
Abstract
Antidepressants often disrupt sleep. Vortioxetine, a multimodal antidepressant acting through serotonin (5-HT) transporter (SERT) inhibition, 5-HT3, 5-HT7 and 5-HT1D receptor antagonism, 5-HT1B receptor partial agonism, and 5-HT1A receptor agonism, had fewer incidences of sleep-related adverse events reported in depressed patients. In the accompanying paper a polysomnographic electroencephalography (sleep-EEG) study of vortioxetine and paroxetine in healthy subjects indicated that at low/intermediate levels of SERT occupancy, vortioxetine affected rapid eye movement (REM) sleep differently than paroxetine. Here we investigated clinically meaningful doses (80–90% SERT occupancy) of vortioxetine and paroxetine on sleep-EEG in rats to further elucidate the serotoninergic receptor mechanisms mediating this difference. Cortical EEG, electromyography (EMG), and locomotion were recorded telemetrically for 10 days, following an acute dose, from rats receiving vortioxetine-infused chow or paroxetine-infused water and respective controls. Sleep stages were manually scored into active wake, quiet wake, and non-REM or REM sleep. Acute paroxetine or vortioxetine delayed REM onset latency (ROL) and decreased REM episodes. After repeated administration, vortioxetine yielded normal sleep-wake rhythms while paroxetine continued to suppress REM. Paroxetine, unlike vortioxetine, increased transitions from non-REM to wake, suggesting fragmented sleep. Next, we investigated the role of 5-HT3 receptors in eliciting these differences. The 5-HT3 receptor antagonist ondansetron significantly reduced paroxetine’s acute effects on ROL, while the 5-HT3 receptor agonist SR57227A significantly increased vortioxetine’s acute effect on ROL. Overall, our data are consistent with the clinical findings that vortioxetine impacts REM sleep differently than paroxetine, and suggests a role for 5-HT3 receptor antagonism in mitigating these differences.
Introduction
Sleep is essential for normal cognitive function and overall health and is an important consideration in the development of novel therapeutics (Dresler et al., 2014). It is well documented that depression is associated with abnormal sleep homeostasis and sleep electroencephalographic (EEG) rhythms, such as disrupted sleep continuity, decreased slow wave sleep, and dysregulated rapid eye movement (REM) sleep (Arfken et al., 2014; Argyropoulos and Wilson, 2005; Kimura and Steiger, 2008; Steiger and Kimura, 2010; Wichniak et al., 2012). Sleep EEG measures such as disturbed REM density and delta sleep ratio are highly replicable and are thought to be useful diagnostic biomarkers for depression (Wichniak et al., 2013). On the other hand it is also well documented that currently used antidepressants such as selective serotonin reuptake inhibitors (SSRIs) are associated with sleep related adverse effects (Wichniak et al., 2012).
Although the complexity of sleep architecture varies between species, it is relatively conserved across mammals and shares features which are comparable (Lesku et al., 2009; Lo et al., 2004; Rial, 2009). In addition, sleep architecture is sensitive to changes in brain neurotransmitters such as serotonin (5-HT) allowing cross-species sleep measurement with pharmacological manipulation to investigate the receptor mechanisms controlling sleep-wake regulation and sleep architecture in response to known and novel agents (Paterson et al., 2011). As reviewed by Pehrson et al. (2015) sleep-EEG changes produced by SSRI antidepressants in rodents, in general, replicate in clinical studies of healthy subjects or depressed patients.
