
Abstract
Aims:
To investigate the safety, tolerability, pharmacokinetics (PK) and pharmacodynamics (PD) of BPL-003, a novel intranasal benzoate salt formulation of 5-methoxy-N,N-dimethyltryptamine (5-MeO-DMT), in healthy participants.
Methods:
In all, 44 psychedelic-naïve participants enrolled in the double-blind, placebo-controlled single ascending dose study (1–12 mg BPL-003). Concentrations of 5-MeO-DMT and its pharmacologically active metabolite, bufotenine, were determined in plasma and urine. PD endpoints included subjective drug intensity (SDI) rating, the Mystical Experience Questionnaire (MEQ-30) and the Ego Dissolution Inventory (EDI).
Results:
BPL-003 was well tolerated at doses up to 12 mg. There were no serious adverse events (AEs), and most AEs were mild; the most common being nasal discomfort, nausea, headache and vomiting. 5-MeO-DMT was rapidly absorbed and eliminated; the median time to peak plasma concentration was approximately 8–10 min and the mean terminal elimination half-life was <27 min. 5-MeO-DMT systemic exposure increased approximately dose-proportionally, while plasma bufotenine concentrations and urinary excretion of 5-MeO-DMT and bufotenine were negligible. The intensity of the SDI ratings was associated with plasma 5-MeO-DMT concentrations. MEQ-30 and EDI scores generally increased with the BPL-003 dose; 60% of participants had a ‘complete mystical experience’ at 10 and 12 mg doses. Profound and highly emotional consciousness-altering effects were observed with BPL-003, with a rapid onset and short-lasting duration.
Conclusion:
The novel intranasal formulation of BPL-003 was well tolerated with dose-proportional increases in PK and PD effects. The short duration of action and induction of mystical experiences suggest clinical potential, warranting further trials.
Clinical trial registration:
NCT05347849.
Introduction
A growing body of evidence from controlled clinical trials indicates psychedelics may have therapeutic benefits in a variety of psychiatric disorders (Goodwin et al., 2022; Ko et al., 2023; Nutt et al., 2023; Raison et al., 2023). Classic serotonergic psychedelics have broad activity at 5-hydroxytryptamine (5-HT) receptors and produce a characteristic altered state of consciousness via partial agonism of the type 2A 5-hydroxytryptamine (5-HT2A) receptor (Nichols, 2016).
5-Methoxy-N,N-dimethyltryptamine (5-MeO-DMT), a tryptamine alkaloid, is a short-acting serotonergic psychedelic that was first synthesized in 1936 (Hoshino and Shimodaira, 1936). 5-MeO-DMT is a 5-HT receptor agonist with affinity to a variety of serotonin receptors. While it has a broad affinity with 5-HT receptors, the highest binding affinity is for the 5-HT1A receptor, with a 300-fold to 1000-fold higher selectivity compared with the 5-HT2A receptor (Halberstadt et al., 2012; Ray, 2010). The behavioural effects and safety margins of 5-MeO-DMT have been best characterized in rodents (Halberstadt et al., 2012). 5-MeO-DMT is rapidly metabolized and inactivated by monoamine oxidase enzymes in the gut and liver (Shen et al., 2010) to generate the pharmacological inactive metabolite, 5-methoxyindoleacetic acid and its only active metabolite, bufotenine (5-HO-DMT) (Yu et al., 2003). The process of O-demethylation is carried out by the cytochrome P450 2D6 (CYP2D6) enzyme and is the primary route of 5-MeO-DMT metabolism. In vitro, bufotenine exhibits around a 5-fold to 10-fold higher affinity to the 5-HT2A receptor than 5-MeO-DMT; however, this drops to a three-fold higher potency when both are present at similar levels in the brain (Shen et al., 2010).
For most psychedelics, effects observed in humans appear to be mediated primarily through the 5-HT2A receptor (Barker, 2018; Madsen et al., 2019; Nichols, 2016; Vollenweider et al., 1998). Contrastingly, the 5-HT1A receptor has been implicated in mood regulation (Kaufman et al., 2016) and autonomic nervous system control (Youn et al., 2013). Pre-clinical data have also shown that decreased locomotor activity, investigatory behaviour and disturbed thalamocortical oscillations can be selectively attenuated using a 5-HT1A antagonist but not a 5-HT2A antagonist (Krebs-Thomson et al., 2006; Riga et al., 2016, 2018).
Compared to N,N-dimethyltryptamine (DMT), which binds with higher affinity to the 5-HT-2A receptor (Barker, 2018), 5-MeO-DMT has a 4-fold to 10-fold more potent affinity for the 5-HT1A receptor in humans (Shen et al., 2010). This difference in preferential binding is suggested to result in the differences between subjective experience, with DMT tending towards vivid and complex visual imagery, whereas marked ego dissolution is more often seen with 5-MeO-DMT (Barker, 2018; Reckweg et al., 2022). The importance of the differences in subjective experience with psychedelics is not yet clear.
5-MeO-DMT is orally inactive (Shulgin and Shulgin, 1997) and therefore usually administered parenterally through smoking or inhalation of vapour, or less commonly via intravenous, intramuscular, rectal, sublingual or intranasal applications (Davis et al., 2018; Metzner, 2013; Weil and Davis, 1994).
A recent narrative synthesis of all published pre-clinical and clinical data (Ermakova et al., 2021) concluded that 5-MeO-DMT is a potentially useful addition to the psychedelic pharmacopoeia because of its short duration of action, relative lack of visual effects and putatively higher rates of ego-dissolution and mystical experiences. A rapid onset and short duration of action logically reduce the duration of treatment sessions and, thus, resource utilization.
There have been no large randomized controlled clinical studies with 5-MeO-DMT in humans and only a handful of observational and small studies are reported in the literature (Rucker et al., 2022). This study aimed to investigate 5-MeO-DMT further using a novel formulation of 5-MeO-DMT benzoate powder applied through an unidose intranasal spray device.
This double-blind, placebo-controlled, phase 1, single ascending dose trial in psychedelic-naïve healthy participants is the first to characterize the pharmacokinetic (PK) and PD effects of 5-MeO-DMT benzoate delivered intranasally.
Methods
Trial participants
In total, 44 healthy adult male and female participants aged between 25 and 55 years with a body mass index of 18.0–30.9 kg/m2 were enrolled in the trial. All participants were psychedelic-naïve. Participants were recruited via online advertisements and word of mouth. A summary of the inclusion criteria and exclusion criteria are summarized below.
Participants were required to meet the following criteria for eligibility; normotensive psychedelic-naïve male or female, deemed healthy based on clinical history, physical examination, electrocardiogram (ECG) and laboratory blood tests. They could not consume more than 14 units of alcohol weekly during the study and were required to abstain from alcohol from 24 h before each outpatient visit until the completion of procedures on Day 3. They were also required to abstain from recreational drug use, including psychedelic drugs, until the end of the study and from nicotine while resident in the unit.
Exclusion criteria included the following: acute or chronic illness or infection, history of epilepsy or seizures, history, or family history of malignant hyperthermia and history of serious reaction to any medicine or excipient. Current, or first-degree blood relative with, a serious mental disorder, or history of attempted or completed suicide. Current suicidal ideation as judged by the Columbia-Suicide Severity Rating Scale (C-SSRS) and any major psychiatric disorders as determined by the Mini International Neuropsychiatric Interview. Current, or history of, alcohol or drug abuse or dependence. Nasal obstruction, blockage or symptoms of congestion. Currently taking monoamine oxidase inhibitors. Pregnant or lactating and unwilling to comply with the contraception requirements of the protocol. Positive test for hepatitis B and C or HIV; taken part in another study or lost more than 400 mL blood (e.g. as a blood donor), within the previous 3 months.
Trial design
This was a phase 1, double-blind, randomized and placebo-controlled trial in psychedelic-naïve healthy participants to evaluate the safety, tolerability, PK and PD of single ascending intranasal doses of a novel 5-MeO-DMT benzoate salt dry powder formulation, hereon referred to as BPL-003 (Beckley Psytech Ltd, Oxford, UK). The study design was advised by a collaboration with the Psychoactive Trials Group at King’s College, and conducted at Hammersmith Medicines Research, Park Royal, London, UK.
Participants were divided into seven cohorts of up to seven participants per cohort. In each cohort, four (Cohorts 1–4) or five (Cohorts 5–7) participants received single intranasal doses of BPL-003 (1, 2.5, 4, 6, 8, 10 or 12 mg in Cohorts 1–7, respectively), and up to two participants per cohort received matching placebo (Supplemental Table 1). BPL-003 or placebo was administered using an Aptar Unidose dry powder intranasal spray device. Sentinel dosing was used with two participants in each cohort (one active and one placebo) who were dosed at least 23 h before the remaining participants. After each dose level, a Safety Review Committee reviewed safety, tolerability, PK and PD (Mystical Experience Questionnaire (MEQ-30)) data and the dose was escalated only if deemed acceptable.
Screening and preparation
Participants were screened within 56 days before their dose of the trial drug. All participants had two psychedelic preparatory visits (one online and one in-person) with a trained psychedelic monitor before being dosed (Day 7 and Day 3) to prepare the participant for trial drug administration, provide information about 5-MeO-DMT and to establish rapport between the participant and the monitor.
