
Abstract
Cortical spreading depression (CSD) is considered a significant phenomenon for human neurological conditions and one of its key signatures is the development of persistent cortical oligemia. The factors underlying this reduction in cerebral blood flow (CBF) remain incompletely understood but may involve locally elaborated vasoconstricting eicosanoids. We employed laser Doppler flowmetry in urethane-anesthetized rats, together with a local pharmacological blockade approach, to test the relative contribution of cyclooxygenase (COX)-derived prostanoids to the oligemic response following CSD. Administration of the non-selective COX inhibitor naproxen completely inhibited the oligemic response. Selective inhibition of COX-1 with SC-560 preferentially reduced the early reduction in CBF while selective COX-2 inhibition with NS-398 affected only the later response. Blocking the action of thromboxane A2 (TXA2), using the selective thromboxane synthase inhibitor ozagrel, reduced only the initial CBF decrease, while inhibition of prostaglandin F2alpha action, using the selective FP receptor antagonist AL-8810, blocked the later phase of the oligemia. Our results suggest that the long-lasting oligemia following CSD consists of at least two distinct temporal phases, mediated by preferential actions of COX-1- and COX-2-derived prostanoids: an initial phase mediated by COX-1 that involves TXA2 followed by a later phase, mediated by COX-2 and PGF2alpha.
Introduction
Cortical spreading depression (CSD) is a massive wave of neuronal and glia depolarization that can be induced experimentally in the cortex of lissencephalic animals.1,2 CSD is now considered a significant phenomenon for human neurological conditions given that similar spreading depolarizations have been recorded following traumatic brain injuries; subarachnoid and intracranial hemorrhage, as well as ischemic strokes. 3 CSD is also considered as a key process underlying the aura phase of migraine,4,5 an important triggering mechanism of migraine pain6–8 and a potential target for migraine headache prophylaxis. 9
CSD is accompanied by cerebrovascular changes that may have significant neurological importance. Most noticeable is a unique series of changes in cerebral blood flow (CBF) that include a brief hyperemia that lasts for 1–2 min followed by persistent decrease in CBF (oligemia) and impairment of cerebral vascular reactivity that lasts for at least 1 h.10–13 The decrease in CBF following CSD is also accompanied by persistent increase in oxygen consumption and persistent decreases in cortical partial pressure of oxygen. 13 In the wake of CSD, the persistent cortical oligemia 13 is likely to compromise cortical function and thus may be of critical importance to neurological disorders such as subarachnoid hemorrhage, 14 stroke 3 and migraine. 15 A better understanding of the factors that contribute to the CSD-evoked persistent oligemia is thus warranted given that increase CBF following CSD is likely to improve local oxygen delivery and cortical function.
An earlier study in rats and rabbits documented increases in cortical and CSF levels of arachidonic acid (AA) following a single episode of CSD.16,17 The metabolism of AA, through numerous enzymatic pathways, yields a variety of eicosanoids, some of which are potent vasoconstrictors. In a recent study, Fordsmann et al. 18 documented in vitro increased cortical synthesis of 20-HETE, an AA metabolite of the cytochrome P450 enzyme (CYP450) pathway following CSD and provided data supporting its role in mediating the post-CSD cortical oligemia in rats. Another major pathway for AA metabolism is through the cyclooxygenase (COX) enzymes, which yield a variety of prostanoids. Following CSD, acute increases in CSF levels of the vasoconstricting prostanoids thromboxane A2 (TXA2) and prostaglandin F2alpha (PGF2alpha) 17 suggests increased COX activation. In addition, the ability of systemic administration of the non-selective COX inhibitor indomethacin to block the post-CSD cortical vasoconstriction COX-derived metabolites points to COX involvement in mediating the post-CSD oligemic response 17 . Because indomethacin also exerts cerebrovascular effects that are non-prostaglandin-related, 19 the role of COX and its related prostanoids in mediating the post CSD oligemia remains unclear.
In the cerebral cortex, prostanoids can be produced by a variety of cells that constitutively express the two main COX isoforms: COX-1 and COX-2. In the present study, using a pharmacological approach, we initially tested the effect of local non-selective COX inhibition, using naproxen, on the oligemic response to CSD. We then examined, using selective inhibitors, the relative contribution of COX-1 vs. COX-2. Finally, we employed a thromboxane synthase inhibitor and an FP receptor antagonist to explore the relative contribution of local TXA2 and PGF2alpha signaling, respectively, into the decrease in CBF following CSD. Our data suggest that COX-1 as well as COX-2 contribute to the oligemia, albeit at different temporal stages of the vascular response and that both TBXA2 and PGF2alpha action contribute to this process.
Materials and methods
Ethics statement
All experimental procedures, followed the guide for Care and use of Laboratory Animal resources (NIH publication No. 85-23 revised 1996), were approved by the Animal Care and Use Committee of the Beth Israel Deaconess Medical Center, and were in compliance with the ARRIVE (Animal Research: Reporting of In Vivo Experiments) guidelines.