The sleep-wake cycle is a highly regulated system involving many cortical and subcortical brain regions and is driven by a homeostatic process, which regulates the amount of sleep, and the circadian process, which regulates the timing of sleep (Borbely 1982; Brown et al., 2012). The interplay between these processes determines when sleep occurs or when the organism is awake (Krystal et al., 2013). Sleep stages or states of wakefulness in rodents are typically defined by EEG as (a) wakefulness, which exhibits low-voltage fast EEG activity and high muscle tone recorded via electromyography (EMG), (b) non-rapid eye movement (NREM) or slow wave sleep, which is characterized by high-amplitude, low-frequency EEG and decreased muscle tone, and (c) REM or paradoxical sleep, which has low voltage fast EEG activity coupled with a complete loss of muscle tone (atonia) and characteristic rapid eye movements (Brown et al., 2012). In the laboratory, wakefulness is further divided into quiet wake or active wake based on the presence or absence of locomotor activity or movement within the test environment (e.g. home cage). Sleep stages are very sensitive to pharmacological manipulations, particularly those of antidepressants, and often it is possible to identify differences between compounds with different pharmacology using sleep-EEG endpoints (Paterson et al., 2011; Wilson et al., 2014). Herein, we focus on how the serotonergic system acting via its different receptors can modulate the sleep-wake cycle.
Much of what we know about the role of 5-HT and sleep has been summarized by Jaime M. Monti (e.g. see Monti, 2011). In short, 5-HT predominantly promotes wakefulness and inhibits REM sleep and during wakefulness the 5-HT system interacts closely with other neurotransmitter systems, including acetylcholine (ACh), glutamate (Glu), dopamine (DA), norepinephrine (NE), histamine (HA) and orexin (hypocretin; OX), to regulate circadian, sleep, and cognitive processes (Leiser et al., 2015; Miyamoto et al., 2012; Monti, 2011; Sebban et al., 1999). This is because 5-HT neurons of the raphe nucleus innervate widespread brain areas, including the cholinergic nuclei of the mesencephalon and the basal forebrain, the dopaminergic neurons of the ventral tegmental area and the substantia nigra compacta, the noradrenergic cells of the locus coeruleus (LC), the gamma-aminobutyric acid (GABA)ergic, histaminergic and orexinergic cells of the hypothalamus and the glutamatergic neurons of the thalamus and the brain stem reticular formation, to name only a subset of brain areas involved in regulation of sleep-wake states (Brown et al., 2012; Datta and Maclean, 2007). Therefore, the sleep-wake cycle is complex and a complete understanding of how it is regulated is still emerging (Zeitzer, 2013). However, to simplify matters, it is thought that during wakefulness serotoninergic tone (as well as that of many other neuromodulator systems mentioned above) gives rise to enhanced cortical activity and arousal, while during sleep the awake-related neurons slow down, thereby withdrawing their effects on so called REM sleep-related cholinergic neurons of the laterodorsal and pedunculopontine tegmental nuclei (LDT/PPT) (Steriade and McCarley, 1990). As 5-HT levels drop, an increase in REM-ON neuronal activity is triggered, which in turn releases ACh onto GABAergic neurons in the locus coeruleus, where GABA inhibits the REM-OFF neurons, resulting in initiation of REM sleep (Aloe et al., 2005; Sutcliffe and de Lecea, 2002). Consequently, the rate of firing of neurons in both the locus coeruleus (Aston-Jones and Bloom, 1981; Curtis et al., 2012) and dorsal raphe nucleus (Wu et al., 2004) falls to almost zero during REM sleep. SSRI-induced increase of extracellular 5-HT consistently results in suppression of REM (see Drago, 2008; Rijnbeek et al., 2003; Wichniak et al., 2012; Wilson and Argyropoulos, 2005 for tables summarizing antidepressant effects on sleep in healthy subjects and patients), which equates to a suppression of REM-ON neurons.