Dosing visit
Participants were at the site from the day before dosing until the morning after dosing. An indwelling intravenous catheter was inserted before trial drug administration. During the session, a nurse and one psychedelic monitor were present in the room, to provide non-directive support and reassurance for participants. To ensure an optimal setting for the psychedelic session, dosing took place in a decorated room with a prepared playlist of relaxing music playing. To ensure psychological comfort and aid relaxation, participants undertook guided breathing exercises before dosing. At the time of dosing, participants were in a semi-supine position on a hospital bed.
For up to 90 min post-dose, participants and the psychedelic monitor each rated overall subjective drug intensity (SDI). The monitor was aware of the participant’s response, but the participant was not aware of the monitor’s response. Approximately 90 min after dosing, participants had a one-to-one guided interview with an independent researcher to discuss their psychedelic experience. Approximately 2.5 h post-dose, psychometric scales were administered to provide quantitative measures of the participant’s psychedelic experience. All participants had their psychological well-being, including measures of suicidality, assessed by the trial psychiatrist before discharge from the ward.
Psychedelic integration visit
Participants had an integration visit 2 days after dosing to discuss their experience on a one-to-one basis with the psychedelic monitor. The session could be in person or remote if this was thought appropriate by the psychedelic monitor.
Follow-up visit
All participants returned to the ward for a follow-up visit around 7 days after administration of the trial drug.
Lifestyle restrictions
Participants were required to adhere to several lifestyle restrictions before the study, during the study and while awaiting follow-up. These restrictions are listed below.
No food or drink containing poppy seeds was allowed from 2 weeks before dosing until the follow-up visit or containing grapefruit from 7 days before dosing until the final follow-up visit.
No alcoholic drinks were allowed from 24 h before admission until completion of procedures on Day 3, and for 24 h before each outpatient visit.
No recreational drugs were allowed from screening until the final follow-up visit.
No caffeinated drinks were allowed during the period of residence. Participants were advised not to drink more than their usual intake 2 days before ward admission.
No strenuous exercise was allowed from screening until follow-up.
Participants were required to fast (no food or drink, except water) for at least 8 h before each laboratory safety blood test. During the inpatient stay, standard meals and drinks were provided before and at approximately 4, 10 and 24 h after dosing. On Day 1, participants were given a light, low-fat breakfast at least 1 h before dosing.
Compliance with ethical standards
This trial was conducted in accordance with the International Council for Harmonization Good Clinical Practice guidelines and ethical principles that have their origin in the Declaration of Helsinki. The study was approved by the London-Brent Research Ethics Committee, London, UK. Protocols were approved by the Medicines and Healthcare Products Regulatory Agency and an independent recognized NHS research ethics committee before eligibility screening. Written informed consent was obtained from each participant before any trial-related procedures were performed.
Trial drug
BPL-003 or a matching placebo was administered as a single intranasal spray into one nostril by a trained member of the research team (usually a registered nurse), in the presence of a psychedelic monitor, using the Aptar Unidose dry powder delivery device.
Safety assessments
Safety and tolerability were evaluated by recording the incidence and severity of treatment-emergent adverse events (TEAEs) throughout the trial, review of clinical laboratory tests, vital signs (blood pressure (BP), heart rate (HR) and temperature), ECGs, physical examinations and C-SSRS questionnaire responses (Oquendo et al., 2003). The C-SSRS was administered and assessed at screening, Day 1, Day 2 and Day 8. A nasal examination was performed pre- and post-dose to determine whether there were any nasal site reactions following study drug administration.
Post-traumatic stress disorder (PTSD) can be caused by a traumatic experience and was assessed at follow-up via the Diagnostic and Statistical Manual of Mental Disorders 5th edition (DSM-5) post-traumatic stress disorder checklist (PCL-5) (Weathers et al., 2013). The PCL-5 is a 20-item self-report measure that assesses the 20 DSM-5 symptoms of PTSD. Participants rate each item from 0 (not at all) to 4 (extremely) to indicate the degree to which they have been affected by that symptom over the past month.
Hallucinogen-persisting perception disorder (HPPD) is a rare clinical condition in which participants who have had previous exposure to a hallucinogenic substance may experience perceptual distortions after cessation of the initial substance use. The persistence of distortions in visual perception in participants after receiving BPL-003 was assessed using the HPPD algorithm, which comprises a series of yes or no questions derived from the standard criteria for HPPD, completed by the study psychiatrist or trained delegate at follow-up.
PK assessments
Plasma and urine 5-MeO-DMT and bufotenine concentrations were quantified with a validated liquid chromatography-tandem mass spectrometry (LC-MS/MS) assay, conducted by Analytical Services International (London, UK). References and internal standards (psilocybin D10) for 5-MeO-DMT and bufotenine bioanalysis were supplied by Chiron or Cerilliant. LC-MS/MS was performed using an Agilent 1290 Infinity IITM with Sciex API 4000TM mass spectrometer. These were used to quantify concentrations of 5-MeO-DMT and bufotenine using 2 μL injections from a 100 μL sample. The limits of quantification for 5-MeO-DMT and bufotenine were 0.5–3.000 ng/mL and calibrated over the range of 0.5–500 ng/mL. Detailed methodology is provided in the Supplemental Materials.
Blood samples were taken pre-dose (dosing day) at 0.5, 2, 4, 6, 10 and 16 min, and 0.5, 1, 1.5, 4 and 24-h post-dose. Urine samples were collected continuously from 0 to 6 h post-dose.
Plasma PK parameters included maximum plasma concentration (Cmax), time to reach Cmax (tmax), area under the plasma concentration–time curve (AUC) from time 0 to time of the last quantifiable concentration (AUClast), AUC extrapolated to infinity (AUCinf) and terminal elimination half-life (t½) for both analytes, plus clearance (CL/F) for 5-MeO-DMT only.
Urine PK parameters included the amount of unchanged drug excreted in urine (Ae), the fraction of administered drug excreted unchanged in urine (fe) and renal clearance of the drug from plasma (CLR), as well as metabolite Ae and CLR.
Pharmacodynamic assessments
Subjective drug intensity
The intensity of BPL-003 subjective effects was rated by the participant and psychedelic monitor using the SDI (Madsen et al., 2019), a Likert scale of 0–10, where 0 was ‘definitely no effect’ and 10 was ‘the strongest effect imaginable for 5-MeO-DMT’. The assessment was performed every 2 min for up to 90 min post-dose, if the trial participants were not responsive, the psychedelic monitor would assess the rating to be 10, as the inability to process or respond to verbal cues was regarded as the maximum subjective intensity. Participants were informed about this prior to dosing.
Mystical Experiences Questionnaire
The MEQ-30 is a 30-item questionnaire to evaluate mystical experiences, with subdomains to measure mystical, positive mood, transcendence and ineffability factors (Barrett et al., 2015). A ‘complete mystical experience’ was defined as ⩾60% of the maximum possible score on each of the four subscales of the MEQ-30. Participants rated the degree to which they experienced each of the 30 phenomena using the following scale: 0 (none; not at all), 1 (so slight cannot decide), 2 (slight), 3 (moderate), 4 (strong (equivalent in degree to any other strong experience)) or 5 (extreme (more than any other time in my life and stronger than 4)). Means were calculated for each subdomain and a total overall score. The percentage of participants experiencing a ‘complete mystical experience’ in each dose cohort was assessed by calculating the number of participants that scored 3 and above (⩾60% of the attainable value) in all four subdomains of the MEQ-30.
Ego Dissolution Inventory
The Ego Dissolution Inventory (EDI) comprises of eight statements (Nour et al., 2016). Participants rated their agreement to each statement by marking on a visual analogue scale from 0 (no, not more than usual) to 100 (yes, entirely or completely). Means were calculated for each statement and a total overall score.
Subjective dose acceptability
Subjective dose acceptability was determined at follow-up by asking participants whether they would refuse to be re-exposed to the same or a higher dose of BPL-003.
Subjective experience data via qualitative interview
A description of the BPL-003 subjective experience data was gained from a qualitative interview utilizing the ‘micro-phenomenology’ interview technique (Petitmengin, 2006). Participants were asked to take part in this optional qualitative interview. If they agreed, the interview commenced on cessation of the psychedelic experience and before any post-dose questionnaires were completed. This was done either face to face on the ward or via video call (the results of this will be reported separately).
Challenging experience questionnaire
The challenging experience questionnaire (CEQ) has been used to characterize challenging experiences with psilocybin (Barrett et al., 2016) and was used in this trial to characterize and quantify any potentially challenging experiences with BPL-003. The questionnaire is grouped into 7 factors with 26 questions rated 0 (none/not at all) to 5 (extreme/more than ever before). Mean percentage scores were calculated for each factor and the total overall percentage score.
Statistical analysis
This trial was exploratory, with no hypotheses tested. No formal sample size determination was made. Summary statistics (arithmetic mean, standard deviation, geometric mean, minimum and maximum) were produced for plasma and urine concentrations of 5-MeO-DMT and bufotenine at each timepoint available. Actual times were used to derive PK parameters, and missing data were not imputed.