Animal anesthesia and surgical preparation
Experiments were conducted on adult male Sprague Dawley rats (9–12 weeks, weighing 300–400 g, Taconic). Rats were deeply anaesthetized with urethane (1.2–1.5 g/kg. i.p.). Throughout the experiment, the level of anesthesia was verified by testing for lack of reflexive responses to pinching of the hind paw. Core body temperature was maintained at 37–38℃, using a temperature-controlled heating pad. A silicone catheter (PE10), filled with heparinized saline, was placed in the left femoral artery to monitor mean arterial pressure (MAP, TA-100 blood pressure monitor, CWE Inc., Ardmore PA, USA). Only animals with stable MAP (90–120 mmHg) were studied. Arterial blood samples were obtained for blood gas analyses using BG blood gas cartridge and IRMA TRU point blood gas analyzer (LifeHealth, Roseville, MN). Throughout the study, animals were mechanically ventilated (SAR-1000, CWE Inc., Ardmore PA, USA) using room air supplemented with O2. Heart rate and oxygen saturation were monitored using pulse oximetry (PhysioSuite, Kent Scientific, Torrington, CT, USA) and maintained at physiological levels (350–450 bpm, >95% O2 saturation). To further assure basic physiological parameters, expired CO2 level was also continuously monitored (CapStar-100, CWE Inc., Ardmore, PA, USA) with end-tidal CO2 values maintained between 3 and 4.5% by adjusting the respiration and airflow rates. These physiological levels corresponded to blood gas levels of pH 7.35–7.45; PaO2 > 110 Torr and PaCO2, 35–45 Torr. A total of five animals displaying abnormal physiological parameters were excluded from the data analyses.
For recording CSD and CBF, the animal was placed in a stereotaxic frame (David Kopf Instrumentation, Tujunga, CA, USA). The skull was exposed by a midline dissection through the skin and removal of the underlying calvarial periosteum. Two craniotomies (see Figure 1(a)) were made to expose parts of the left hemisphere using a saline cooled dental micro drill equipped with a 0.7-mm burr (FST, Foster city, CA, USA). A small craniotomy (1 mm × 1 mm) was made above the frontal cortex to induce CSD. A more caudal and larger craniotomy (2 mm × 2 mm) was performed above the visual cortex to allow recording of CSD and resultant changes in CBF. The dura mater was not removed, to avoid injury to pial vessels, meningeal inflammation and possible mechanical induction of CSD. The dura was kept moist before and during baseline testing using a cortex buffer containing (in mM) 135 NaCl, 5 KCl, 1 MgCl2, 5 CaCl2, 10 glucose and 10 HEPES at pH 7.3.
(a) Experimental preparation. CSD was elicited using a pin prick (PP) in the left frontal cortex. CSD propagation was monitored by recording the shift in DC potential using a glass microelectrode placed in the visual cortex. Changes in CBF were recorded using the LDF probe placed epidurally adjacent to the DC recording electrode. (b) Data from one rat treated epidurally with vehicle and subjected to CSD showing changes in CBF and DC current (top traces). The gray-filled areas represent the time points used to calculate the hyperemic response the initial transformation to oligemia (“dip”) and the persistent oligemia. The two bottom traces are recording of end-tidal CO2 (ETCO2, blue trace) and mean arterial pressure (MAP, red trace). Note the lack of change in these physiological parameters following CSD.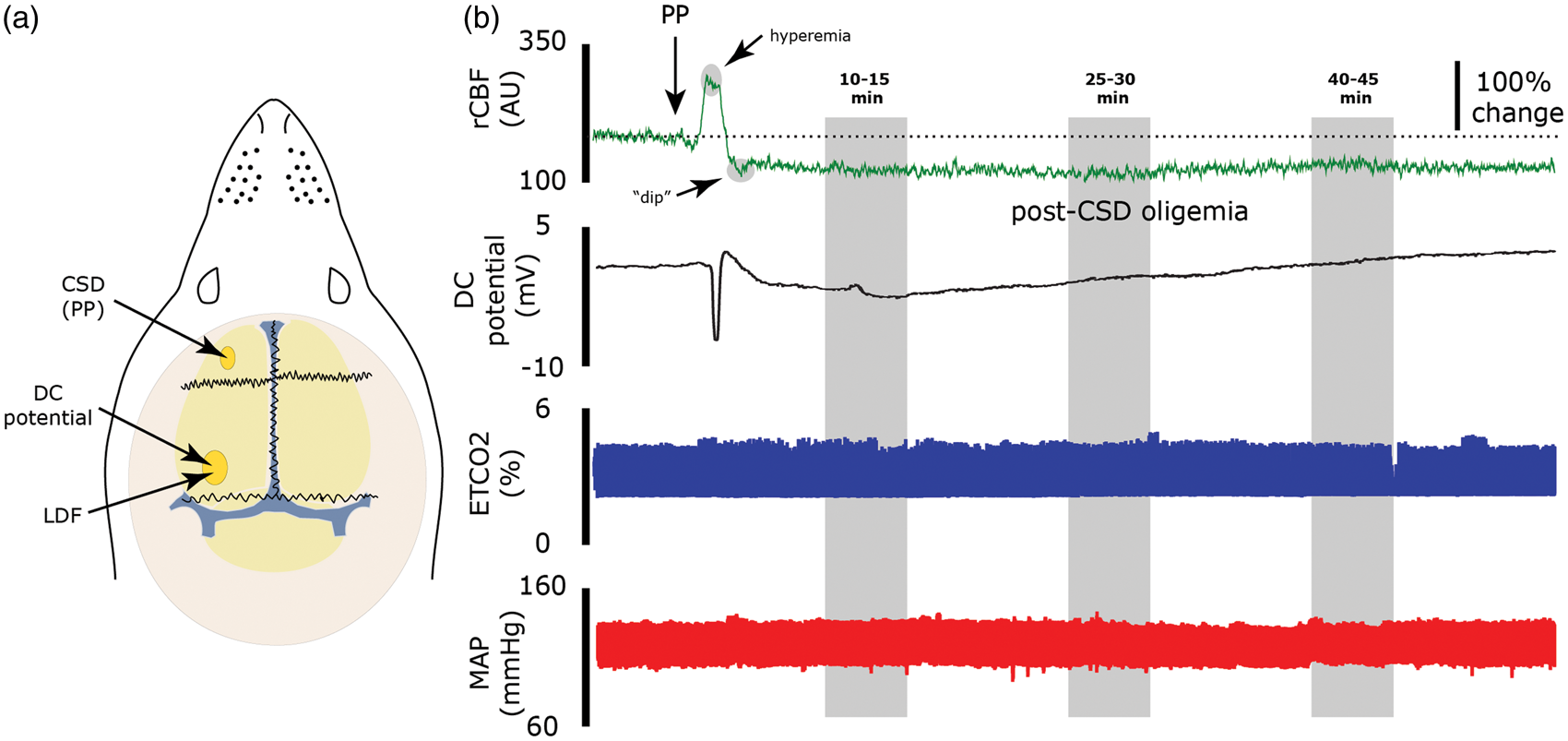
Measurements of changes in direct current potential and cerebral blood flow