Selective activation of specific 5-HT receptor subtypes influences sleep and REM, in particular, in opposing ways (Artigas, 2013; Montgomery and Fineberg, 1989; Staner et al., 2001) (Table 1). 5-HT1A receptor agonists typically promoted wakefulness and suppressed REM sleep, while antagonists promoted REM sleep (Bjorvatn et al., 1997; Bjorvatn and Ursin, 1998; Boutrel et al., 1999, 2002; Dzoljic et al., 1992; Gillin et al., 1996). Also, evidence indicates that selective activation of the somatodendritic 5-HT1A receptor in the dorsal raphe nucleus (DRN) increased REM, however activation of the postsynaptic 5-HT1A receptor at the level of the laterodorsal or pedunculopontine tegmental nuclei decreased REM (Monti and Monti, 2000). One explanation for this is that 5-HT1A receptor-responsive neurons in the pedunculopontine tegmental nucleus become maximally active immediately before and during REM sleep and activation of these neurons likely contributes to the generation of REM sleep (Gillin et al., 1996; Grace et al., 2012; Wilson et al., 2005). 5-HT1B receptor agonism typically induced waking and suppresses REM as well (Bjorvatn and Ursin, 1994; Boutrel et al., 1999; Monti et al., 1995, 2010) and 5-HT1B receptor antagonism induced REM sleep (Boutrel et al., 1999). 5-HT1D receptor agonism has been reported to inhibit REM sleep and increase wakefulness (Bruni et al., 2011). 5-HT2A/2C receptor antagonism induced NREM sleep (Monti, 2010; Monti and Jantos, 2006; Morairty et al., 2008), while 5-HT2A/2C receptor agonism reduced REM sleep (Amici et al., 2004; Monti and Jantos, 2006). There is evidence for the action of 5-HT3 receptor antagonism in maintaining REM sleep, while 5-HT3 receptor agonism is suppressing REM (Staner et al., 2008). For example, 5-HT3 receptor agonism was found to reduce total REM and the number of REM periods (Monti and Jantos, 2008; Monti et al., 2011) and increase wakefulness (Gyongyosi et al., 2010). Moreover, demonstrating the translatability of preclinical rodent data to clinical observations, in healthy subjects, 5-HT3 receptor agonism produced a suppression of REM (Staner et al., 2001, 2008). 5-HT3 receptor antagonism increased NREM sleep in the rat (Adrien et al., 1992; Ponzoni et al., 1993), and reduced effects of other agents on wakefulness and sleep. Very little is known about 5-HT4 or 5-HT5 receptor mediated effects on sleep. 5-HT6 receptor agonism significantly increased wakefulness and reduced NREM and REM (Monti et al., 2013) while 5-HT6 receptor antagonism increased sleep and decreased waking (Morairty et al., 2008). 5-HT7 receptor antagonism reduced the total amount of REM sleep (e.g. less frequent episodes of REM sleep or decreased bout length; synonymous with induced REM suppression) and/or significantly increased the latency to onset of REM sleep consistently across several studies in rat and human (Bonaventure et al., 2007, 2012; Hedlund et al., 2005; Matthys et al., 2011; Monti et al., 2008, 2012, 2014; Shelton et al., 2009; Thomas et al., 2003). Paradoxically, 5-HT7 receptor agonists have also been shown to increase wakefulness and reduce REM (Monti et al., 2008, 2014; Shelton et al., 2009). Additional studies are required to fully understand the role of 5-HT7 receptors in sleep.
Effects of selective serotonin (5-HT) receptor agonism or antagonism on sleep.