Results
Baseline characteristics
In all, 44 participants enrolled in the trial, of which 13 received a placebo and 31 received a single dose of BPL-003 (Supplemental Figure 1). All 31 participants (100%) who received a dose of BPL-003 were included in the PK and PD populations. Participant demographic characteristics are summarized in Supplemental Table 1; most participants were male (73%) and white (59%).
Safety and tolerability
Overall, 21 participants (47.7%) had TEAEs; 19 participants (61.3%) received BPL-003 and 2 participants (15.4%) received a placebo. The incidence of TEAEs in participants who received BPL-003 did not appear to correlate with the dose. Most TEAEs, 34 out of 38 (89.5%), were mild in severity; 4 out of 38 (10.5%) were moderate in severity. There were no severe or serious TEAEs, or any TEAEs leading to withdrawal from the trial.
All TEAEs and the number of times each event occurred are shown in Table 1. The most frequently reported TEAEs were nasal discomfort nausea and headache.
Summary of treatment-emergent adverse events.
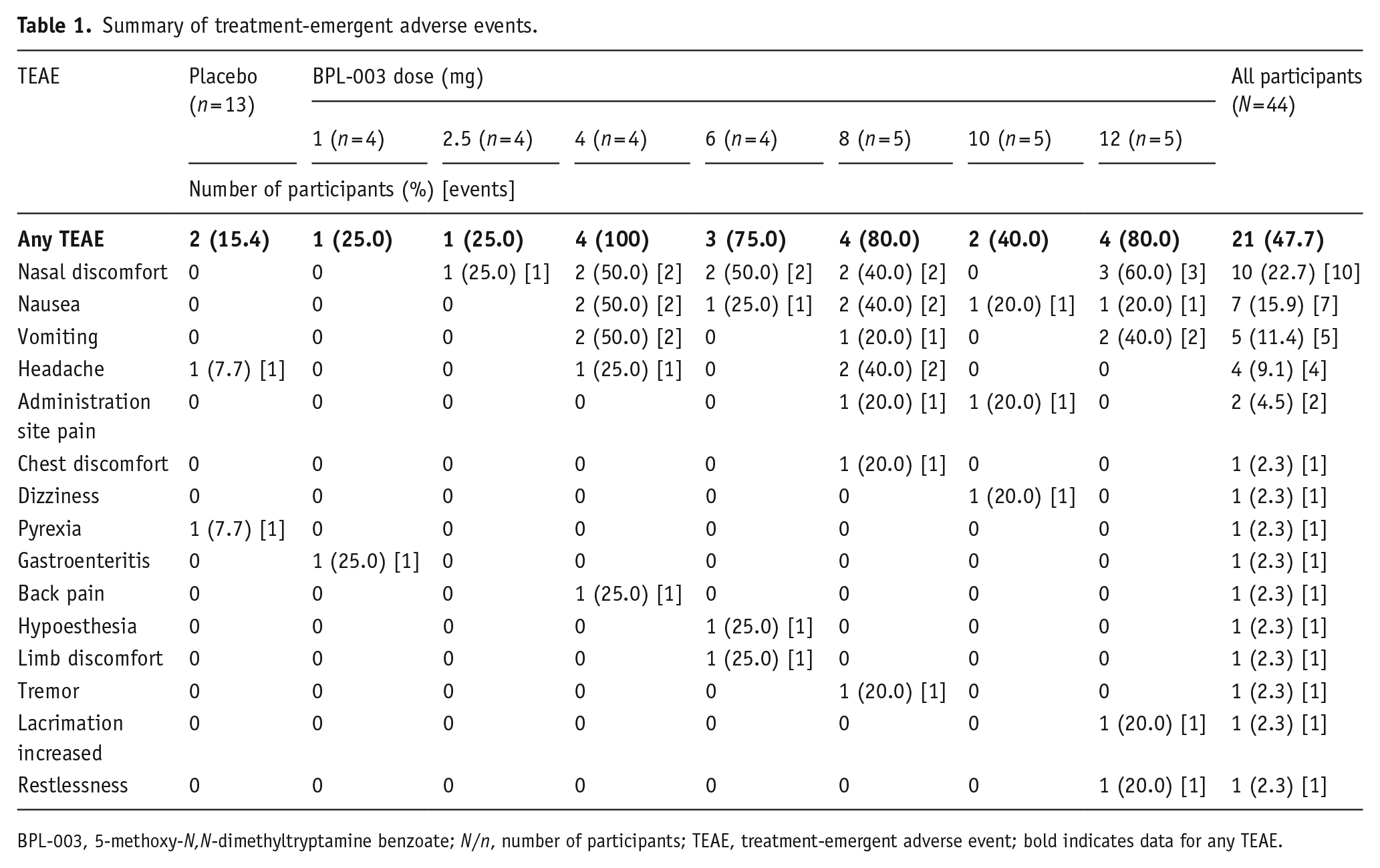
BPL-003, 5-methoxy-N,N-dimethyltryptamine benzoate; N/n, number of participants; TEAE, treatment-emergent adverse event; bold indicates data for any TEAE.
There were no clinically significant findings for laboratory parameters, vital signs, ECGs or physical examinations. There were transient increases in BP and HR which began soon after BPL-003 treatment but recovered within the 90-min observation period without intervention. None were considered clinically significant or assessed as AEs by the investigator.
No post-dose HR measurements outside of the reference range (40–100 beats per minute (bpm)) were recorded in a placebo participant. Two or more post-dose HR readings above the reference range (>100 bpm) were recorded in seven (22.6%) participants dosed with BPL-003 (range 101–166 bpm). Post-dose increases in systolic BP above the reference range were recorded in 3 (23.1%) placebo participants (ranging 141–155 mmHg) and 15 (48.4%) BPL-003-treated participants (ranging 141–181 mmHg). Post-dose diastolic BP readings above the reference range were recorded in 0 (0%) placebo participants and 17 (54.8%) BPL-003 participants (ranging 91–113 mmHg). Systolic BP over time is shown in Table 2 and the peak post-dose systolic BP in Table 3; diastolic BP and HR data are listed in Supplemental Table 2. There were transient increases in systolic BP >160 mmHg in five participants (Table 3).
Mean systolic blood pressure over time.
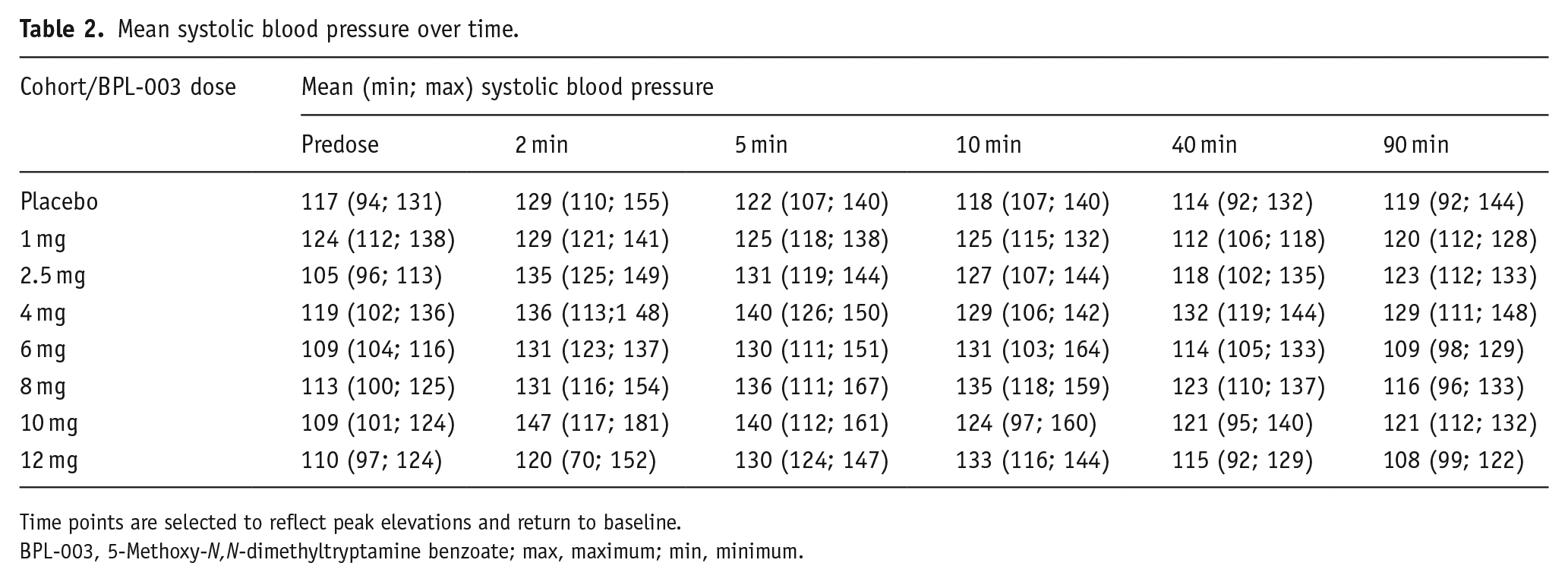
Time points are selected to reflect peak elevations and return to baseline.
BPL-003, 5-Methoxy-N,N-dimethyltryptamine benzoate; max, maximum; min, minimum.
Post-dose peak systolic blood pressure.