To detect the induction of CSD, a glass micropipette (tip ∼10 µm) filled with 150 mM NaCl (impedance 70–120 kΩ) was lowered into the caudal craniotomy, through a small dural incision, and positioned about 1 mm below the cortical surface to record direct current (DC) potential (Figure 1(a)). DC potential was continuously recorded using a reference Ag/AgCl pellet ground electrode (A-M Systems) placed in the right temporal muscle. CSD was elicited experimentally by mechanical stimulation (pin prick) that was delivered by inserting a glass micropipette (diameter 10–20 µm) about 2 mm into the frontal cortex for 2 s. A wave of CSD was usually registered by the recording electrode, as a negative DC deflection, within 1–2 min of cortical stimulation. To record CBF changes induced by CSD, a needle probe (VP4, 0.8 mm diameter) connected to a laser Doppler flowmeter (LDF, Moor instruments, Wilmington, DE, USA) was positioned just above the intact dura, as close as possible to the DC recording electrode and in an area devoid of large blood vessels (>100 µm). Such LDF system (using a near IR 785 nm probe and fiber separation of 0.25 mm) is expected to record CBF changes at a cortical depth of ∼1 mm. 20 LDF recordings were obtained with the ambient lights turned off. DC and CBF data, as well MAP and ETCO2, were digitized and continuously sampled via an A/D interface (micro1401 and spike 2 software, CED, Cambridge, UK).
Experimental protocol and pharmacological treatments
Baseline DC potential and LDF/CBF values were established in animals with stable physiological parameters for at least 30 min. The inhibitors, or their vehicle, were then topically applied to the dura (∼50 μl), at the site of CBF recording, and a second pre-CSD baseline (baseline + drug) trial was recorded for 30 min. A single CSD was then induced and CBF changes were recorded for at least 50 min, during which the pharmacological agents were left on the dura to maintain continuous inhibition. Animals were randomly treated with the following agents: (1) the non-selective COX inhibitor (S)-6-Methoxy-α-methyl-2-naphthaleneacetic acid sodium salt (naproxen, 100 µM); (2) the selective COX-1 inhibitor 5-(4-Chlorophenyl)-1-(4-methoxyphenyl)-3-trifluoromethyl pyrazole (SC-560, 500 µM,); (3) the selective COX-2 inhibitor, N-[2-Cyclohexyloxy-4-nitrophenyl]methanesulfonamide (NS-398, 1 mM); (4) the selective thromboxane synthase inhibitor (2E)-3-[4-(1H-Imidazol-1-ylmethyl)phenyl]-2-propenoic acid hydrochloride (Ozagrel, 1 mM); (5) the selective FP receptor antagonist 5Z,13E)-(9S,11S,15R)-9,15-Dihydroxy-11-fluoro-15-(2-indanyl)-16,17,18,19,20-pentanor-5,13-prostadienoic acid (AL-8810, 1 mM). All agents, except naproxen, were purchased from Cayman Chemicals (Ann Arbor, MI, USA). Naproxen was purchased from Sigma-Aldrich (St. Louis, MO, USA). SC-560, NS-398 and AL-8810 were first diluted in DMSO and, for local application, were further diluted with the cortex buffer (final DMSO concentration <1%). Doses chosen were based on pilot dose response experiments and previous studies.21–24
Data analysis
Data are expressed as means ± SEM. Raw LDF data were processed with a custom-written script made for Spike 2. Baseline values were based on the average of the LDF values collected during the 300 s prior to the application of the drug (baseline) and the induction of CSD (baseline + drug). For the CBF data collected after the CSD, the following data points were analyzed: maximum CBF during the hyperemia, minimum (dip) CBF, obtained during the initial stage of the oligemia, immediately following the reversal of the hyperemic response. Three additional data points were collected during the persistent oligemia stage. These were based on averaged LDF data obtained during the 10–15, 25–30 and 40–45 min time points post CSD. Statistical analyses were conducted using Statview (SAS). Sample size for analysis was based on the expected differences between groups. Time-course comparisons between vehicle and treatment groups were analyzed using two-factor, repeated measures analysis of variance (ANOVA) followed by post-hoc unpaired two-tailed Student's t test. Within-group analyses employed paired two-tailed student's t test. For the ANOVA, p < 0.05 was considered statistically significant. For multiple t tests, Bonferroni corrections were employed and p < 0.0125 (0.05/4) was considered as the level of significance.
