACAP38 has a 100-fold higher affinity to the PAC1-receptor than VIP. 134 The functional interaction extends beyond the action on the same receptors as PACAP38-positive nerve fibers innervate VIP-positive neurons135,136 and PACAP38 induces VIP gene expression136,137 as well as VIP release. 138
Beyond their role in parasympathetic communication, VIP and PACAP38 induce vasodilation. Cluster headache and, in some patients, migraine are accompanied by cranial autonomic symptoms such as lacrimation, conjunctival injection and rhinorrhea all of which are associated with local vasodilation. VIP has long been suspected to be one of the main mediators of these symptoms as it has been identified, among others, in nerves innervating the large cerebral vessels, nose, and eyes139,140 and from a clinical perspective migraine attacks show elevated VIP levels only in the case of accompanying autonomic symptoms, 84 while cluster headache attacks always show elevated VIP plasma concentrations. 86
Despite their structural similarity, in the context of migraine and cluster headache, the functional effects of PACAP38 extend beyond those induced by VIP. First, in preclinical in vivo models of trigeminal activation, VIP and PACAP38 induce a transient meningeal vasodilation but only PACAP38 facilitates neuronal activation and sensitization in the TCC, accompanied by neuronal hypersensitivity to somatosensory cranial stimulation, most likely through an activation of neurally located PAC1-receptors.141,142 In line with these results, local application of PACAP38 onto meningeal afferents induces the release of CGRP from the TNC and not from the TG, whereas VIP exposure had no effect on CGRP release. 143 Second, clinical studies confirm these results as attacks of migraine without aura can only be induced by PACAP38 but not by VIP infusion.144–146 Interestingly, these PACAP38-induced attacks are not accompanied by an increase in plasma CGRP 138 reflecting the preclinical observations that identified this mechanism only in the TNC. 143 During spontaneous migraine attacks without autonomic symptoms, an elevation of PACAP38 but not VIP is observed in the cranial circulation.84,147,148 However, as stated above, if migraine attacks are accompanied by autonomic symptoms, an increase in VIP plasma levels is observed. 84 In cluster headache, which is characterized by its accompanying autonomic symptoms, elevations of both neuropeptides, PACAP38 and VIP, are observed.86,149 Taken together, these results suggest that PACAP38 has the ability of activating the trigeminal and the parasympathetic system, whereas the action of VIP seems to be limited to parasympathetic neurons. However, by activating the system in the opposite direction, namely through stimulation of the first branch of the trigeminal nerve, parasympathetic activation with the release of VIP can be induced. 150 These interactions represent the anatomical and functional basis of the trigemino-autonomic reflex, a key component in the pathophysiology of cluster headache.10,17 Based on these findings, targeting PACAP38-driven mechanisms, in particular the PAC1-receptor, may offer a novel target for the treatment of migraine and cluster headache. In a preclinical in vivo model of trigeminal activation, the use of a PAC1-receptor antibody has already provided promising results. 97
SP
In preclinical animal models, trigeminal activation, via TG stimulation, causes the release of SP. 85 The same effect can be observed in humans when the TG is activated during a thermocoagulation procedure. 85 SP binds to the NK1 receptor and induces vasodilation as well as plasma protein extravasation and an inflammatory response in the meninges. 151 The release of SP (in dorsal root slices) and meningeal plasma protein extravasation can both be inhibited by sumatriptan.152,153 In contrast, trigeminovascular activation via chemical and electrical stimulation of dural meningeal afferents did not induce SP release,150,154 and neurogenic meningeal vasodilation is not mediated by SP as the NK1 receptor antagonist RP67580 does not attenuate this vascular reaction. 30 These findings can be explained by the fact that in principle SP is localized in the TG and may co-localize with CGRP within that structure155,156 but that only a small number of trigeminal neurons projecting to the meningeal vessels contains SP.157–159
However, clinical studies analyzing the role of SP in migraine and cluster headache did not support the promising results obtained from preclinical studies. In spontaneous attacks of migraine and cluster headache, no elevations of SP plasma concentration were observed.84,86 Even so, several antagonists of the NK1 receptor, the receptor to which SP binds to exert its actions, have been developed and tested for the treatment of migraine. All these antagonists have been tested in Phase II trials and failed in demonstrating therapeutic efficacy.160–164 In light of the available evidence. it is highly unlikely that SP may play a significant role in migraine and cluster headache.
