
Abstract
Background:
Vortioxetine is efficacious and well tolerated in patients with major depressive disorder (MDD) and is available as an immediate-release tablet and oral drop solution. The oral drop solution may offer clinical benefits versus a tablet, such as the reduced risk of nausea, personalised dosing and ease of administration.
Aims:
To investigate the bioequivalence of vortioxetine 20 mg/mL oral drop solution versus a 20 mg immediate-release tablet.
Methods:
Healthy adults were randomised 1:1 to receive vortioxetine 20 mg oral drop solution or immediate-release 20 mg tablet after fasting on days 1 and 29 in an open-label, single-centre, single-dose crossover study. The area under the plasma concentration–time curve from 0 to 72 h (AUC0–72h) and maximum plasma concentration (Cmax) were analysed. Bioequivalence was concluded if the 90% CI for the oral drop solution-to-immediate-release tablet ratio for AUC0–72h and Cmax were contained within a range of 0.80−1.25.
Results:
Vortioxetine oral drop solution was bioequivalent to the tablet (n = 26; estimated AUC0–72h ratio 1.06 (90% CI: 1.03–1.10); Cmax ratio 1.01 (90% CI: 0.97–1.05)). A similar proportion of participants reported adverse events in each study arm but more headache events (7 vs 1) were reported with the oral drop solution versus tablet. The most common adverse event was nausea (16–23% of participants; all mild intensity).
Conclusions:
Vortioxetine oral drop solution is bioequivalent to immediate-release tablets. Oral drop solution provides an alternative to tablets and facilitates clinical benefit through individualised treatment, including gradual dose up-titration, for patients with MDD.
Introduction
Antidepressant therapy is generally offered as a tablet formulation but a more readily titratable oral drop solution offers many clinical benefits in the treatment of patients with major depressive disorder (MDD), including personalised dosing, reduced pill burden and ease of administration for patients with difficulty swallowing (both physical or anxiety based). An oral drop solution may also be an option for patients with MDD who have an aversion or stigma associated with antidepressant pills.
Vortioxetine is a multimodal antidepressant shown to be efficacious and well tolerated for the treatment of MDD (Christensen et al., 2022; Fagiolini et al., 2021; McIntyre et al., 2023; Thase et al., 2016) and is available as an immediate-release tablet formulation in 5-, 10-, and 20-mg strengths (in some countries a 15-mg tablet formulation is also available) (European Medicines Agency, 2018). An alternative formulation, an oral drop solution of 20 mg/mL vortioxetine (where one drop is equivalent to 1 mg of vortioxetine), is also currently authorised in 34 countries, including in the European Economic Area, the United Kingdom, Singapore, Switzerland and Argentina.
Vortioxetine oral drop treatment may be initiated at one drop on day 1 and up-titrated by one drop each day until the recommended starting dose of 10 mg has been reached to lessen the risk of nausea, the most commonly reported adverse event with treatment, which is generally reported within the first week of treatment (European Medicines Agency, 2018); Baldwin et al., 2016a; Christensen et al., 2023a). If 10 mg vortioxetine is well tolerated, the dose should be further up-titrated to 20 mg based on the individual patient’s response to achieve the greatest therapeutic benefit (Christensen et al., 2023b). A titratable oral drop formulation allows increased personalisation of dosing to optimise efficacy and safety compared with a fixed-dose tablet formulation.
Vortioxetine is one of the few antidepressants shown to have a clear dose–response relationship across the spectrum of symptoms experienced by patients with MDD, including depressive, cognitive and physical symptoms, anxiety and functional impairment (Adair et al., 2023; Christensen et al., 2023b; Florea et al., 2017; Iovieno et al., 2021; Thase et al., 2016). An analysis of data from six pivotal clinical trials in patients with MDD demonstrated that vortioxetine 20 mg/day was significantly more effective than vortioxetine 10 mg/day in improving depressive symptoms as assessed by the Montgomery-Åsberg Depression Rating Scale, including response at week 2, sustained response and proportion of patients achieving remission without compromising tolerability. The rate of reported adverse events in patients up-titrated to 20 mg at day 7 was similar to that in patients randomised to 10 mg. Therefore, rapidly increasing vortioxetine dosing up to 20 mg once daily after 1 week of treatment with 10 mg is recommended to achieve the maximum therapeutic benefit (Adair et al., 2023; Christensen et al., 2023b).