Results
Effects of pharmacological treatments on baseline and hyperemic responses to CSD
Baseline, resting CBF was not affected following treatment with vehicle (n = 13), naproxen (n = 6), SC-560 (n = 7), NS-398 (n = 6), Ozagrel (n = 7) or AL-8810 (n = 7; Figure 2(a)). Following the induction of CSD, as defined by the cortical large direct current shift, vehicle treated animals displayed a robust acute increase in CBF (185.5% ± 9.6%) in agreement with previous studies.13,25 As Figure 2(b) demonstrates, none of the inhibitors tested had a significant effect on this CSD-evoked hyperemic response.
Effect of the inhibitors on resting CBF and peak hyperemic response to CSD. Local application of the non-selective COX inhibitor naproxen, selective COX-1 inhibitor SC-560, selective COX-2 inhibitor NS-398, the thromboxane A synthase inhibitor ozagrel and the FP receptor antagonist AL-8810 neither affected the resting CBF (a), nor the peak hyperemic response following CSD (b).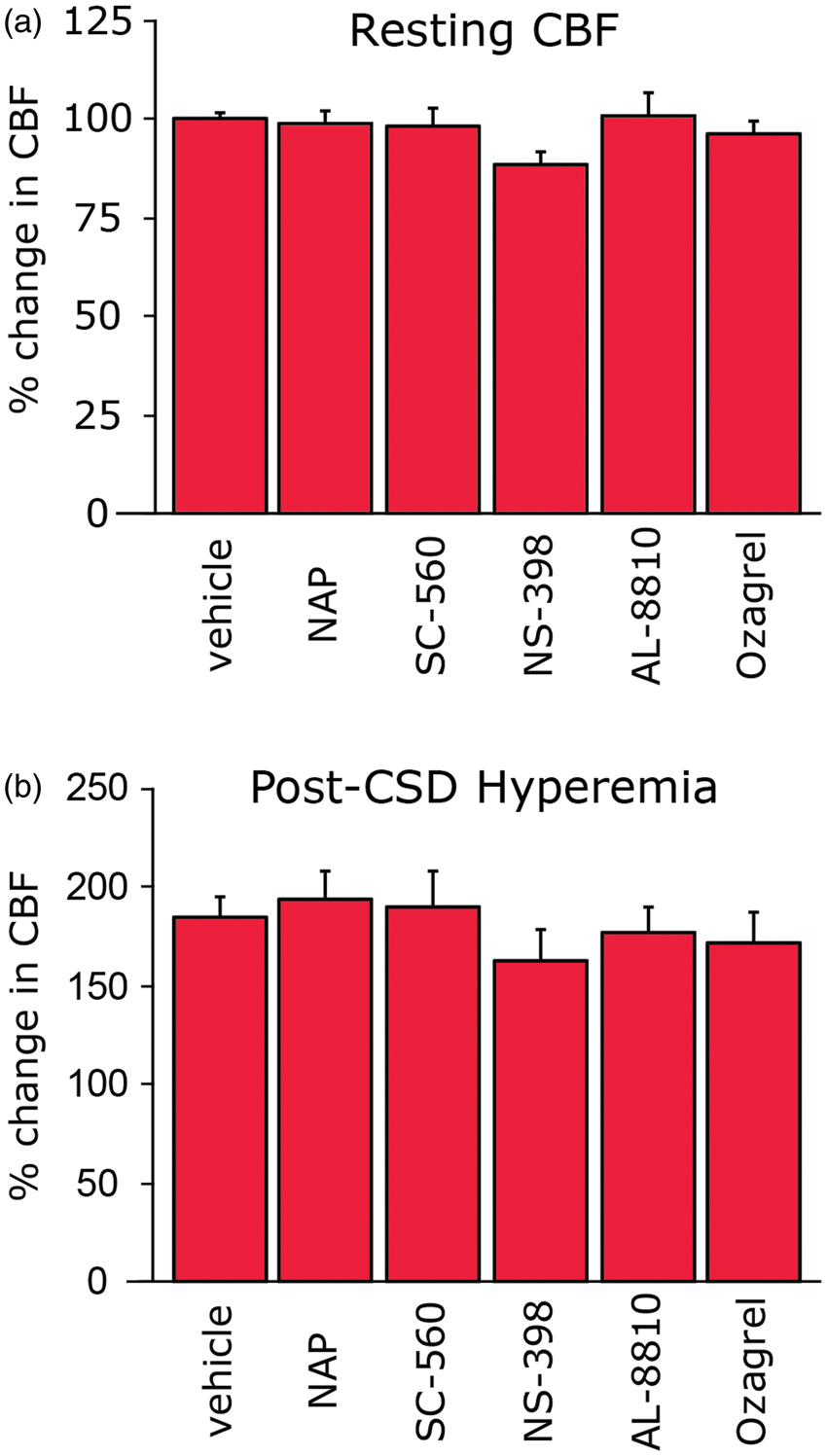
Effect of non-selective and selective COX inhibition on the post-CSD cortical oligemia
Effect of non-selective COX inhibition
In vehicle-treated animals, the increase in CBF during the arrival of the CSD wave was followed by a precipitous dip in CBF (−38.7% ± 1.6%, Figure 3(a)). The reduction in CBF was maintained for at least 45 min following post CSD (−26.2% ± 2.9% at 40–45 min, p < 0.0001 vs. baseline + vehicle). When compared to vehicle treatment, non-selective COX inhibition with naproxen significantly reduced the CSD-evoked oligemia (F(1,17) = 48.7, p < 0.0001). As shown in Figure 3(b), the significant inhibition exerted by naproxen was noted for the early dip in CBF as well as for the 10–15, 25–30 and 40–45 min post-CSD time points (p < 0.0001 vs. vehicle, for all time points). At all times tested, naproxen treatment exerted a complete inhibition of the oligemic response, with LDF values not different than those observed during the baseline + drug period (p > 0.05).