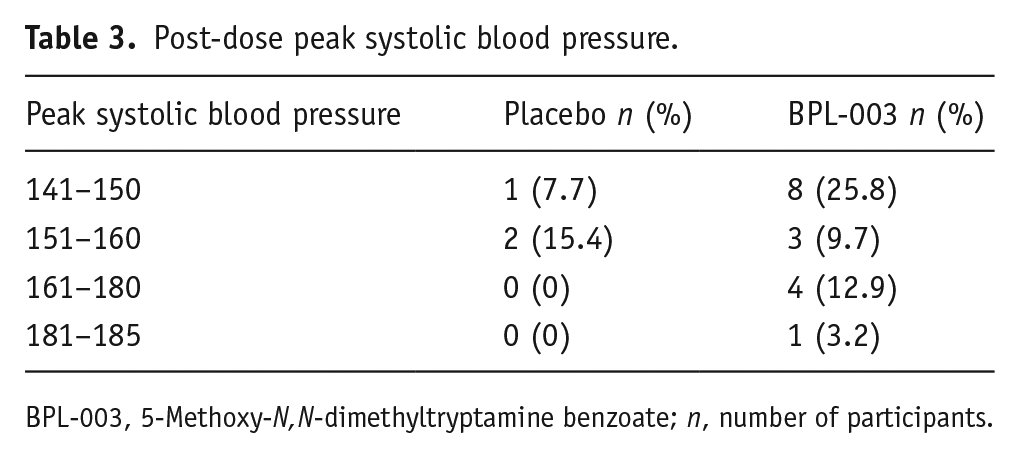
BPL-003, 5-Methoxy-N,N-dimethyltryptamine benzoate; n, number of participants.
No participant reported any suicide-related thoughts or behaviours. No HPPD (clinical assessment) or PTSD symptoms (PCL-5 scale) were evident at follow-up for any participant treated with a placebo or BPL-003.
Pharmacokinetics
Plasma 5-MeO-DMT PK
The toxicokinetic threshold was determined from intranasal 5-MeO-DMT in animal toxicology studies (rat and dog), administered daily for 14 days followed by a 14-day recovery period. Based on the parameters examined, the reported no observed AE level was determined to be 1.5 mg/kg/day, corresponding to a Cmax of 421 ng/mL, and AUC0-Tlast (AUCINF obs) of 213 (220) h × ng/mL (combined for both sexes).
Mean plasma 5-MeO DMT concentration–time curves are presented in Figure 1 and PK parameters are summarized in Table 4.
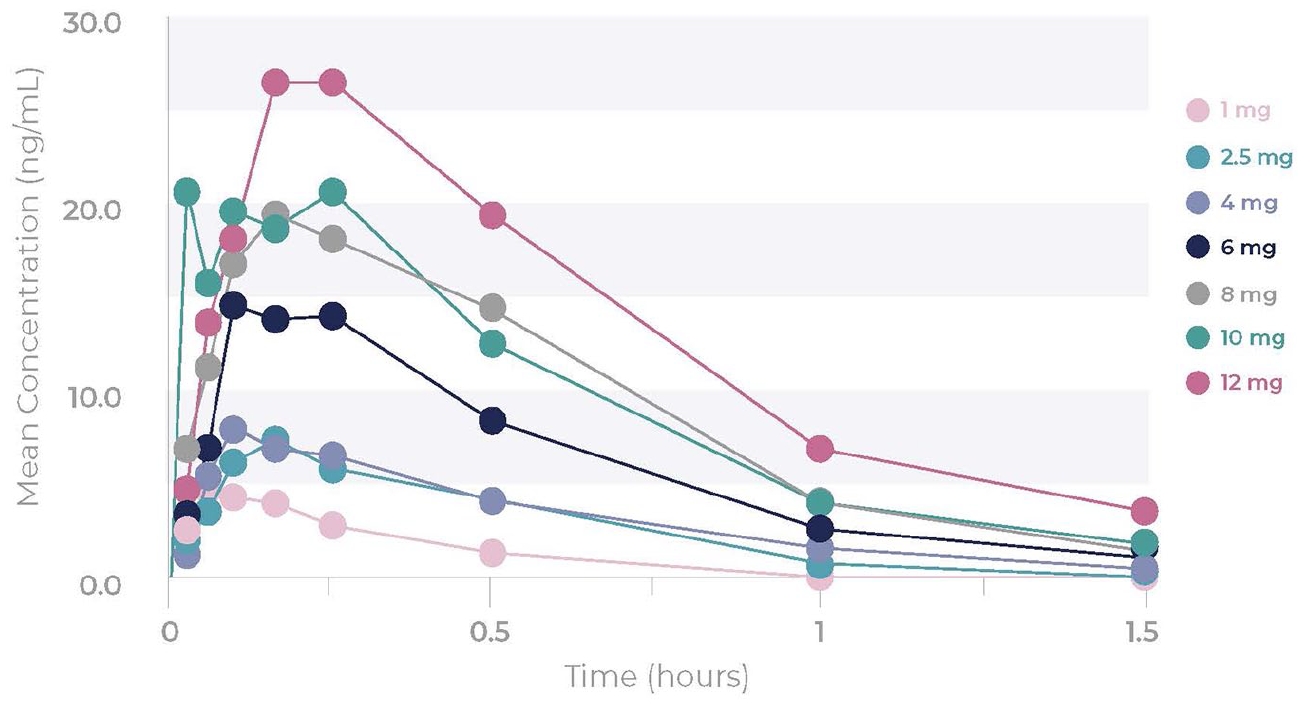
Mean plasma concentration–time curves of 5-MeO-DMT. Plasma BLQ concentration values were imputed as zero and included as such in the calculation of means.
Summary of 5-MeO-DMT PK parameters.
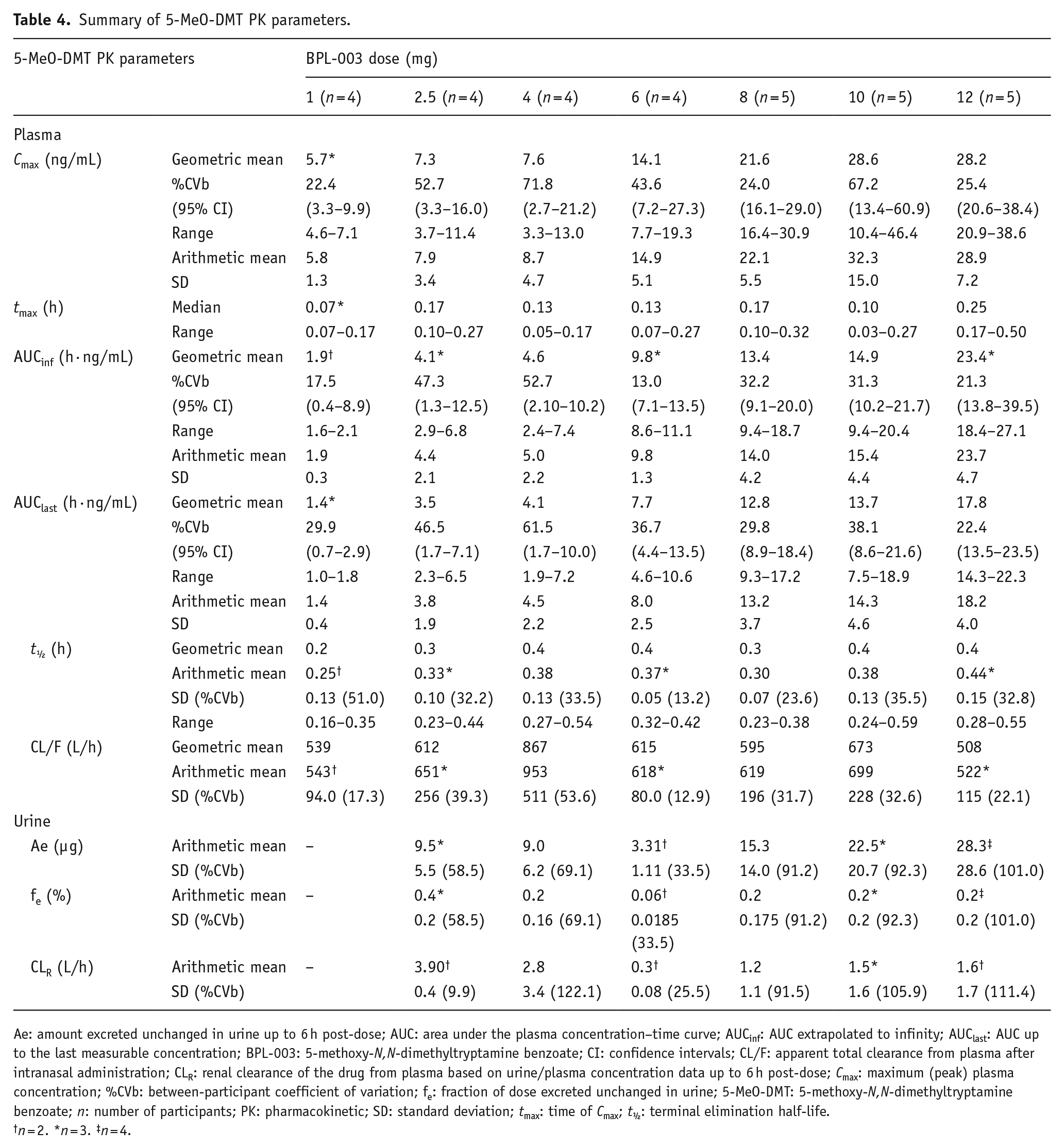
Ae: amount excreted unchanged in urine up to 6 h post-dose; AUC: area under the plasma concentration–time curve; AUCinf: AUC extrapolated to infinity; AUClast: AUC up to the last measurable concentration; BPL-003: 5-methoxy-N,N-dimethyltryptamine benzoate; CI: confidence intervals; CL/F: apparent total clearance from plasma after intranasal administration; CLR: renal clearance of the drug from plasma based on urine/plasma concentration data up to 6 h post-dose; Cmax: maximum (peak) plasma concentration; %CVb: between-participant coefficient of variation; fe: fraction of dose excreted unchanged in urine; 5-MeO-DMT: 5-methoxy-N,N-dimethyltryptamine benzoate; n: number of participants; PK: pharmacokinetic; SD: standard deviation; tmax: time of Cmax; t½: terminal elimination half-life.
n = 2. *n = 3. ‡n = 4.