Physicians estimate that 45% of patients with anxiety and/or depression have difficulty swallowing tablets (Llorca, 2011). While many of these cases may be related to psychological rather than physical barriers to swallowing, psychological concerns can relate to choking on tablets and linking tablets to adverse events (Llorca, 2011). Difficulty swallowing tablets can also lead to patients being less adherent to treatment than patients who do not experience discomfort (Llorca, 2011).
Accordingly, delivery of vortioxetine via an oral drop solution offers an opportunity to improve outcomes for patients with MDD by offering personalised treatment and minimising barriers to adherence. Therefore, this study aimed to investigate the bioequivalence of a 20 mg dose of vortioxetine administered as a 20 mg/mL oral drop solution formulation compared with an immediate-release tablet formulation of the same dosage.
Methods
An open-label, single-centre, single-dose crossover study was performed to investigate the bioequivalence of vortioxetine administered as an oral drop solution versus an immediate-release tablet formulation. The study was performed in compliance with the International Conference on Harmonisation and accordance with Good Clinical Practice guidelines and the principles of the Declaration of Helsinki. The study was conducted in the United Kingdom (UK), and institutional review board approval was obtained from the Plymouth Independent Ethics Committee (Plymouth, UK). All participants provided written informed consent prior to enrolment.
Healthy adults aged 18–55 years with a body mass index (BMI) of 19–30 kg/m2 and blood pressure within normal ranges who agreed to use adequate contraception for the duration of the study and 4 months after completion were enrolled in this study. Participants also had not taken any prescribed or over-the-counter medication (apart from combined oral contraceptives), or any herbal medicine known to interfere with the metabolic cytochrome P450 pathways within 1 week or 5 half-lives (whichever was longer) of dosing.
Participants were randomised 1:1 to receive a single dose of open-label vortioxetine 20 mg in a single immediate-release tablet or 1 mL of oral drop solution (20 mg/mL). Tablets were ingested with 240 mL of water. The oral solution was administered directly into the participant’s mouth in a 1-mL syringe, followed by ingestion of 240 mL of water. The study drug was administered on day 1, followed by washout until crossover on day 29 and administration of the alternate formulation (i.e. participants randomised to oral drop solution on day 1 were administered the immediate-release tablet on day 29 and vice versa). Participants were confined to the study centre from days –1 to 4 and from days 29 to 32. A follow-up visit occurred on day 37 (±2 days).
The study drug was administered when participants were in a fasted state (overnight; ~8 h) and participants did not drink water for 2 h pre-dose until 2 h post-dose. Food was allowed 4 h post-dose and participants were served standard meals while confined to the clinic.
Concomitant paracetamol (2 g/day for a maximum of 3 days) was permitted, according to the investigator’s discretion. Participants refrained from drinking alcohol for 48 h prior to the first study drug dosing until after the completion of each study period but could drink a maximum of 2 units of alcohol per day during the washout period. Smoking or using nicotine-containing products was prohibited during the study. Participants also refrained from consuming caffeine, xanthine, grapefruit, grapefruit-containing products or marmalade from 48 h prior to dosing until discharge from the clinic but were free to consume caffeine or xanthine during the washout period. In addition, participants must not have done strenuous exercise (defined as >70% of the maximal pulse rate for ⩾1 h or more) from 7 days before the first dose of the study drug until after the final follow-up visit (day 37 ± 2 days).