Effect of non-selective and selective, COX inhibition on the cerebral oligemia following CSD: (a) In animal treated locally with vehicle, CSD gave rise to persistent oligemia (##p < 0.001 compared to LDF values obtained during the pre-CSD baseline + drug treatment period. (b) Local application of naproxen had a robust inhibitory effect on the post-CSD oligemic response at all time points tested. **p < 0.01 compared with vehicle. (c) Local application of SC-560 preferentially inhibited the early post-CSD oligemic response, the “dip” and the 10–15 time points. **p < 0.01 compared with vehicle, #p < 0.0125 compared with pre-CSD, baseline + drug values. (d) Local application of NS-398 inhibited only the late oligemic response starting at the 10–15 time point. **p < 0.01 compared with vehicle, #p < 0.0125 compared with pre-CSD, baseline + drug values.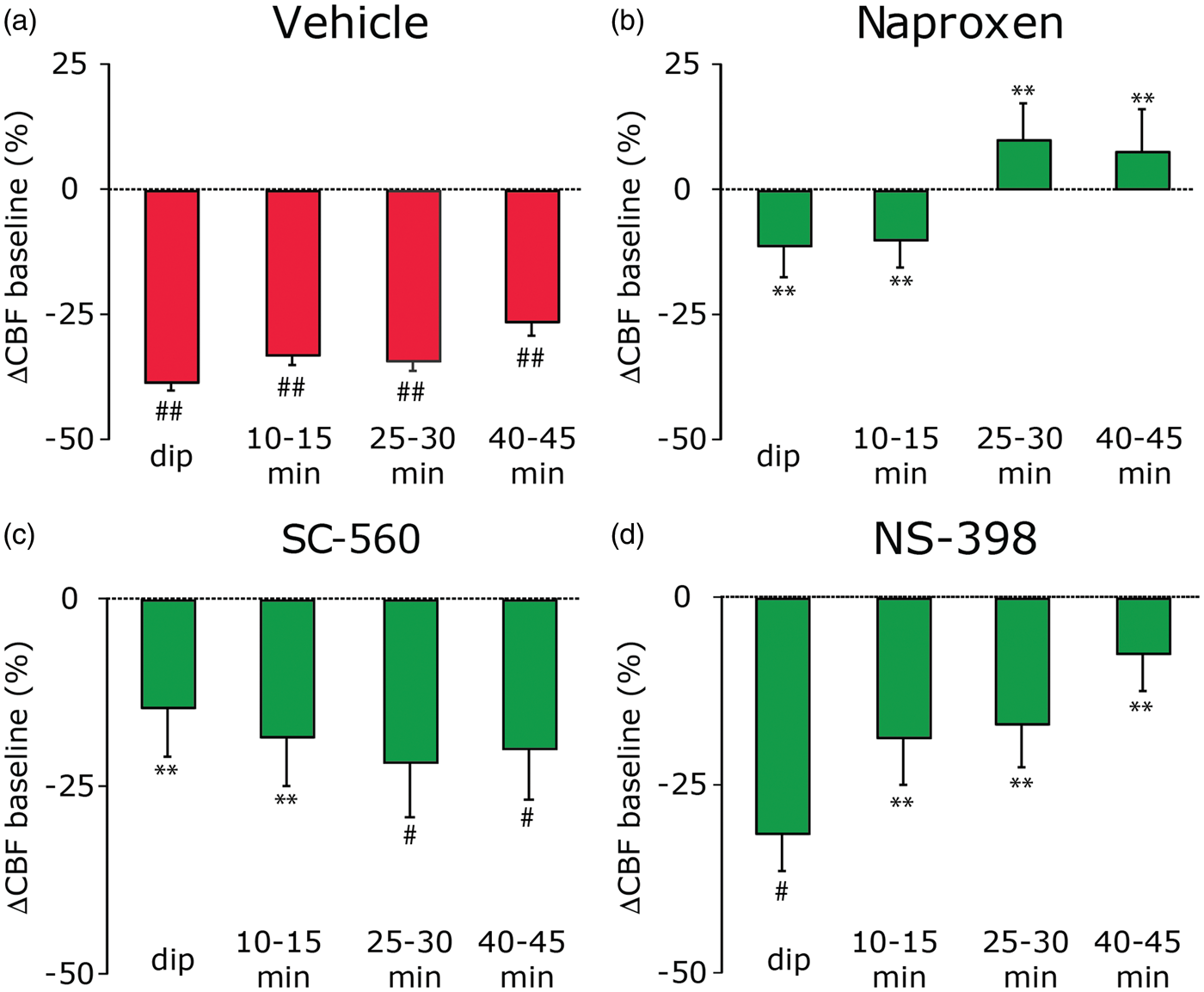
Effect of selective COX-1 inhibition
The local anti-oligemic effect exerted by naproxen treatment corroborates the previous data of Shibata et al. 17 in rabbits, which employed systemic administration of indomethacin, and thus provides further support for the role of locally elaborated COX vasoactive metabolites in mediating the persistent cortical oligemia following CSD. Because naproxen, such as indomethacin, is a non-selective COX inhibitor and may also have pharmacological effects that are COX-independent,26,27 we next examined the effect of SC-560, which inhibits COX-1 1000 times more potent than COX-2. 28 Similar to the effect of naproxen, when compared to vehicle, local treatment with SC-560 exerted overall an inhibitory effect on the oligemic response (F(1,18) = 10.3, p < 0.01). Nevertheless, unlike the effect of naproxen, the inhibition exerted by SC-560 was time dependent and became significant only for the early phase of