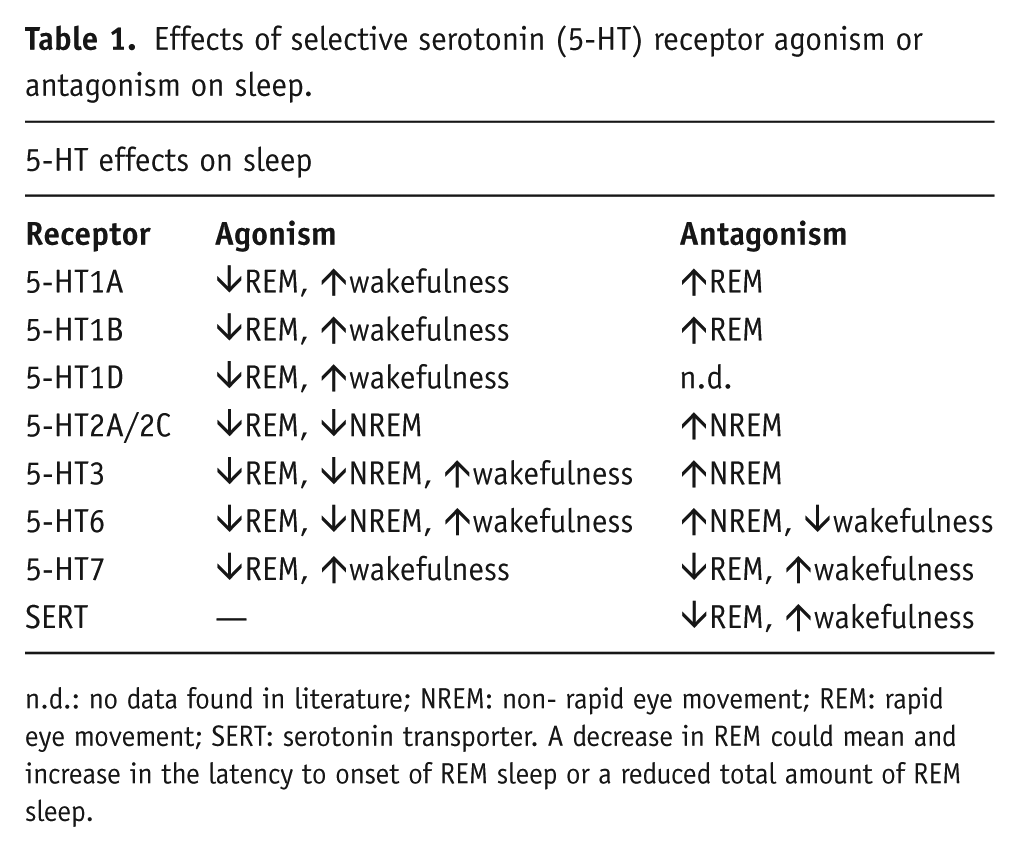
n.d.: no data found in literature; NREM: non- rapid eye movement; REM: rapid eye movement; SERT: serotonin transporter. A decrease in REM could mean and increase in the latency to onset of REM sleep or a reduced total amount of REM sleep.
The multimodal antidepressant vortioxetine (Lu AA21004; 1-[2-(2,4-dimethylphenyl-sulfanyl)-phenyl]-piperazine) is an antagonist at the 5-HT3 receptor ligand-gated ion channel, a 5-HT7 and 5-HT1D receptor antagonist, 5-HT1B receptor partial agonist, 5-HT1A receptor agonist and serotonin transporter (SERT) inhibitor in vitro (Bang-Andersen et al., 2011; Sanchez et al., 2015). Microdialysis studies in rats have shown that vortioxetine enhances extracellular levels of 5-HT, ACh, NE, DA, and HA in brain regions involved in the regulation of sleep, emotional and cognitive functions as well as having a modulatory role on GABA and glutamate function (Leiser et al., 2014b; Mørk et al., 2012; Pehrson et al., 2013; Pehrson et al., 2015; Pehrson and Sanchez 2015). In depressed patients treated with vortioxetine, the incidence of spontaneously-reported sleep-related adverse events was found to be 2.0–5.1% compared to 4.4 % in patients who had received placebo (Baldwin et al., 2012, 2013). Furthermore, the recent data by Wilson et al., (2015), (preliminary findings published by Wilson et al., 2013), demonstrate in healthy subjects that vortioxetine produces different effects on REM sleep than paroxetine: namely, that despite the same SERT occupancy, vortioxetine seemed to affect REM sleep to a lesser degree than paroxetine. The present sleep-EEG study in rats was used to back-translate these clinical findings and further investigate the role of 5-HT3 receptor antagonism in mitigating this effect. To mimic the steady state conditions of the human study, a sub-chronic dosing regimen was chosen for the rat study.
Methods
Animals
Male Sprague-Dawley rats (250–500 g) from Charles River were individually housed under a 12-hour light/dark cycle and temperature (21±2°C) and humidity (60±10%) control with chow and water ad libitum. Animals were used in accordance with guidance on care and use of laboratory animals by Lundbeck Research USA and the National Research Council (2011).