NKA
NKA is another tachykinin that in contrast to SP binds to the NK1 and NK2 receptors. NKA is located in meningeal afferents and preclinical studies have demonstrated that the release of NKA into the meningeal circulation induces meningeal vasodilation.24,27,151 However, the vasodilating effect of NKA in the meninges appears to be mediated mainly through NK1 receptors. 151 Given that the meningeal effects of NKA and SP share the same molecular mechanism and NK1 receptor antagonists have proven to be ineffective in migraine, as outlined above, it may be concluded that NKA does not play a significant role in this disorder.
Neuropeptide Y
Neuropeptide Y (NPY) is another parasympathetic neuropeptide that has been investigated in the context of migraine for several reasons. NPY binds at several NPY receptors of which the NPY Y1 and Y2 receptors can be found in the TG 165 and the TCC. 166 In a rodent animal model, NPY induces a NPY Y1 receptor-mediated inhibition of nociceptive neuronal activity in the TCC 167 suggesting a potential role in migraine. Beyond that, NPY is found in orexinergic neurons in several areas of the hypothalamus including the arcuate, lateral, and paraventricular hypothalamic nuclei. Neurons containing NPY that originate in the arcuate nucleus project to several pain modulating structures of the CNS including the PAG, NRM, and LC. 168 Beyond its neuroanatomical association, the orexinergic system and NPY interact functionally as stimulation of orexinergic neurons with orexin A inhibits nociceptive neuronal activity in the TCC through the OX1 receptor and additionally induces the release of NPY.75,169 In addition to its effects on nociceptive neuronal transmission, NPY induces a NPY Y2 receptor-mediated vasoconstriction of the middle meningeal and cerebral arteries170,171 as well as a meningeal plasma protein extravasation. 172
In contrast to the results from preclinical experiments, clinical studies on migraineurs are inconclusive as one study shows reduced NPY levels in peripheral blood, while another study shows the opposite result.84,173 Given the interaction with the orexinergic system, a dual orexin receptor antagonist failed to show preventive efficacy in migraine, despite showing promising results in preclinical studies.169,174 Given the existing evidence, further research, in particular with a NPY receptor antagonist, is needed to clarify the relevance of NPY in migraine.
Nitric oxide
The role of nitric oxide (NO) in the pathophysiology of migraine and cluster headache has been discussed for many years. NO is involved in a large number of physiological mechanisms with its vasodilating action being the most prominent feature. In this context, NO may modulate nociceptive neuronal activity in several regions of the nervous system such as the TG, TCC, and the PAG.175–177 In animal models, NO may potentiate the neuronal responses to facial stimuli, 178 whereas the local application of a NO synthase (NOS) inhibitor inhibits the response to peripheral stimulation.175,179,180 In addition to its role in eliciting trigeminal activation, NO is involved in mediating trigeminal sensitization,177,181 the pathophysiological correlate of cutaneous allodynia and probably an important element in the chronification of migraine. The facilitating effect of NO is largely related to its functional interaction with CGRP. Experimentally, the administration of NO leads to a local release of CGRP and inducing neuronal activation and sensitization in several in vivo and in vitro models of migraine.129,182–186 Moreover, NO may activate TRPV1 channels thereby increasing the release of CGRP. 182 However, the interaction between NO and CGRP is bidirectional as CGRP may induce vasodilation through the synthesis and release of NO. 107 In this interaction, the different isoforms of NOS play synergistic roles as the neuronal NOS (nNOS) increases NO production at the neuronal level inducing the release of CGRP from trigeminal fibers and the endothelial NOS (eNOS)-mediated NO production is activated by CGRP. 186
These preclinical studies support a large number of clinical observations that highlight the potential role of NO in migraine and cluster headache. In patients suffering from migraine or cluster headache, NO donors have the ability to trigger their attacks.187–190 In migraineurs, NO may even trigger premonitory symptoms of a migraine attack. 191 However, the detailed mechanism behind this triggering effect remains to be elucidated, in particular as the NO-donor triggered attacks show a remarkable delay between the administration of the NO donor and the initiation of the migraine or cluster headache attack. Despite the findings that suggest a role for a vascular or an inflammatory mechanism, these mechanisms are highly unlikely to play a causative role as several NOS-inhibitors against iNOS (GW274150)192,193 and nNOS (NXN-188) 194 have been investigated for the treatment of migraine and failed to demonstrate efficacy in multiple Phase II trials. The results of these trials and the observation that NO-donors do not trigger attacks of migraine with aura195,196 raise substantial questions on the role of NO in migraine which will have to be addressed in future studies.