oligemia phase (p < 0.01 vs. vehicle for the immediate dip in CBF and 10–15 min time points, Figure 3(c)). The inhibitory effect of SC-560 on the dip response as well as at the 10–15 min was complete (p > 0.05, compared to baseline + drug), suggesting a key role for COX-1 in mediating the initial decrease in CBF following CSD.
Effect of selective COX-2 inhibition
To examine the relative contribution of COX-2 in mediating the post CSD oligemic response, we tested the effect of local treatment with NS-398, which is 3000 times more potent in inhibiting COX-2 than COX-1. 29 Similar to SC-560, local administration of NS-398 also exerted an overall inhibitory effect on the post-CSD oligemic response (F(1,17) = 13.1, p < 0.001). The anti-oligemic effect exerted by NS-398 was also time-dependent, but unlike that of SC-560, was limited to the more delayed oligemic response, with significant inhibition noted only for the 10–15, 25–30 and 40–45 min time points (p < 0.01 for all, Figure 3(d)). At these time points, the inhibitory effect of NS-398 was complete, yielding LDF values not significantly different than those observed at baseline (p > 0.05), indicating a role for COX-2 in mediating the delayed and persistent CSD-related cerebral oligemia.
Is there a role for the prostanoids TXA2 and PGF2alpha in the post-CSD cortical oligemia?
Effect of ozagrel
We next tested the role of TXA2, a vasoconstricting prostanoid generated from the COX metabolite prostaglandin H2 by thromboxane-A synthase. Administration of the selective thromboxane-A synthase inhibitor ozagrel exerted overall an inhibitory effect on the post-CSD oligemia (F(1,18) = 5.1, p < 0.05). However, when compared to vehicle, the inhibition elicited by ozagrel was limited to the immediate dip in CBF (p < 0.01, Figure 4(a)). This inhibition, however, was not complete, with CBF values at this time point lower than those observed at baseline (p < 0.0125).