Blood samples for pharmacokinetic assessments were drawn pre-dose and at 1, 2, 4, 6, 8, 10, 12, 16, 24, 36, 48 and 72 h post-dose following both study drug administrations, centrifuged (2000g, for 10 min at 4° to 8°C) within 30 min of collection and frozen within 2 h of collection in an upright position below –70°C. Plasma vortioxetine concentrations were assessed using a validated analysis method that had a lower limit of quantification of 0.2 ng/mL.
The area under the plasma vortioxetine concentration–time curve from 0 to 72 h (AUC0–72h) and maximum plasma vortioxetine concentration (Cmax) were analysed separately for each formulation using analysis of variance with treatment, period, sequence, and participant nested within the sequence as fixed effects. Bioequivalence was concluded if the 90% CI for the oral drop solution-to-immediate-release tablet ratio for both AUC0–72h and Cmax were contained entirely within an interval ranging from 0.80 to 1.25.
The study planned to enrol 26 participants based on a predicted within-participant coefficient of variation percentage (CV%) of the area under the plasma concentration–time curve from 0 to infinity (AUC0–∞) of 10.81% and Cmax of 14.28%, which indicated that a sample size of 20 would offer 99.9% and 98.2% power for correctly concluding bioequivalence for AUC0–∞ and Cmax, respectively, with a true ratio of 1.05. To account for a 30% withdrawal rate, an additional six participants were enrolled. Assuming that AUC0–∞ and Cmax are independent, the overall power to detect bioequivalence was 98%.
The pharmacokinetic analysis set included all participants with evaluable data for both dosing periods (including no vomiting within 12 h of dosing and a pre-dose vortioxetine concentration in the second dosing period <5% of Cmax). Adverse events were classified according to the Medical Dictionary for Regulatory Activities (MedDRA®), version 14.0 (International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use), and assessed in the all-participants-treated set, which included all randomised participants who were administered at least one dose of study drug.
The principal statistical software used was SAS® version 9.2 (SAS Institute Inc.). Pharmacokinetic analysis was conducted using WinNonlin version 5.2 (Pharsight Corporation).
Results
In total, 26 participants were enrolled in this study between 7 September and 17 November 2011. All participants were randomised and received at least one dose of the study drug, with 25 participants completing the study. One participant withdrew because of a protocol violation (positive test for substances of abuse on day 28 prior to dosing).
The study population was evenly split between male and female participants (n = 13 each), and the majority were White (n = 22; 84.6%). Two participants (7.7%) were Black, one was Asian (3.8%) and one was classified as Other (3.8%). Participants had a mean age (SD) of 32 (11.1) years (range 20–53) and a mean BMI of 24 (2.9) kg/m2.
Pharmacokinetics
The average AUC0–72h and Cmax for vortioxetine 20 mg oral drop solution was found to be bioequivalent to the immediate-release vortioxetine 20 mg tablet, with the estimated AUC0–72h ratio (1.06; 90% CI: 1.03–1.10) and Cmax ratio (1.01; 90% CI: 0.97–1.05) within the prespecified bioequivalence range of 0.8–1.25 (Table 1 and Figure 1). The arithmetic mean AUC0–72h of 308 h ng/mL with the oral drop solution was similar to the arithmetic mean AUC0–72h of 290 h ng/mL with the immediate-release tablet. The arithmetic mean Cmax was also similar for both groups (7.62 ng/mL with the oral drop solution vs 7.52 ng/mL with the immediate-release tablet). Low-to-moderate interparticipant variability in pharmacokinetic parameters was observed with both the oral drop solution and immediate-release tablet formulations (AUC0–72h CV%, 27.4 and 28.5%, respectively; Cmax CV%, 22.6 and 22.4%, respectively). The median time to maximum plasma concentration (tmax) was 6 h (range 6–16 h) for both formulations.
Pharmacokinetic parameters of vortioxetine 20 mg following single-dose administration (pharmacokinetic analysis set).

AUC0–72h: area under the plasma vortioxetine concentration–time curve from 0 to 72 h; Cmax: maximum concentration of plasma vortioxetine; CV%: coefficient of variation percentage; h: hour; tmax: time to maximum plasma concentration of vortioxetine.