Neurovascular mechanisms of cranial autonomic symptoms
Cranial autonomic symptoms, such as conjunctival injection, lacrimation, nasal congestion, rhinorrhea, eyelid edema and forehead/facial sweating, can be hugely debilitating, and they are a prominent and defining feature of TACs, such as cluster headache.4,197,198 They are also prevalent in up to 50% of migraine patients.199–201 Their presence is thought to exacerbate the general migrainous phenotype.200,201 These data suggest there is likely some overlap of pathophysiology between migraine and TACs, as they relate to cranial autonomic symptoms.
Cranial autonomic symptoms are thought to be mediated, in part, by activation of the trigeminal autonomic reflex, and the parasympathetic autonomic projection to the cranial vasculature.
10
A reflex connection from the TNC to the pontine SuS nucleus, is thought to connect these two important somatosensory and autonomic pathways. The SuS is the origin of cells of the parasympathetic vasodilator pathway, and it projects to the cranial vasculature, including the dura mater and the lacrimal gland, predominantly via the greater petrosal nerve (green nerve), and its synapse with the sphenopalatine ganglion (SPG), and the VIIth (facial) nerve (light blue nerve).
202
Symptoms of Horner’s syndrome; facial sweating, miosis, and ptosis, are thought to be mediated by local third-order cranial sympathetic (orange nerve) lesioning due to carotid swelling.203,204 This is largely because fibers of the superior cervical ganglion are compromised by carotid dilatation as they pass through the carotid canal. Thus, there is clearly a neurovascular link to these craniofacial symptoms (Figure 2).
Anatomy of the neurovascular pathways that constitute the trigeminal-autonomic reflex. Schematic representation of neurovascular pathways of the trigeminal-autonomic reflex involved in cranial autonomic symptoms in migraine and cluster headache. They are thought to result, in part, from activation of the trigeminal-autonomic reflex; a reflex connection from the trigeminal nucleus caudalis (TNC; grey neuron), via the superior salivatory nucleus (SuS; green diamond), which provides an autonomic parasympathetic projection to the cranial vasculature. This is predominantly through the greater petrosal nerve (green neuron) and its relay with the sphenopalatine ganglion (SPG), but also via the facial (VIIth cranial) nerve (sky blue neuron). Descending projections from hypothalamic nuclei (red and yellow neurons) including the posterior (PH), paraventricular (PVN), lateral (LH), dorsomedial (DMH) and pre-optic hypothalamic nuclei (PON), to the TCC (red projections) and SuS (yellow projections) neurons, and the sympathetic nervous system (yellow projections), are thought to modulate and control both trigeminovascular nociceptive transmission (purple network of neurons) and parasympathetic (green)/sympathetic (orange) autonomic projections to the cranial vasculature that result indirectly or directly, respectively, in cranial autonomic symptoms ipsilateral to head pain. A third-order sympathetic nerve lesion (orange projection), in part mediated by internal carotid artery (ICA) vasodilation, is thought to result in Horner’s syndrome (see text for full description). VMH: ventromedial hypothalamus; SON: supra-optic nerve: TG: trigeminal ganglion: SPG: sphenopalatine ganglion; SCG: superior cervical ganglion: PAG: periaqueductal gray; LC: locus coeruleus; RVM: rostral ventromedial medulla.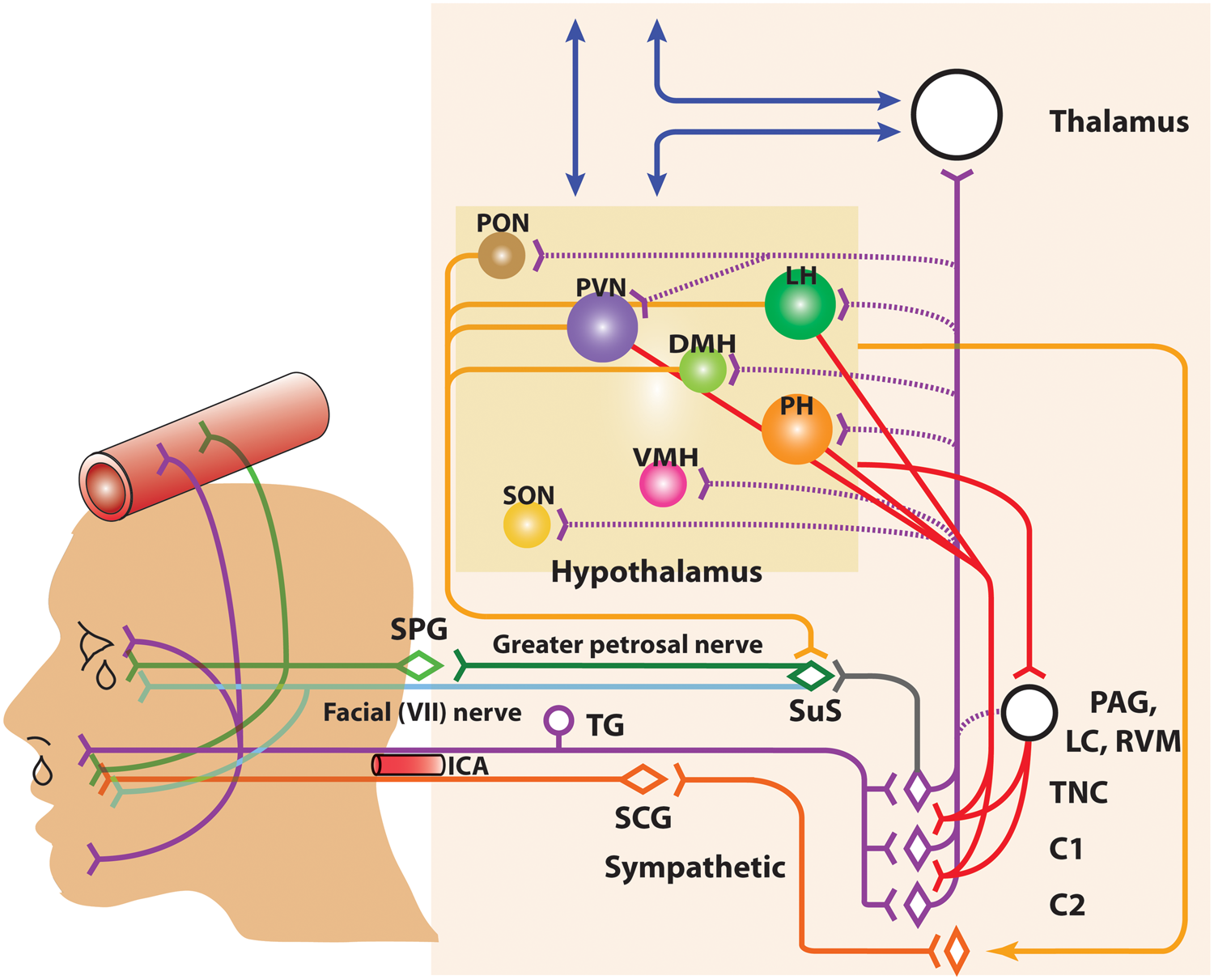
Stimulation of various components of the cranial parasympathetic projection; the SuS, facial nerve, and SPG, all mediate increases in cerebral blood flow,205–207 indicating a direct link between neural activation and cerebrovascular changes. Extracranial vascular changes of the common carotid artery, mediated by activation of the LC is via the facial nerve, and the SPG and otic ganglia.208–210 Furthermore, direct stimulation of the SuS produces blood flow changes in the lacrimal gland, indicative of autonomic features.211,212 These changes are specifically mediated by activation of the cranial parasympathetic pathway, as a specific SPG blocker attenuates these responses, as does the cluster headache treatment, inhaled oxygen.211,212 It is believed this parasympathetic outflow to the extracranial vasculature, responsible for cranial autonomic symptoms, uses VIP as one of its main neurotransmitters. 140 In experimental clinical studies in migraineurs, both VIP and PACAP cause severe cranial autonomic symptoms,144,146,213 as well as extracranial vasodilation. VIP is also a major transmitter released during cluster headache, alongside CGRP, 125 and VIP levels are increased in the extracranial vasculature in migraine patients who specifically experienced severe cranial autonomic symptoms. 84 These data suggest both VIP and PACAP are important in the mechanisms of cranial autonomic symptoms. While extracranial vasodilation might not be obligate in the generation of migraine and cluster headache, activation of the parasympathetic neurovascular pathway is essential to autonomic symptoms.