EEG, surgical procedure, recording and analysis
Bipolar (differential) EEG screw electrodes for fronto-parietal EEG and wire electrodes for EMG of dorsal neck muscles were implanted in each animal under anesthesia as described previously (Bastlund et al., 2004; Ebert et al., 2008; Leiser et al., 2014b; Sanchez et al., 2007; Vogel et al., 2002) and connected to a sterile multi-channel telemetric device (TL10M3-F50-EEE; Data Sciences International (DSI)) that was implanted subcutaneously (s.c.) on the flank. After 10 days of recovery, EEG recordings were initiated in their home cages.
For sub-chronic pharmacological evaluation, recordings using Dataquest A.R.T. software (DSI) at a sampling rate of 500 Hz were started at 15:00 the day prior to the acute injection that occurred on “Day 1” at 09:00 (three hours after lights came on). Animals received a s.c. injection of either vortioxetine or paroxetine or vehicle and were then returned to their home cage with either vehicle or drug-infused chow or water (see below) and recorded for the entire 24 h for 10 days. A daily health check and husbandry occurred each day between 09:00–09:30 when the recordings were momentarily suspended. For acute pharmacological evaluation, recordings were started 90 min before injection (09:00; 3 h into the light phase) and continued for up to 4 h post injection.
Offline, using NeuroScore (DSI), artefacts were removed from the data and sleep stages assigned manually for every 10-second epoch using EEG, EMG, and locomotor activity (LMA) counts derived from transmitter signal by conventional methods as previously described (Ivarsson et al., 2005; Leiser et al., 2014b; Parmentier-Batteur et al., 2012): active wake (less regular, low amplitude EEG with high EMG and LMA); quiet wake (less regular, low amplitude EEG, with low EMG, and no LMA); NREM sleep, consisting of high-amplitude waves with predominant delta (1-4 Hz), low EMG and no LMA; paradoxical or REM sleep exhibited stable, low-amplitude waves dominated by theta (4–8 Hz) with near absent EMG and no LMA.
Compounds and dosing regimen
Vortioxetine (DL-lactate salt) and paroxetine were synthesized by H Lundbeck A/S. The selective 5-HT3 receptor agonist SR57227A was synthesized by H Lundbeck A/S. The selective serotonin 5-HT3 receptor antagonist ondansetron was purchased from Sigma-Aldrich (St. Louis, Missouri, USA). For acute dosing vortioxetine, paroxetine, ondansetron, and SR57227A were dissolved in sterile distilled water and administered subcutaneously (s.c.) in a volume of 2.0 mL/kg. For the subchronic vortioxetine experimental group, the regular rat chow was switched to vortioxetine-enriched diet (600 mg vortioxetine per kg rat chow, Open Source Diets, Purina Rodent Chow with Blue Dye) and for the vehicle group, the regular chow was switched to repelleted rodent chow (Purina 5001 Rodent Chow, Open Source Diets), which had exactly the same nutritional content as in the vortioxetine-infused chow. Food was replenished as needed throughout the experiment. Animals demonstrated food consumption by dye presentation in the feces. Moreover, this vortioxetine administration paradigm was previously shown to result in full SERT occupancy in rats (Wallace et al., 2014).
For the subchronic paroxetine experimental group, paroxetine at a dose of 2.5 mg/30 mL drinking water was prepared fresh every three days and replenished as necessary. Fluid intake was recorded and all animals consumed at least 30 mL of fluids daily. Regular drinking water in the vehicle groups was changed at the same time as the paroxetine group. A total of eight animals were used per treatment group unless otherwise noted, however some animals were excluded prior to data analysis due to EEG device failure or poor EEG or EMG quality making it difficult to properly score sleep stages. This rendered the number of animals for the vortioxetine chow group to n=7, 7, 6, and 5, and the control group to n=7, 6, 6, and 5 for days 1, 3, 7, and 10 respectively. Animals were euthanized at the end of each study.