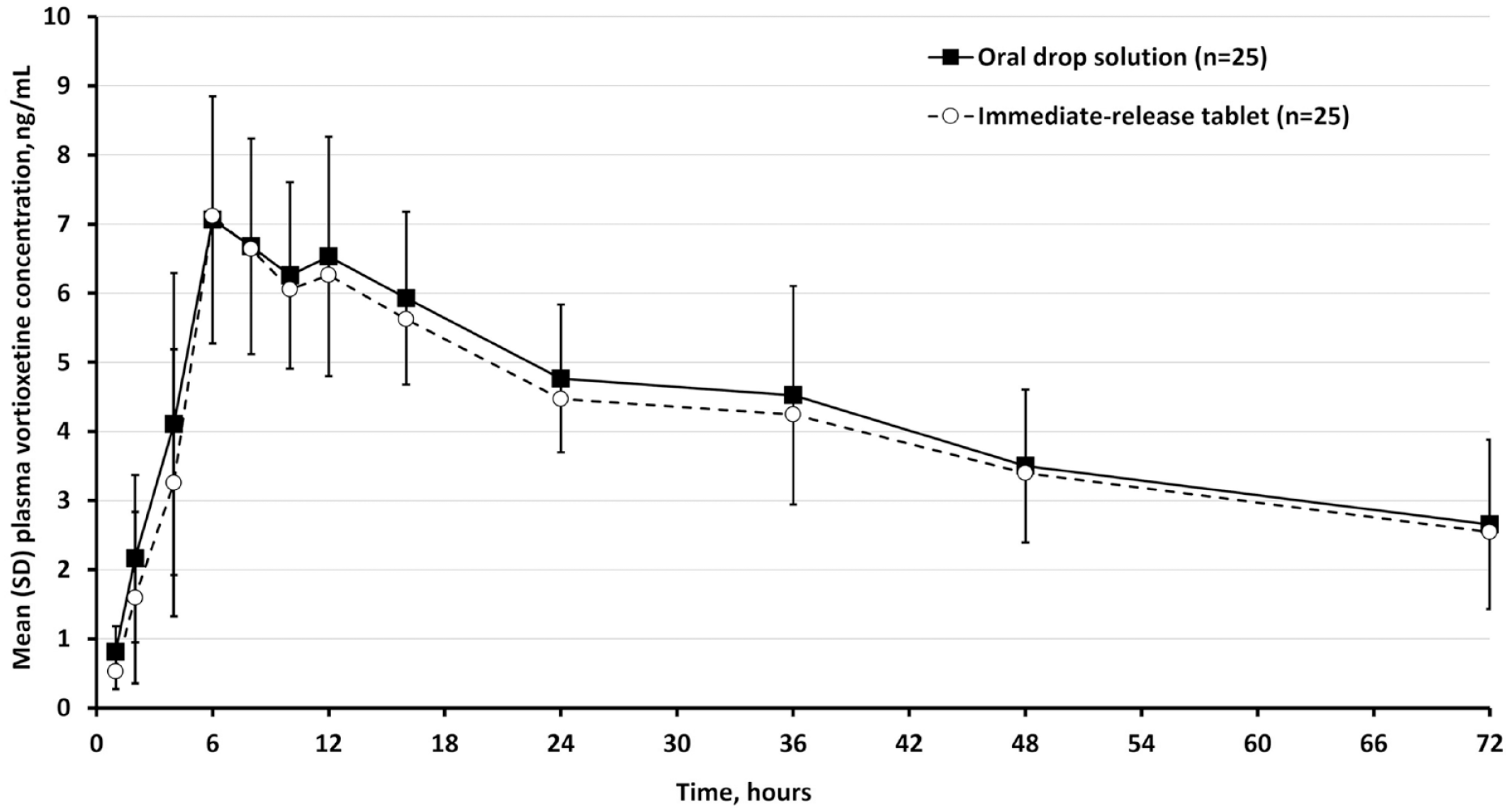
Plasma vortioxetine concentrations following single-dose administration of vortioxetine 20 mg.