BPL-003 was rapidly absorbed, with a median tmax of 4–15 min post-dose across all dose levels. Elimination of 5-MeO-DMT was rapid across all dose levels. Arithmetic mean t½ ranged from 15 to 27 min across dose levels. Plasma 5-MeO-DMT concentrations were below the limit of detection by 4 h after administration in all participants. All participants who received BPL-003 had quantifiable 5-MeO-DMT plasma concentrations.
Systemic exposure to 5-MeO-DMT (Cmax and AUC parameters) generally increased with BPL-003 dose. The geometric mean Cmax was 5.7 ng/mL after 1 mg BPL-003 and 28.2 ng/mL after 12 mg. Arithmetic mean CL/F was 522–953 L/h across all dose levels. There did not appear to be a correlation between the BPL-003 dose and CL/F. No dose exceeded the pre-defined toxicokinetic thresholds of 421 ng/mL for Cmax and 213 h∙ng/mL for AUCinf.
Plasma bufotenine PK
All bufotenine plasma concentrations were below the limit of quantification (BLQ) (<0.5 ng/mL) except in one sample for one participant.
Urine 5-MeO-DMT PK
Urinary PK parameters for 5-MeO-DMT are summarized in Table 4.
Urine excretion of 5-MeO-DMT was negligible. The arithmetic mean fraction of 5-MeO-DMT excreted unchanged (fe) at 6 h post-dose ranged between 0.06% and 0.38% across dose levels. Due to the low levels of 5-MeO-DMT detected in urine, PK parameters derived were highly variable.
Urine bufotenine PK
Urine levels of bufotenine were all BLQ (<0.5 ng/mL).
Pharmacodynamics
Effects by SDI
Overall, both monitor and participant SDI ratings increased with the BPL-003 dose, indicating a stronger subjective experience of BPL-003 with increasing dose. SDI onset was rapid, generally within 2–10 min and short-lasting, generally resolving completely within 45–90 min. The mean subjective intensity ratings of participants in the placebo and the highest three BPL-003 doses are shown in Figure 2. The time course of the SDI ratings was similar to that of the plasma 5-MeO-DMT concentrations (Figures 1 and 2). The time of peak SDI rating occurred at approximately tmax for plasma 5-MeO-DMT. Furthermore, the SDI ratings declined as plasma 5-MeO-DMT concentrations declined. These results indicated an association between systemic exposure to BPL-003 and the intensity of the subjective experience.
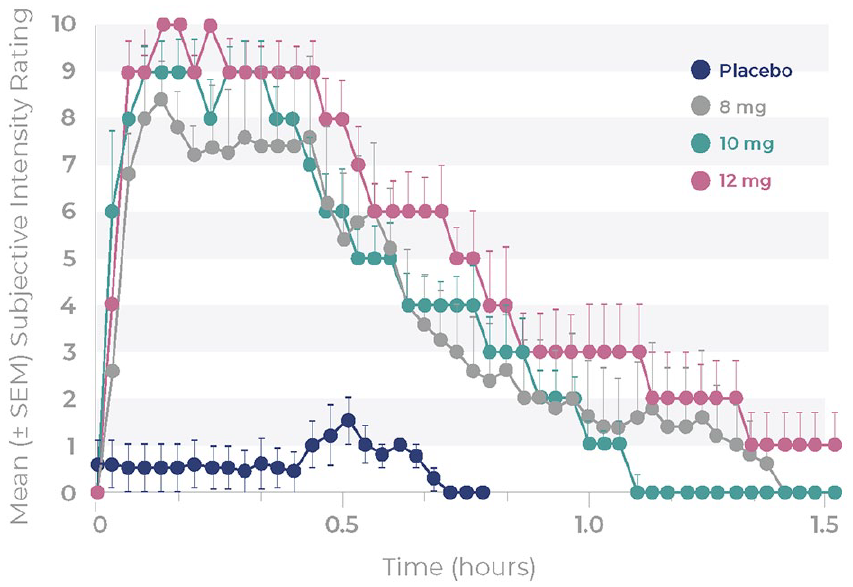
Mean (±SEM) subjective drug intensity ratings after single doses of placebo or 8, 10 and 12 mg BPL-003.
Effects by MEQ
The MEQ-30 subdomain and total scores increased with the BPL-003 dose, with the highest total score of 3.7 at the maximum (12 mg) BPL-003 dose level and the lowest total score of 0.6 with placebo (Figure 3 and Table 5). A complete mystical experience, defined by reaching or exceeding a score of 3 on all four sub-domains of the scale, was reported in three out of five participants (60%) at both the 10 and 12 mg doses; for lower doses, a maximum of one participant had a complete mystical experience (⩽25%).
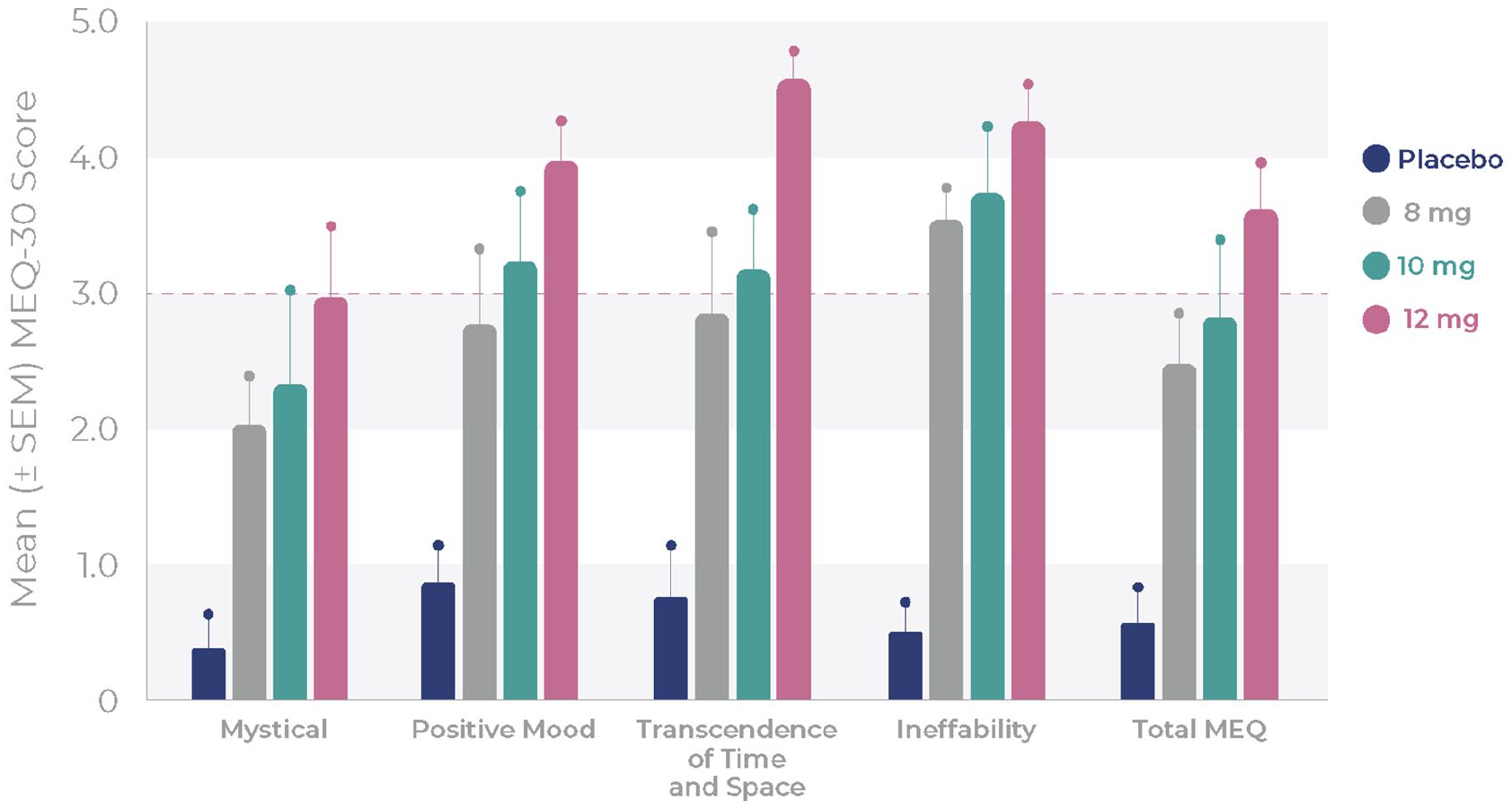
Mean (±SEM) MEQ-30 scores after single doses of placebo or 8, 10 and 12 mg BPL-003.