The cranial parasympathetic vasodilator pathway also has a huge influence on both central trigeminovascular neurons, and the dural microenvironment. It has been suggested that activation of this neurovascular pathway, and cranial autonomic symptoms, may also affect the neurophysiology related to head pain in migraine and cluster headache. The dural blood vessels are richly innervated by parasympathetic nerve fibers 214 and activation of this pathway causes the release of ACh, VIP, and NO, from dural vascular terminals of post-ganglionic sphenopalatine neurons, also containing PACAP. Activation can lead to dilation of intracranial vessels, plasma protein extravasation and local dural release of inflammatory mediators. 215 Experimentally, activation of the cranial parasympathetic pathway, via electrical stimulation of the SuS, causes neuronal action potentials in the TCC via two separate neural pathways. Firstly, via retrograde activation of the trigeminal autonomic reflex, within the brainstem. Secondly, however, by activation of the parasympathetic outflow to the cranial vasculature, which indirectly activates trigeminal afferents from the dura mater to the TCC, as well as producing cranial autonomic symptoms.211,212 These latter responses are attenuated by the specific SPG blocker, as well as oxygen treatment. Interestingly, this is not accompanied by dilation of meningeal blood vessels. 211 Again, these data support the suggestion that activation of this neurovascular pathway is important, but vasodilation, in this case of intracranial vessels, is not necessary to mediate activation of noxious trigeminal-autonomic neurons.
Similar to head pain in migraine and cluster headache, it is very likely that the primary trigger for cranial autonomic symptoms is away from the autonomic nervous system, and more likely under the influence of central structures that have control over these peripheral neurovascular pathways. Cluster headache in particular is defined by its seasonal variation, with ‘clustering’ of bouts over several months. 4 This suggests the biological clock – the hypothalamus – is primarily involved in the origins of this disorder. Clinical studies have demonstrated that hypothalamic activation is relatively specific to cluster headache, and other TACs,12,45–47,216 although hypothalamic activation in migraine has been reported.11,16 Preclinical data confirm that several specific hypothalamic nuclei, including the PVN and lateral hypothalamus, send descending projections to the TNC and SuS cell bodies.70,202,217–219 It is not known whether hypothalamic nuclei independently influences trigeminal somatosensory and SuS-autonomic pathways to produce headache and cranial autonomic symptoms. However, the reflex connection between trigeminal and parasympathetic neurons, and the projection of parasympathetic neurons to the cranial vasculature, which we know influence trigeminal neuronal responses, suggests that each pathway can influence and exacerbate responses of the other, via the release of vasoactive neuropeptides, such as VIP and PACAP. What does seem clearer is that hypothalamic neurons are ideally placed to influence and trigger somatosensory and autonomic neurovascular mechanisms via the trigeminal nucleus and SuS, respectively, if they are not functioning properly. Further, it is likely that cranial autonomic symptoms in migraine and cluster headache are mediated by secondary activation of the trigeminal autonomic reflex, indirectly via a trigemino-hypothalamic, or even more directly by a SuS-hypothalamic, pathway.
Neurovascular mechanisms of CSD (migraine aura)
Both imaging studies220–224 and animal model-based research225–229 show profound, complex, and long-lasting changes in cortical neurovascular relationships during migraine aura in humans and in animal models of CSD, respectively. Although these neurovascular events are unlikely to be the causative factor of head pain during either migraine or cluster headache, many of them may be involved in disabling symptoms and in the continuance of the disorders.230–232 For example, CSD is the likely underlying cause of visual aura in humans220–222,224 and thus provides a compelling link between human brain dysfunction and animal studies that aim to establish both therapeutic interventions and a mechanistic understanding of CSD. Moreover, animal studies suggest that CSD may also be involved in activating mechanisms of peripheral233–235 and central sensitization 236 involved in migraine headache, and several drugs that are prophylactically effective in migraine lessen CSD susceptibility in animal models.237,238 Migraine-associated genes also affect CSD dynamics when expressed in murine models.239–241 Although further work is necessary to establish the precise role(s) of CSD in migraine and headache, detailing the neurovascular changes during CSD and human imaging studies may be instructive because endogenous peptides, like PACAP and CGRP that appear to be involved in migraine pathophysiology and headache, exert powerful vascular effects with complex interactions with the surrounding neural and glial cell populations. Understanding the aberrant neurovascular responses in migraine aura and headache may thus point to processes that may be targeted for therapeutic intervention.