Safety
A similar proportion of participants reported adverse events in each study arm but a higher number of adverse events was reported in participants administered the 20 mg oral drop solution compared with the 20 mg tablet (Table 2). The majority of adverse events were mild in intensity (77%), with the remainder being of moderate intensity. Seven of 14 participants (50%) who received the oral drop solution in period 1 reported 14 adverse events in period 1, and 8 of 12 participants (67%) who received the oral drop solution in period 2 (i.e. after crossing over from the tablet formulation) reported 13 adverse events; for tablets, 6 of 12 participants (50%) reported 10 adverse events in period 1 and 5 of 13 (38%) reported 6 adverse events in period 2.
Adverse events (all-participants-treated set). a
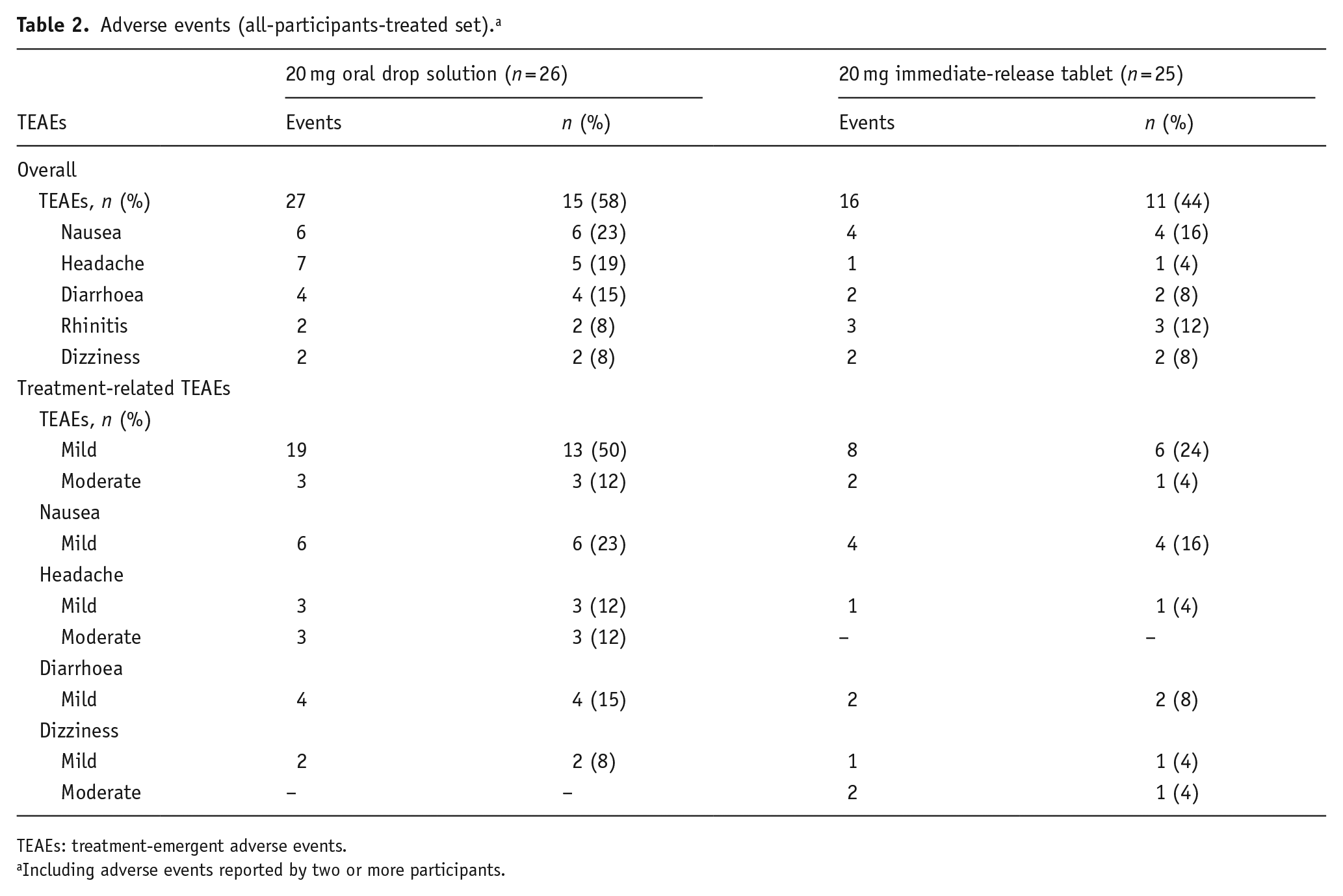
TEAEs: treatment-emergent adverse events.