Summary of EDI and MEQ-30 scores.
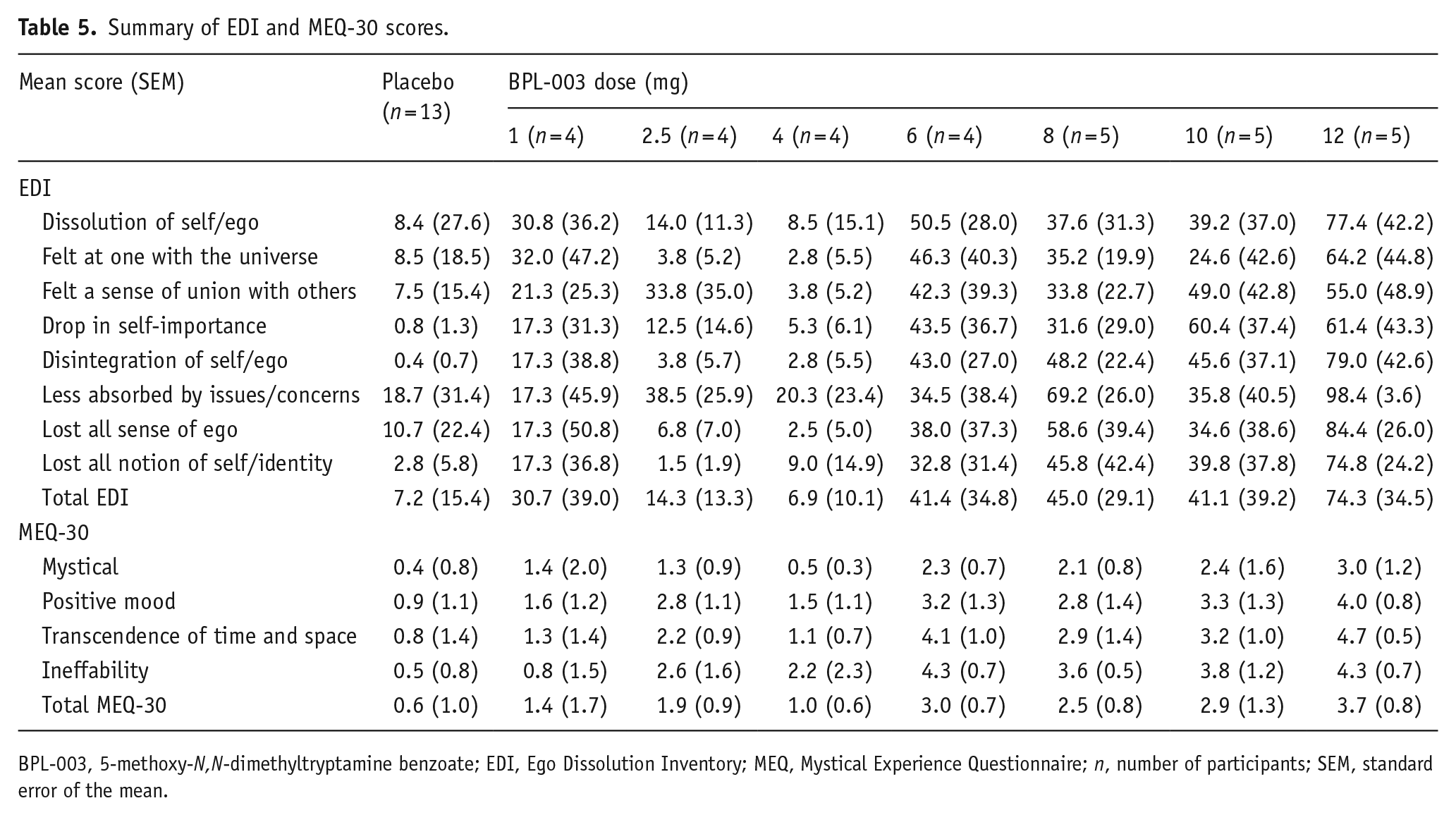
BPL-003, 5-methoxy-N,N-dimethyltryptamine benzoate; EDI, Ego Dissolution Inventory; MEQ, Mystical Experience Questionnaire; n, number of participants; SEM, standard error of the mean.
Effects by EDI
The mean EDI subdomain and total scores tended to increase with dose (Figure 4 and Table 5). The mean total EDI score was 7.2 for placebo (n = 13), 45.0 for 8 mg (n = 5), 41.1 for 10 mg (n = 5) and 74.3 for 12 mg BPL-003 (n = 5).
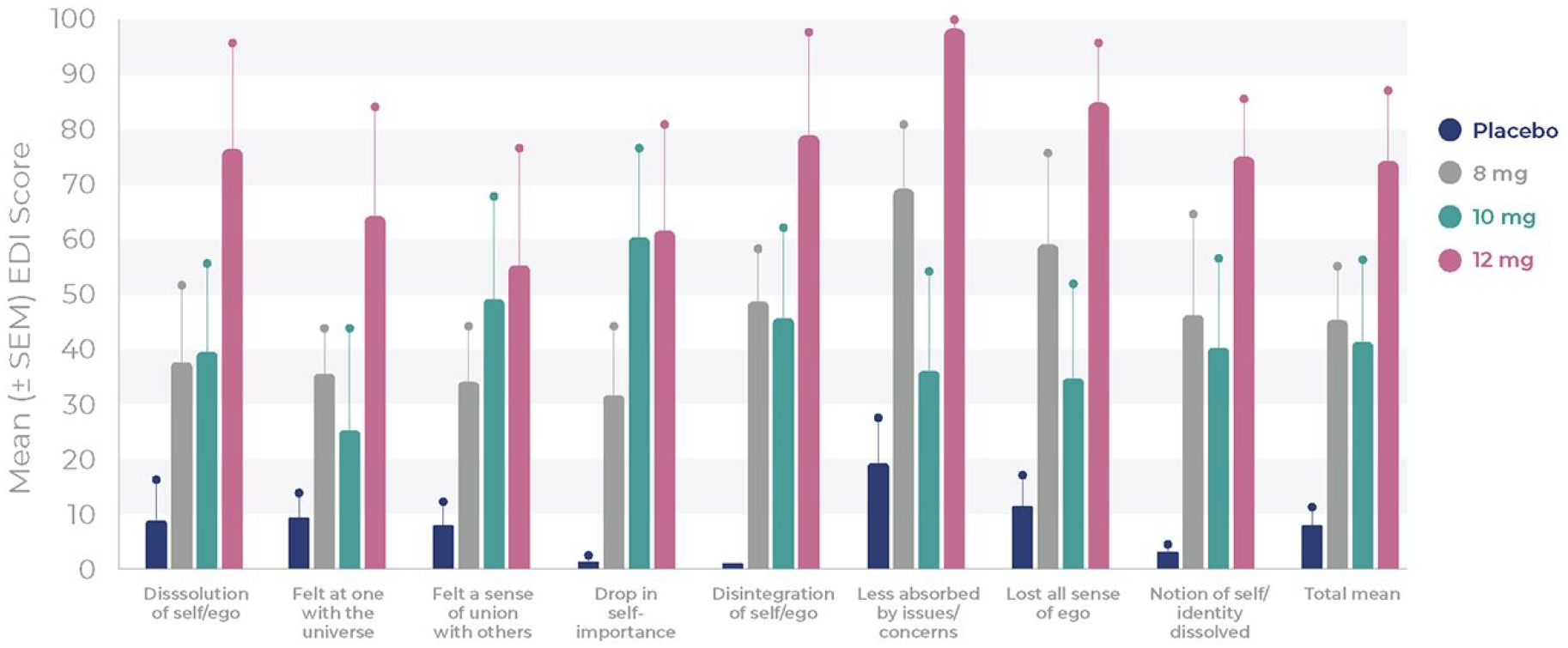
Mean (±SEM) EDI scores after single doses of placebo or 8, 10 and 12 mg BPL-003.
Effects by CEQ
CEQ scores were generally less than 40% across the seven subdomains, with total CEQ scores ranging from 20% to 35% for BPL-003 of 4–12 mg (Table 6).
Summary of CEQ scores.