Effect of the thromboxane-A synthase inhibitor ozagrel and the FP receptor antagonist AL-8810 on the cerebral oligemia following CSD. (a) Ozagrel had a partial inhibitory effect on the very early “dip” post-CSD oligemic response and did not affect CBF at the later time points. **p < 0.01 compared with vehicle, #p < 0.0125 compared with pre-CSD, baseline + drug values. (b) AL-8810 inhibited the late oligemic response at the 10–15, 25–30 and 40–45 min time points, but not the immediate “dip”. **p < 0.01 compared with vehicle; #p < 0.0125; ##p < 0.001 compared with pre-CSD, baseline + drug values.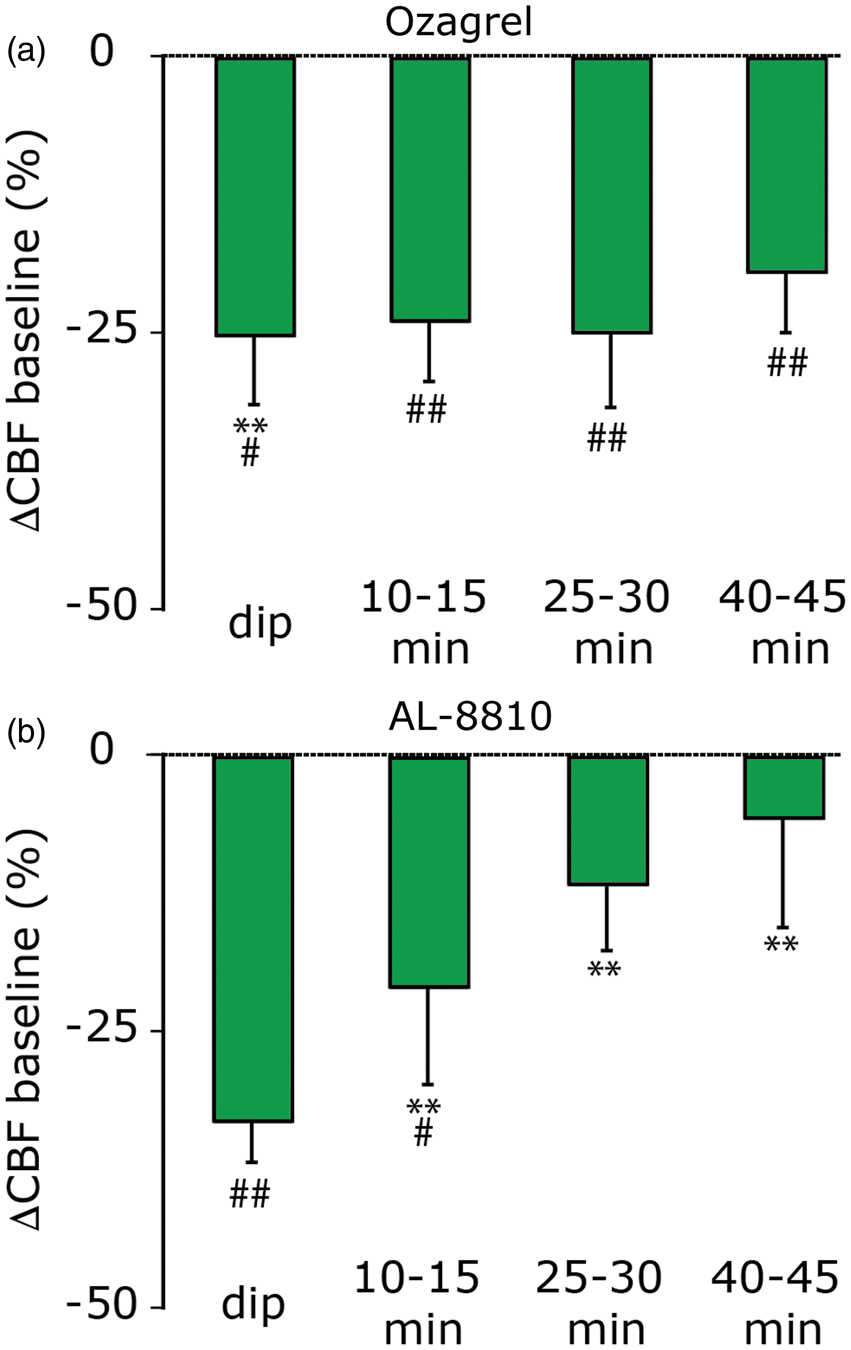
Effect of AL 8810
Finally, we tested the role of the prostaglandin PGF2alpha, a vasoconstricting prostanoid generated from PGH2 by prostaglandin F synthase that acts upon vascular FP receptors. Administration of the selective FP receptor antagonist AL-8810 exerted overall an inhibitory effect on the post-CSD oligemia (F(1,18) = 13.2, p < 0.001). When compared to vehicle, AL-8810 inhibited the oligemic response at the 10–15, 25–30 and 40–45 min time points (all p < 0.01, Figure 4(b)). At the 25–30 and 40–45 min time points, the inhibition was complete, with CBF values not different that those observed at baseline.
Discussion
The key new findings from this inhibitor-based study are (1) A concerted local action of vasoactive prostanoids produced by COX-1 and COX-2 contribute to the post-CSD cortical oligemia; (2) COX-1 related prostanoids mediate the initial phase of the decrease in CBF, while COX-2 related prostanoids mediate the later stage of this vascular response; (3) Local action of the vasoconstrictor prostanoid TXA2 plays a partial role in mediating the initial decrease in CBF following the hyperemia; (4) The prostanoid PGF2alpha, by acting upon FP receptors, contributes to the post-CSD oligemic response, in particular to its maintenance.