CSD
CSD, first described by Leao in 1944,226,227 is a mass depolarization of neurons and glia where ionic homeostasis is lost, i.e. there are profound shifts in extracellular potassium, sodium, calcium, and glutamate. Changes propagate at a peculiarly slow rate across the cortical surface (∼3–5 mm), and the changes can also propagate to subcortical regions240,242 and transhemispherically (Eftekhari, Charles, personal communications). CSD can be elicited by numerous stimuli, including physical damage to the brain or high frequency electrical stimulation to the cortex. Chemically, CSD is generally elicited by KCl delivered topically to the meninges or pressure injected into superficial layers of the cortex, although high concentrations of glutamate, oubain, and glutamatergic agonists can also result in CSD. 228 More recently, optogenetic methods have been used to target specific populations of neurons, and CSD can be reliably elicited by this less invasive approach. 243 Another prominent feature of CSD is a multiphasic and propagative wave of hyperperfusion (hyperaemia) followed by hypoperfusion (oligaemia). Additional phases of changes in regional cerebral blood flow have been documented including a third prolonged hypoperfusion, where normal neuronal and blood flow dynamics take approximately an hour to return to baseline levels. 244 In animal models, the changes to arterial caliber can precede the changes in optical intrinsic signal (OIS) reflectance and argue for separable neural and vascular processes. 229 CSD has been observed in humans, in compromised brain,245–247 and diverse animal models (e.g., rat, mouse, pig, cat, rabbit, hamster) and is the likely mechanism for producing migraine aura symptoms. CSD is also a proposed mechanism for activating meningeal afferents235,248 and corticothalamic loops that may be involved in central mechanisms of allodynia and the sensitization of sensory input. 236 These mechanisms remain intriguing but speculative, and it should be noted that many migraineurs do not experience aura, and some that do experience distortions in visual percepts do not experience headache.
Neurovascular coupling and uncoupling in CSD
Neurovascular coupling is the regular interplay between neural activity changes and corresponding vascular changes to alter cerebral blood flow to maintain metabolic support. An intriguing possibility is that the disruption of appropriate vascular responses to changes in neural activity may lead to nociceptive activations and that repetitive neurovascular de-couplings may facilitate further neurovascular dysfunction. In anesthetized rodent models, normal EEG recordings, and OIS reflectance changes from the cortical surface show regular oscillations that are synchronized with vascular dilatations and constrictions. During CSD, and for an extended period thereafter, the coherence between these two signals is disrupted suggesting a de-coupling or reconfiguration between neural activity (as measured by EEG) and the normally coherent vascular response. 244 Studies have shown interesting links between absolute CSD duration and corresponding changes in blood flow and oxygenation249–251 that indicate that restorative processes are active post-CSD, but these changes are on a slower time-scale and appear insufficient to quickly restore the normal neurovascular coupling that is present before the CSD event.
It should be noted that neurovascular responses are known to be anesthesia and anesthesia-level dependent, 252 but similar multiphasic vascular responses have been observed in awake mice using Doppler flowmetry following optogenetic activation of neurons leading to CSD. 243 These neurovascular changes are long-lasting after the initial CSD and may be involved in the delayed activation of nociceptive pathways.