SERT occupancy
Doses were chosen based on previous studies of SERT occupancy-dose relations measured by ex vivo autoradiography. Vortioxetine-infused chow was administered as previously described; Chronic administration of vortioxetine at 0.6 g/kg of food led to 85–90% occupancy of the 5-HT transporter, and 55–60% occupancy of the 5-HT1B receptor (Wallace et al., 2014). The target of >80% SERT occupancy was chosen since this is generally considered to be the minimally effective dose for SSRI therapeutic effect (Meyer et al., 2004) and the vortioxetine dose of 10 mg/kg produced a similar level of SERT occupancy (~90%) in rodents as the highest clinically used dose, 20 mg/day (Areberg et al., 2012; Stenkrona et al., 2013).
Rats were treated with paroxetine at 1, 2.5, 3, or 5 mg/kg/day per os (p.o.) for 14 days (n=4–8/treatment group). Rats in the 2.5 mg/kg/day paroxetine-treated group were the same animals in which sleep-EEG experiments were conducted; all other groups were conducted in a study prior to initiation of the sleep-EEG study for dose finding purposes. SERT occupancy was determined as previously described (Betry et al., 2013; Du Jardin et al., 2014; Leiser et al., 2014b; Pehrson et al., 2013; Wallace et al., 2014). Briefly, rats were anaesthetized using CO2, decapitated and their brains quickly harvested, flash-frozen, and then sectioned coronally at 20 μm thickness. Slices were mounted on slides with a minimum of three replicate slices per brain and stored at −20°C until being used in autoradiography experiments. Slides were defrosted at room temperature under a constant stream of air for 30–45 min and then incubated for 90 min with buffer (50 mM Tris-HCl, 150 mM NaCl and 5 mM KCl, pH 7.4) containing 0.5 nM [3H]DASB. Non-specific binding was determined using 1 µm escitalopram. Slides were washed three times in buffer at 4°C for 5 min, briefly dipped in distilled water, air-dried, and then placed in a desiccator for at least 60 min. Finally, the slides were exposed at room temperature in a Beta imager (Biospace Lab, France) for 16 h prior to analysis (Betry et al., 2013). Surface radioactivity (cpm/mm2) was measured using Beta Vision+ software version 2.0 (Biospace Lab, France) from a region of interest defined a priori on the basis of mapping experiments conducted by this laboratory.
Polysomnographic data analysis
Vivarium lights turned on at 06:00. and off at 18:00, thus the Light phase of the experiment when rats were typically quiescent or sleeping was from 06:00–18:00. and the Dark phase of the experiment when rats were typically awake and active was from 18:00–06:00. Figure 1 illustrates the time in REM for each 30-minute bin in zeitgeber time, which refers to the time relative to the light/dark schedule with 0 corresponding to lights on and 12 to lights off. Significance was determined by multifactorial analysis of variance (ANOVA) for each day and treatment group comparisons (e.g. vortioxetine vs its control and paroxetine vs its control) separately with a Fisher least significant difference (LSD) post-hoc test for multiple timepoint comparisons.