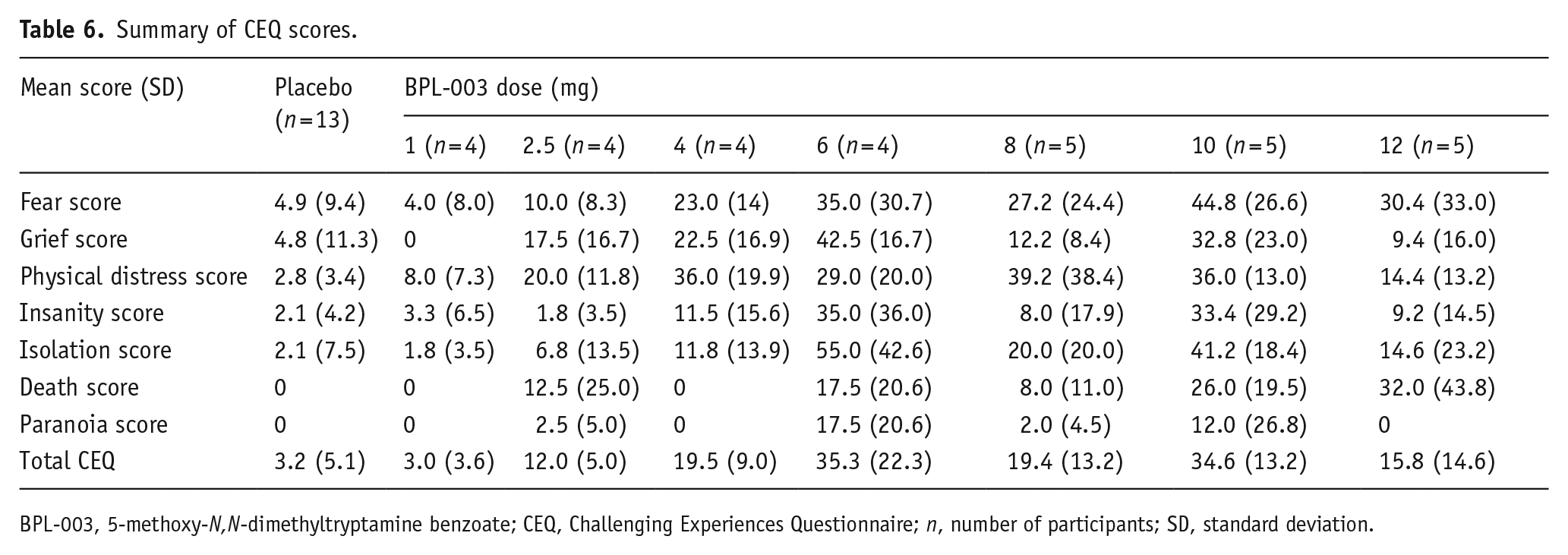
BPL-003, 5-methoxy-N,N-dimethyltryptamine benzoate; CEQ, Challenging Experiences Questionnaire; n, number of participants; SD, standard deviation.
BPL-003 subjective dose acceptability
In all, 27 (87%) participants who received BPL-003 stated they would accept the same or higher dose. However, acceptability was not dose related; all participants who received the highest dose would accept the same or a higher dose. Four participants stated they would not want to receive the same or higher dose of BPL-003 (one participant that received 6 mg, two that received 8 mg and one that received 10 mg) for varied reasons. One participant re-experienced painful memories and although they gained insights from this, they would not want to repeat the experience. The other three participants stated that their experience was unpleasant, or they did not feel they had personally meaningful experiences. AEs were reported in three of the four participants who would not want to receive the same or a higher dose.
BPL-003 subjective experiences by qualitative interview
The common psychedelic experiences that were reported during the qualitative interviews highlighted the dynamic temporal progression of the subjective effects. Generally, participants described a rapid onset of the psychedelic effects, followed by inwards focused attention, altered sense of time, intense emotions, various degrees of fear or discomfort, as well as a psychological struggle to ‘let go and surrender’ to the experience. If participants were able to relax into the experience and ‘let go’, the sensation of floating in space, an ocean or void often followed, with feelings of peacefulness, calmness, relaxation or bliss. At the higher doses (10 and 12 mg BPL-003), most participants appeared to experience a complete mystical experience. Gradually external awareness returned, and participants stated this allowed them to reflect on their experiences, often recounting meaningful insights.
Many participants emphasized the importance of the psychological preparation, and the rapport and trust they built with the psychedelic monitors which they said helped participants to feel safer during the experience.
Discussion
Several psychedelic compounds are undergoing clinical trials to assess their utility for various neuropsychiatric indications, and some have shown encouraging efficacy signals in larger controlled clinical trials (Goodwin et al., 2022). The compounds differ not only in their receptor occupancy profiles but also in their PK and PD profiles and subjective effects. There is a wealth of evidence that implicates the five HT1A receptors in the pathophysiology of depression (Kaufman et al., 2016; Yohn et al., 2017) and it is a therapeutic target for many antidepressants (Smith et al., 2023). 5-MeO-DMT has a unique profile that, in addition to the activation of the 5-HT2A receptor, also shows a preferential receptor affinity to the 5-HT1A receptor.
The mode of administration of psychedelic compounds influences the variability and absorption characteristics, and for parenteral applications, the intranasal delivery route seems to be a promising approach (Daly et al., 2019). This study investigated a proprietary formulation of synthetic 5-MeO-DMT benzoate salt given as a dry powder using an intranasal Unidose spray device. Single intranasal doses of 1–12 mg BPL-003 were found to be safe and well tolerated in the healthy psychedelic-naïve participants of this trial. Results demonstrated a dose-proportional increase in exposure levels, a rapid absorption with a Tmax of around 10 min, and a short t½ of around 20 min, which corresponded with profound and short-lasting consciousness-altering effects of 45–90 min in duration.
Most TEAEs were mild in severity (89.5%) and resolved within minutes after BPL-003 exposure, and all other TEAEs were moderate in severity (10.5%). There were no serious adverse events (SAEs), otherwise significant TEAEs, or TEAEs leading to participant withdrawal during the trial. The most frequently reported TEAEs (⩾5%) were nasal discomfort, nausea, vomiting and headache. Generally, less AEs were observed in this study when compared to psychedelic use in uncontrolled circumstances (Barrett et al., 2016; Breeksema et al., 2022; Johnson et al., 2008), reinforcing 5-MeO-DMT administration is safer in a clinical setting.
There were no clinically significant laboratory, vital signs, ECG or physical examination findings. Results from the C-SSRS showed participants experienced no increase in suicidal thoughts, intentions or behaviour. Additionally, there were no significant findings on the assessment of HPPD and PTSD at 1 week, although no further follow-up assessments were conducted after this. Transient increases in HR and BP were observed with BPL-003 treatment; however, these resolved rapidly without treatment. This is in line with literature reports of other short-lasting psychedelics, for example, trials with dimethyltryptamine in healthy volunteers (Vogt et al., 2023). The greatest increase in HR and BP co-occurred with arm and body movements that may also have affected the measurements, in addition to the pharmacological effects of the drug, and emotional response to the psychedelic experience.
One of the objectives of this study was to identify and explore a well-tolerated dose of BPL-003 that would reliably elicit mystical experiences in most trial participants, to evaluate whether progression into patient studies was appropriate. A dose range of 10–12 mg was found to meet this requirement, with 60% of participants experiencing a complete mystical experience and/or ego dissolution events. The elicitation of full mystical experiences may be an indicator of future therapeutic efficacy. One study examining the inhalation of 5-MeO-DMT in a naturalistic group setting of 362 participants found spontaneous and unintended improvements in self-reported depression and anxiety (Davis et al., 2019). Participants who reported an improvement in their depression or anxiety had significantly higher MEQ-30 scores during their first 5-MeO-DMT session compared to those reporting no improvements in their depression. Similar findings were reported in a study of 42 participants given a single administration of vapour from toad secretion containing 5-MeO-DMT (Uthaug et al., 2019). Assessments were made at baseline, 24 h and 4 weeks post-inhalation and results found significant improvements in ratings of depression, anxiety and stress at 4 weeks. Participants who experienced high levels of ego dissolution or oceanic boundlessness during the session displayed higher ratings of satisfaction with life and lower ratings of depression and stress. This study did not have a control group and the dose range of toad secretion ranged from 20 to 120 mg. Here, we found a variability in participants’ response, with some having intense psychedelic effects at 8 mg BPL-003, and others not achieving a full mystical experience at a dose of 12 mg. This contrasts with the results of a phase 1 study assessing the impact of four different dose levels of 5-MeO-DMT administered via inhalation as single doses of 2, 6, 12 and 18 mg (Reckweg et al., 2021). Results found all doses elicited significantly higher MEQ-30 ratings compared to the lowest dose of 2 mg. However, eight participants reported a peak experience at varying dose levels, possibly suggesting that the 5-MeO-DMT dose needed to achieve a peak experience varies considerably between individuals, in line with findings from this study.
Since this trial was undertaken, a phase 1/2 trial assessing the safety and efficacy of inhaled 5-MeO-DMT in 16 patients with treatment-resistant depression (TRD) has been published (Reckweg et al., 2023). Phase 1 (n = 8) investigated two single doses of 5-MeO-DMT: 12 and 18 mg, with a primary endpoint of safety. Phase 2 (n = 8) investigated an individualized dosing regimen with up to three increasing doses of 5-MeO-DMT: 6, 12 and 18 mg within a single day, with a primary endpoint of efficacy, assessed by the proportion of patients in remission (MADRS ⩽10) on Day 7. Results showed the proportion of patients in remission at Day 7 was 50% and 25% in the 12 and 18 mg groups of phase 1, respectively, and 87.5% in the individualized dosing regimen of phase 2.