Neurovascular complexities arising from animal models
Despite the utility of animal models for migraine and headache-related research, there are some important caveats to consider. First, many of the experimental data depend on anesthetized animals and the precise brain dynamics may be altered in awake, behaving animals. Recent innovations to record from conscious animals and to perform multimodal investigations to measure both neural and vascular parameters will be of continued importance in determining which results are generally applicable to the awake state. As mentioned above, the multi-phasic CSD vascular response observed in anesthetized animals appears to be preserved although further work is warranted to determine the nature of the neurovascular response in conscious rodents. Modern neuroscientific methods allow for continuous behavioral monitoring while also simultaneously recording from and stimulating diverse brain regions. Utilizing these methods in conjunction with ultrasound, laser Doppler flowmetry, high-speed multi-spectral imaging,253,254 will allow a deeper understanding of neurovascular processes as they relate to migraine pathophysiology.
Another issue with CSD animal studies is that the neurovascular and neural response appear to differ in lissencephalic and gyrencephalic animals.255,256 For example, CSD usually spreads across the entire cortical surface in lissencephalic animals. Although there appear to be areas of rodent cortex especially susceptible to CSD, and slight asymmetries in how CSD spreads, 257 there are a priori no gyral boundaries that limit the spread of CSD. In contrast, psychophysical work in humans suggest that the mechanism underlying visual aura is likely to be spread within a gyrus and to not radiate across gyral boundaries. 258 Because CSD is more limited in scope, the degree of neurovascular derangement and CSD associated edema would also likely be constrained in brains with gyri and sulci.
Neurovascular relations are also heavily state and history dependent. The changes observed in vascular caliber for a first CSD can be dramatically different in subsequent CSDs in the same animal. Little is known about chronic CSDs and their impact in awake, moving animals for extended longitudinal studies. This becomes more problematic when one also considers that the surgical procedures and the implants used to monitor neural and neurovascular activities can in fact produce CSD, making it difficult to determine when the first CSD occurred or to control for repeated or stimulus-evoked CSDs. Another subtler point is that many drugs have direct effects on the vascular system but those same drugs can also alter neurons that in turn lead to opposite changes in the vascular response (Charles et al., personal communication, submitted to this special issue). Which drug effect predominates to determine the vascular response may not be known.
A final point about neurovascular relations is that they are not well understood even in presumably normal brain function and can differ developmentally.259,260 For example, the same sensory stimulus can produce different vascular responses in juvenile versus adult rodents. 260 Further, how this translates to the developing human brain and primary headache disorders is not known. Moreover, there are strong non-linear relations between blood flow and neural activities (for examples, see Hillman 261 ). Observing mismatches between neural activity and blood flow responses may thus not signal a derangement of neurovascular coupling at all. The degree to which cells such as astrocytes or vascular endothelial cells contribute to a neurovascular response (e.g. ‘local’ vs. ‘propagated’) 262 is still an open area of research. 263
Cortical neurovascular mechanisms are an important component of the pathophysiology of primary headaches, particularly in migraine aura. It is clear there are cortical neurovascular perturbations that coincide with aura symptoms in migraine, and CSD is the most likely explanation for this. The exact relationship of both cortical neural and vascular components in migraine aura is still not well understood, or indeed the relationship of these cortical neurovascular changes to other symptoms, including migraine headache. However, it is undeniable these neurovascular changes do occur in the migrainous brain, and these changes affect the generalized migraine experience for the patient. Unravelling the relationship of this neurovascular coupling is likely to increase our understanding of migraine pathophysiology further.
Summary
There is a large body of evidence that highlights the importance and complexity of neurovascular mechanisms relevant to migraine and cluster headache. Despite the initial notion that only vascular mechanisms represent the causal basis of these disorders, this view has now been largely refuted. The lack of clinical efficacy of all pharmacological approaches that target vascular mechanisms confirms this view. However, the existence and the prominent role of neurovascular mechanisms in migraine and cluster headache are unquestionable. While it is now believed that the causative mechanisms related to these primary headaches lean far more heavily towards a neural basis, predisposing patients to these disorders, it is still thought that the relationship and delicate coupling between neural and vascular mechanisms are crucial to their pathophysiology in triggering and maintaining individual attacks.
Footnotes
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Jan Hoffmann is consulting for and/or serving on advisory boards for Allergan, Autonomic Technologies Inc. (ATI), Chordate Medical and Novartis. He received honoraria for speaking from Allergan, Novartis, and Teva. These activities are not related to the submitted work. Serapio M. Baca declares no conflicts. Simon Akerman reports an unrestricted grant, honoraria, and travel reimbursements from electroCore LLC, unrelated to the submitted work.