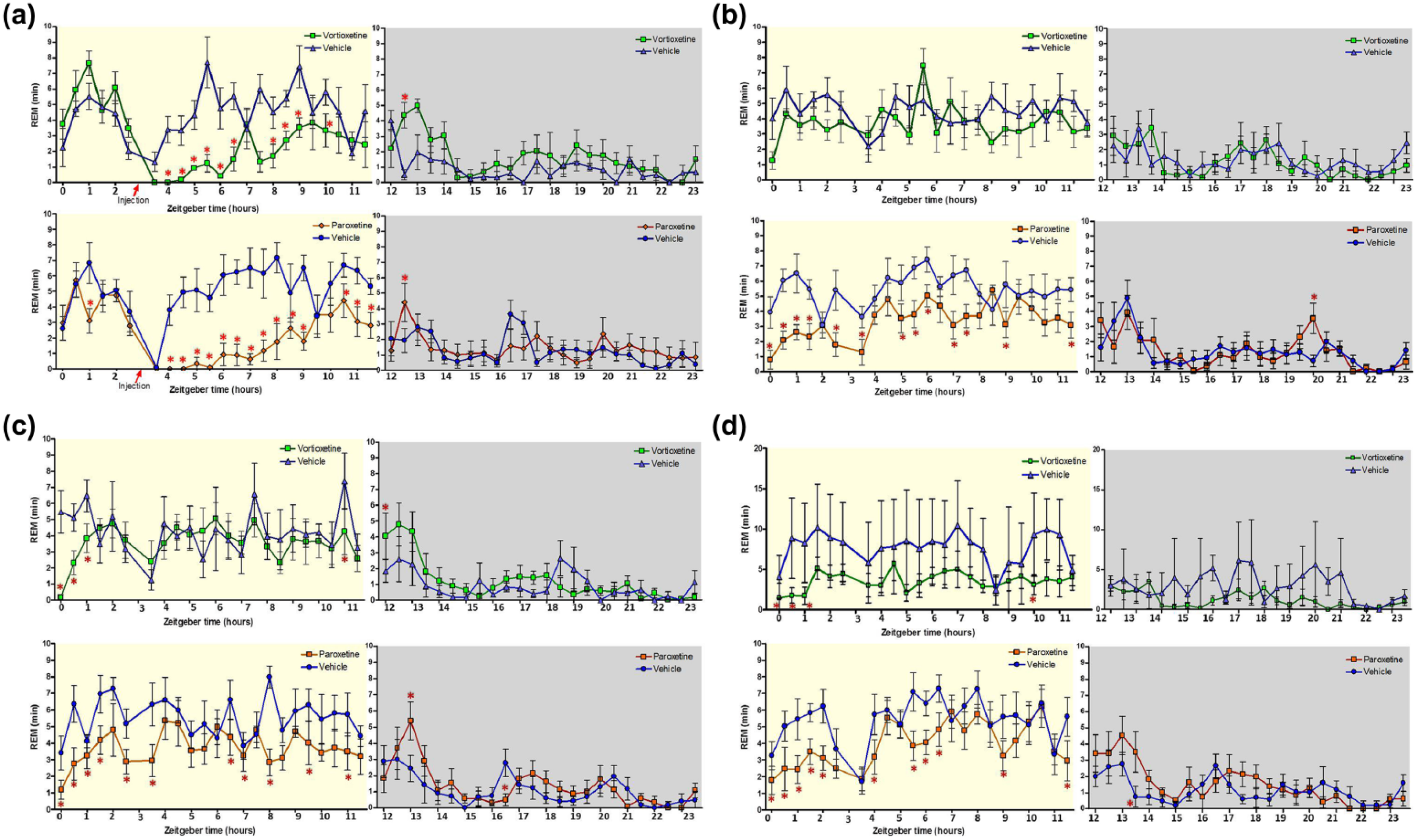
Time spent in rapid eye movement (REM) (min) for vortioxetine and paroxetine before and after acute drug administration on day 1 and for 24 h on days 3 and 7. Data shown reflect mean±standard error of the mean (SEM) of the sum of time spent in REM for each 30-minute epoch after drug administration (marked with a red triangle on x-axis at 3 h after lights on) for day 1 or the whole light phase for all other days. The x-axis is plotted using zeitgeber time, which refers to the time relative to the light/dark schedule with time 0 corresponding to lights on and 12 to lights off. Paroxetine and its respective control, n=8 for all days; vortioxetine chow group had n=7, 7, 6, and 5, and control group had n=7, 6, 6, and 5 for days 1, 3, 7, and 10 respectively due to electroencephalographic (EEG) device failure. High variance on Day 10 (d) in the vehicle group is due to this lower animal number, however no change was noted in the vortioxetine group compared to day 7 (c). Significance was determined by multifactorial analysis of variance (ANOVA) with a Fisher LSD post-hoc test for multiple timepoint comparisons (*p<0.05; Figure 1(a), Day 1: vortioxetine (VOR) vs vehicle (VEH) F(46, 595)=3.0618, p<0.001; paroxetine (PAR) F(46, 754)=4.0164, p<0.001; Figure 1(b), Day 3: VOR vs VEH F(46, 595)=0.85, p=0.75; PAR vs VEH F(46, 754)=1.9986, p<0.001; Figure 1(c), Day 7: VOR vs VEH F(46, 542)=1.5352, p=0.016; PAR vs VEH F(46, 754)=1.8757, p<0.001; Figure 1(d), Day 10: VOR vs VEH F(46, 436)=1.3977, p=0.05; PAR vs VEH F(46, 754)=1.8572, p<0.001).