Including adverse events reported by two or more participants.
Nausea was the most common adverse event in each study arm, occurring in 23% of participants administered the 20 mg oral drop solution and 16% of those administered the 20 mg tablet. Nausea occurred within 1 h post-dose and generally resolved within 2 h. All nausea events were mild in intensity.
The incidence of headache and diarrhoea was 19% and 15%, respectively, in participants administered the oral drop formulation compared with 4% and 8%, respectively, for those administered the immediate-release tablet. Headache occurred between 1.5 h and 14.5 days post-dose and lasted between 1 and 12 h, while diarrhoea occurred between 50 min and approximately 2 days post-dose, lasting between 1 min and 1 day. Four of the seven headache events in participants administered the oral drop formulation were moderate in intensity (three were treatment related and one was not treatment related). All other headache events, including in participants administered the tablet formulation, were mild in intensity. All diarrhoea events were mild in intensity. All other adverse events of moderate intensity were not considered to be treatment related.
No serious adverse events were reported, and no participant discontinued the study because of an adverse event.
Discussion
In this study, healthy participants were administered single doses of vortioxetine as a 20 mg tablet or 20 mg oral drop solution to investigate the bioequivalence between these formulations. In addition to investigating bioequivalence, this study offered an opportunity to further understand the tolerability profile of vortioxetine when initiated at doses higher than the recommended 10 mg starting dose (tablets or drops) for patients with MDD.
With regard to bioequivalence, plasma vortioxetine parameters following the administration of 20 mg vortioxetine oral drop solution in a fasting state met the prespecified criteria for bioequivalence versus immediate-release vortioxetine 20 mg tablets. While this study was performed in a controlled environment with healthy participants who fasted before being administered vortioxetine, there is no reason to believe that the real-world efficacy or pharmacokinetic parameters of vortioxetine oral drop solution or immediate-release tablets in patients with MDD would deviate from the observations made during this study, especially given that the absorption and bioavailability of vortioxetine are not affected by food (Chen et al., 2018). Furthermore, the pharmacokinetic parameters of vortioxetine immediate-release tablets have been shown to be linear, dose proportional and time independent up to the highest investigated dose (75 mg) (Areberg et al., 2014). Therefore, the bioequivalence between the immediate-release tablet and oral drop formulation shown for 20 mg in this study may be extrapolated to the lower doses of 10 mg and 5 mg.
In this study, healthy participants were administered vortioxetine 20 mg, which is higher than the recommended starting dose of 10 mg. Nevertheless, 16%−23% of participants reported mild nausea after administration. No new safety concerns were observed, most adverse events were mild in intensity and there were no dropouts during the crossover period due to adverse events. Vortioxetine oral drop solution may also expand the clinical utility of vortioxetine for patients with MDD by allowing for a more personalised, incremental titration of vortioxetine dosing (Christensen et al., 2023b; Llorca, 2011) alone or in combination with an immediate-release tablet.