We observed a clear relationship between plasma 5-MeO-DMT concentration and the reported SDI. The onset, length and intensity of the psychedelic experience after administration of 8, 10 and 12 mg BPL-003 followed the plasma concentration closely (Figures 1 and 2), indicating a close relationship between PD and PK measures. Higher doses of BPL-003 (8, 10 and 12 mg) showed an increase in the MEQ-30 and EDI, without untoward negative effects as measured by scales that were implemented to monitor for challenging experiences, post-traumatic events and the willingness to be re-exposed to the drug. Qualitatively, there appeared to be some differences in the activation of the MEQ-30 subdomains when the 5-MeO-DMT dose was increased, with ineffability, then transcendence of time, then positive mood and finally mystical subdomains being engaged to a greater degree at the higher dose/exposure levels. Some participants did experience challenging effects, particularly during the first 10–15 min after dosing. Qualitatively, at the onset, most participants felt some degree of fear and anxiety and a number of participants felt overwhelmingly intense emotional experiences.
There have been reports of SAEs from the use of serotonergic hallucinogens in the literature. Hallucinogen-induced psychosis was reported in a 3-year longitudinal study of early-phase psychosis and substance use comorbidity (Caton et al., 2005) and suicidal ideation or behaviour or self-injury was reported in phase 2 investigating psilocybin in TRD (Goodwin et al., 2022). Therefore, careful consideration is needed when progressing to trials using clinically depressed patients, to ensure appropriate eligibility and medical monitoring to mitigate these risks. In this study, BPL-003 was found to be well tolerated, safe and no SAEs were reported.
Our study has some strengths. All participants enrolled were evaluated and no participants were lost to follow-up, resulting in a complete data set for analysis. None of the participants had experience with using psychedelics prior to the study; thus, problems associated with de novo psychedelic experiences should be highlighted, while pre-existing positive bias towards psychedelics minimized.
Limitations are, firstly, that this study was conducted in a highly controlled hospital-like setting with healthy volunteers; participants in different environments and patients with psychiatric disorders may respond differently to 5-MeO-DMT. Secondly, functional unblinding is inevitable in studies of drugs with a strong psychoactive effect and undoubtedly occurred here. Subjectively determined observations, in particular, may be exaggerated (both positively and negatively) by raters being aware of allocation.
Conclusion
The results of this phase 1 clinical trial of BPL-003 demonstrated that single intranasal doses of up to 12 mg 5-MeO-DMT were safe and well tolerated, with predictable PK. Psychedelic effects increased with BPL-003 dose, with a rapid onset of profound psychedelic effects that were short (45–90 min) in duration. The results of this single ascending dose study indicate that BPL-003 doses between 8 and 12 mg might be suitable for further pharmaceutical development in neuropsychiatric conditions with high unmet medical needs.
Supplemental Material
sj-docx-1-jop-10.1177_02698811241246857 – Supplemental material for Phase 1, placebo-controlled, single ascending dose trial to evaluate the safety, pharmacokinetics and effect on altered states of consciousness of intranasal BPL-003 (5-methoxy-N,N-dimethyltryptamine benzoate) in healthy participants
Supplemental material, sj-docx-1-jop-10.1177_02698811241246857 for Phase 1, placebo-controlled, single ascending dose trial to evaluate the safety, pharmacokinetics and effect on altered states of consciousness of intranasal BPL-003 (5-methoxy-N,N-dimethyltryptamine benzoate) in healthy participants by James Jonathan Rucker, Claire Roberts, Mathieu Seynaeve, Allan H. Young, Ben Suttle, Takahiro Yamamoto, Anna O. Ermakova, Fiona Dunbar and Frank Wiegand in Journal of Psychopharmacology
Supplemental Material
sj-docx-2-jop-10.1177_02698811241246857 – Supplemental material for Phase 1, placebo-controlled, single ascending dose trial to evaluate the safety, pharmacokinetics and effect on altered states of consciousness of intranasal BPL-003 (5-methoxy-N,N-dimethyltryptamine benzoate) in healthy participants
Supplemental material, sj-docx-2-jop-10.1177_02698811241246857 for Phase 1, placebo-controlled, single ascending dose trial to evaluate the safety, pharmacokinetics and effect on altered states of consciousness of intranasal BPL-003 (5-methoxy-N,N-dimethyltryptamine benzoate) in healthy participants by James Jonathan Rucker, Claire Roberts, Mathieu Seynaeve, Allan H. Young, Ben Suttle, Takahiro Yamamoto, Anna O. Ermakova, Fiona Dunbar and Frank Wiegand in Journal of Psychopharmacology
Supplemental Material
sj-docx-3-jop-10.1177_02698811241246857 – Supplemental material for Phase 1, placebo-controlled, single ascending dose trial to evaluate the safety, pharmacokinetics and effect on altered states of consciousness of intranasal BPL-003 (5-methoxy-N,N-dimethyltryptamine benzoate) in healthy participants
Supplemental material, sj-docx-3-jop-10.1177_02698811241246857 for Phase 1, placebo-controlled, single ascending dose trial to evaluate the safety, pharmacokinetics and effect on altered states of consciousness of intranasal BPL-003 (5-methoxy-N,N-dimethyltryptamine benzoate) in healthy participants by James Jonathan Rucker, Claire Roberts, Mathieu Seynaeve, Allan H. Young, Ben Suttle, Takahiro Yamamoto, Anna O. Ermakova, Fiona Dunbar and Frank Wiegand in Journal of Psychopharmacology
Footnotes
Acknowledgements
The authors would like to thank the study participants and acknowledge the contribution of the study team (including the BPL Clinical Project Manager Ian Macleod) and site team members, including the psychedelic monitors and the medical writers at Alchemy Medical Writing. This work presents independent research part-funded by the National Institute for Health Research (NIHR) Biomedical Research Centre at South London and Maudsley NHS Foundation Trust and King’s College London. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health. No award/grant number is applicable. For open access, the author has applied a Creative Commons Attribution (CC BY) licence to any Accepted Author Manuscript version arising from this submission.
Data availability statement
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship and/or publication of this article: C.R. is an employee of Beckley Psytech Ltd. M.S. is currently an employee of Beckley Psytech Ltd but was employed by King’s College London (until July 2022) and is currently completing a PhD with King’s College London. F.W. was an employee of Beckley Psytech Ltd when this study was undertaken. F.D. is a medical advisor to Beckley Psytech Ltd. A.O.E. is a consultant to Beckley Psytech Ltd. J.J.R. has received payments for advisory boards for Clerkenwell Health (Past), Beckley Psytech Ltd (Past), Delica Therapeutics (Past) and articles for Janssen. He has received financial assistance for attendance at conferences from Compass Pathways (past) and Janssen. He has been awarded grant funding (received and managed by King’s College London) from Compass Pathfinder, Beckley Psytech Ltd, Multidisciplinary Association for Psychedelic Studies, National Institute for Health Research, Wellcome Trust, Biomedical Research Centre at the South London and Maudsley NHS Foundation Trust. He has no shareholdings in pharmaceutical companies and no shareholdings in companies developing psychedelics. A.H.Y. is employed by King’s College London and he is an Honorary Consultant of South London and Maudsley NHS Foundation Trust (NHS UK). His independent research is funded by the National Institute for Health and Care Research (NIHR) Maudsley Biomedical Research Centre in South London and Maudsley NHS Foundation Trust and King’s College London. He has previously received funding from Beckley Psytech Ltd. He has received payments for lectures and advisory boards for Flow Neuroscience, Novartis, Roche, Janssen, Takeda, Noema Pharma, Compass, Astrazenaca, Boehringer Ingelheim, Eli Lilly, LivaNova, Lundbeck, Sunovion, Servier, Livanova, Janssen, Allegan, Bionomics, Sumitomo Dainippon Pharma, Sage and Neurocentrx. He has received grant funding from the following companies: NIMH (USA); CIHR (Canada); NARSAD (USA); Stanley Medical Research Institute (USA); MRC (UK); Wellcome Trust (UK); Royal College of Physicians (Edin); BMA (UK); UBC-VGH Foundation (Canada); WEDC (Canada); CCS Depression Research Fund (Canada); MSFHR (Canada); NIHR (UK). Janssen (UK) EU Horizon 2020. He is the Editor of the Journal of Psychopharmacology and Deputy Editor of BJPsych Open. He has no shareholdings in pharmaceutical companies. T.Y. is an employee of Hammersmith Medicine’s Research. The clinical trial, performed at Hammersmith Medicine’s Research, was sponsored by Beckley Psytech Ltd. B.S. is a consultant to Beckley Psytech Ltd.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship and/or publication of this article: This trial was funded by Beckley Psytech Ltd. J.J.R. received salary support (100%) from a fellowship (CS-2017-17-007) funded by the National Institute for Health Research (NIHR) from 2018 to 2023, when this study was undertaken.
Research involving human participants
This trial was conducted in accordance with the International Council for Harmonisation Good Clinical Practice guidelines and ethical principles that have their origin in the Declaration of Helsinki. Protocols were approved by the Medicines and Healthcare Products Regulatory Agency and an independent recognized research ethics committee before eligibility screening.
Informed consent
Written informed consent was obtained from each participant before any trial-related procedures were performed.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.