Previous studies implicated local action of vasodilatory prostanoids produced by COX-1 30 and COX-231,32 in mediating neurovascular coupling (i.e. functional hyperemia). The hyperemia associated with CSD, although occurs following a massive wave of cortical excitation, was suggested to be COX-independent because systemic administration of indomethacin, a non-selective COX inhibitor, did not block it. 33 Our findings showing a similar lack of inhibition following local application of naproxen, a non-selective COX inhibitor with a different structure than indomethacin (a propionic acid derivative vs. an indole compound), as well as by the more selective COX-1 and COX-2 inhibitors support this view.
In the wake of CSD, the prolonged oligemia that follows the brief hyperemia may be attributed to a decrease in basal vasodilating influence. Such a mechanism is supported by the finding that the decrease in CBF is associated with impaired cerebrovascular reactivity, including impaired vasodilatory response to hypercapnia and neurovascular coupling, mediated in part by diminished endogenous signaling through the NO-cGMP signaling cascade.13,34 Another mechanism that has been suggested to mediate the oligemia is increased vasoconstricting influence, mediated by the elaboration of AA-derived vasoconstricting eicosanoids.17,18 Our data are in agreement with this vasoconstrictor hypothesis and provide further support for the involvement of AA-related vasoconstricting prostanoids that are synthesized by both COX-1 and COX-2.
Our data suggest that the oligemia following CSD has at least two phases—an early phase mediated by COX-1 followed by a later phase mediated by COX-2. The finding that the COX-1 and COX-2 pathways are responsible for different parts of the post-CSD oligemic response points to the possibility that different cortical cell types are involved in controlling the post-CSD cortical vasoconstriction. Cerebral COX-1 is expressed primarily in neurons, glia and endothelial cells35–37 and has been suggested to play a role in maintaining resting vascular tone.30,38 Constitutive expression of cerebral COX-2, however, is limited to excitatory neurons where, unlike COX-1, it is thought to mediate neurovascular coupling.31,32,39 The reliance on COX-1 metabolites for the transformation of the hyperemic response to oligemia following CSD suggests that despite their massive depolarization, excitatory cortical neurons which primarily express COX-2,40,41 are unlikely to contribute. The finding that local action of TXA2 is involved in mediating, at least in part, the initial oligemic response points potentially to the role of cortical astrocytes, which become activated during CSD 42 and are capable of releasing this vasoconstricting prostanoid. 43 Release of TXA2 from activated vascular endothelial cells 43 may also contribute to this vascular response. Interestingly, reduced NO signaling in cerebral arteries, which occurs in CSD, 32 has been linked to increased TBXA2 signaling. 44
Pyramidal neurons, which are the main source of COX-2-derived prostanoids, are likely to contribute to the delayed stage of the oligemia. The finding that this subset of cortical neurons is silenced for at least an hour following CSD 45 —when the cerebral oligemia is present—suggests that the activation of COX-2 and ensuing generation and release of vasoconstricting prostanoids from these neurons is not activity dependent. The quiescence of these neurons following the CSD may also suggest that the release of vasodilating prostanoids from COX-2 at the same time is limited, thus potentially potentiating the oligemic response. The earlier finding of increased PGF2alpha release following the CSD 17 together with our current finding that blockade at the FP receptor inhibits this stage of the oligemia points to PGF2alpha as a key COX-2-related vasoconstrictor prostanoid that mediates of the prolonged oligemic response.
We applied the inhibitors topically to the dura to minimize potential pial and parenchymal inflammation associated with the process of dural removal. This approach, although less efficient than direct parenchymal application, has been used previously to manipulate cortical function:46–48 it is consistent with the notion that agents with molecular weights similar to the inhibitors used in our study undergo rapid transmeningeal diffusion into all layers of the cortex following topical epidural application, with a dilution factor of 1:100–500.47–49 Such dilution could potentially explain why in our study epidural SC-560 treatment (at 500 µM) did not reduce resting CBF while previous studies noted decreased resting CBF and cerebral vasoconstriction following parenchymal superfusion of this inhibitor at concentrations ≥10 µM.30,38,50 Because SC-560, as well naproxen, affected CBF only during the oligemic stage, it is also tempting to speculate that these COX inhibitors are more efficient in blocking the synthesis of vasoconstricting prostanoids that mediate the post-CSD oligemia than inhibiting the formation of vasodilating prostanoids that control basal CBF. Nonetheless, given the uncertainty about the precise parenchymal concentration of the inhibitors in our study, the effects elicited by these agents should be interpreted with caution given the possibility that the inhibition was only partial.
The development of the persistent cerebral oligemia following CSD coincides with the activation of meningeal pain fibers, a mechanism thought to mediate the genesis of the headache during migraine. Because proinflammatory COX products can promote a prolonged activation of meningeal afferents, 51 it will be of interest to explore the relative contribution of the oligemia to the CSD-evoked meningeal afferent activation and particularly whether COX inhibition can ameliorate this nociceptive response.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The study was supported by grants from the NIH/NINDS (NS077882, NS086830, NS078263 to DL).
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author contributions
HG performed experiments, conducted statistical analyses of the data and edited the manuscript; JZ performed experiments, conducted data analysis, and edited the manuscript; DL designed the experiments, performed statistical analysis of the data and wrote the manuscript.