In clinical practice, vortioxetine treatment may be initiated at any dose level using the oral drop solution before titrating dosing to reach the recommended starting dose of 10 mg. We have consistently observed a significant reduction in the prevalence of adverse events, particularly nausea, in clinical practice when vortioxetine is titrated using the oral drop solution diluted in water or (even better, when possible) in fruit juice, as opposed to being administered directly into the participant’s mouth in a 1-mL syringe followed by ingestion of 240 mL of water, as in this study. Treatment is initiated at one drop per day (vortioxetine 1 mg) and increased by one drop per day up to 10 drops (vortioxetine 10 mg) after 10 days. Up-titration then continues by increasing dosing by one or two drops per day, up to 20 drops (equivalent to vortioxetine 20 mg), the dose at which maximum therapeutic benefit is achieved (Adair et al., 2023; Christensen et al., 2023b). Likewise, dosing can be titrated up or down using an oral drop solution to achieve an optimal balance of efficacy, safety and tolerability when treating a patient with vortioxetine. However, accurate measurement of small volumes of oral drop solution in a real-world setting may be difficult (Llorca, 2011).
In this study, all nausea events were mild in intensity, with an incidence of 16% and 23% in the 20 mg tablet and 20 mg oral drop solution groups, respectively. In a recent analysis of data pooled from six short-term vortioxetine studies, the incidence of nausea was 13.3% for the 10 mg/day group and 17.9% for the 20 mg/day group during the first week of treatment (when patients in the vortioxetine 20 mg/day group were receiving the starting dose of 10 mg/day), which decreased substantially to 3% for the 10 mg/day group and 4.4% for the 20 mg/day group during week 2 (when patients in the vortioxetine 20 mg/day group were up-titrated to the target 20 mg dose) (Christensen et al., 2023a). Although the findings of this study in healthy adults may not be generalisable to the MDD population, there is an increasing body of evidence that suggests vortioxetine dosing can be quickly titrated to 20 mg after 1 week of treatment with 10 mg, without compromising tolerability.
In general, elderly patients are more likely to be accepting of oral drops than younger patients because of the increased prevalence of barriers to consuming tablets, such as comorbidities, polypharmacy, dysphagia and phagophobia (Liu et al., 2014; Llorca, 2011). Accordingly, the ease of swallowing an oral drop solution compared with a tablet can potentially remove a barrier to initiating and adhering to antidepressant treatment for significant subgroups of patients with MDD, particularly those with anxiety.
These results must be considered in the context of the small sample size of healthy participants, and the use of a single dose and descriptive statistics being reported, which means that it is not possible to draw firm conclusions about the statistical significance of any differences in the safety profile of each formulation. However, the long-term efficacy, safety and tolerability of vortioxetine have been established in other studies (Baldwin et al., 2016b; Vieta et al., 2017), although clinical studies evaluating the use of oral drop vortioxetine in patients with MDD would provide more insight into the use of an oral drop solution in clinical practice.
In conclusion, 20 mg vortioxetine oral drop solution is bioequivalent to 20 mg of the immediate-release tablets, offering an additional formulation that allows individualised treatment and ease of administration for patients with MDD. In addition, the 20 mg dose of vortioxetine was well tolerated, with a safety profile consistent with that previously seen with 10 mg during the first week. The most common adverse event was nausea, and only of mild intensity.
Footnotes
Acknowledgements
Under the direction of the authors, medical writing assistance was provided by Leandra Dang, PharmD, and Blair Hesp, PhD, CMPP, on behalf of Syneos Health Medical Communications. H. Lundbeck A/S provided funding to Syneos Health for support in writing this manuscript.
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: A. Fagiolini is/has been a consultant and/or a speaker and/or has received research grants from Angelini, Apsen, Boehringer Ingelheim, Daiichi Sankyo, Doc Generici, Italfarmaco, Janssen, Lundbeck, Mylan, Neuraxpharm, Otsuka, Pfizer, Recordati, Viatris and Vifor. M. Adair, M.C. Christensen, K.B. Petersen and J. Areberg are employees of H. Lundbeck A/S.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was funded by H. Lundbeck A/S.
Data Availability Statement
The data supporting the findings of this study are included in the article; further inquiries can be directed to the corresponding author.