Quantitative analysis of each sleep stage, REM, NREM, and wake (quiet wake and active wake), was calculated from the sum of time spent in each 30-minute epoch during the light or dark phase, except for D1 in which only the epochs after drug administration were added. Vortioxetine or paroxetine treatment group was compared to its corresponding vehicle treatment group at days 1, 3, 7, and 10. Significant differences between compound and vehicle were determined using Student’s unpaired t-test when data presented a normal distribution or Mann-Whitney test if data were not normally distributed as determined by the D’Agostino and Pearson omnibus normality test. Data are presented as mean± standard error of the mean (SEM) and levels of statistical significance as *p<0.05; **p<0.01; ***p<0.005.
For the acute experiments, the REM onset latency was determined as the first epoch of REM lasting greater than 20 s following drug treatment. The REM onset latency was not used in the subchronic dosing study because rats alternate sleep and wakefulness throughout the day and night (i.e. polyphasic) unlike humans, who tend to sleep and transition into sleep phases in one recording session overnight thereby making it easier to calculate the first true REM onset. The nonstationary (ultradian) periodic rhythms in rats precludes calculating true first REM onset (Stephenson et al., 2012).
Results
Comparisons of subchronic vortioxetine and paroxetine
Figure 1 shows, in 30-minute bins, the duration of REM-sleep over 24 h on days 1, 3, 7, and 10. During the light phase (06:00–18:00) vortioxetine decreased the duration of REM sleep throughout the day on day 1, whereas paroxetine produced a decrease of REM sleep throughout the day on all days. There were no significant drug effects on REM sleep duration during the dark phase. Specifically, on Day 1 (Figure 1(a)), following the acute dose of vortioxetine and paroxetine, there was a suppression of REM in agreement with our previously reported results for acute administration in rats (Leiser et al., 2014b; Sanchez et al., 2007) and consistent with clinical findings (see companion paper Wilson, et al., this issue). This REM sleep suppression after acute vortioxetine was dose-dependent in rats (Leiser et al., 2014b) and humans (Wilson, et al., this issue). Yet, the amount of REM sleep returned to vehicle levels for the vortioxetine-treated group well before the onset of the dark phase (15:30–16:00), while the paroxetine-treated group did not recover from the REM suppression until after the onset of the dark phase (18:00–18:30). On Day 3 (Figure 1(b)), the vortioxetine group exhibited normal REM sleep compared to its control group. However, the paroxetine group continued to demonstrate a disruption in REM sleep throughout the day. Day 7 (Figure 1(c)) and Day 10 (Figure 1(d)) exhibited similar effects, showing that vortioxetine induced few changes to REM except for the first 1.5 h of the light phase, while paroxetine’s disruption to REM sleep lasted throughout the day. The high variance on Day 10 observed in the vehicle control group of the vortioxetine study was due to low animal number (n=5; see Methods).
REM sleep changes were quantified by summating the total amount of REM sleep during the light phase (Figure 2(a) and (b)). Paroxetine significantly suppressed REM for the duration of the experiment in relation to its control group (p<0.05, unpaired t-test). No noticeable changes in REM were observed in dark phase (Figure 3(a) and (b)). NREM sleep was also quantified for both the light (Figure 2(c) and (d)) and the dark phase (Figure 3(c) and (d)). During the light phase, both the vortioxetine- and paroxetine- treated groups demonstrated an elevated NREM sleep compared to their respective control groups on Day 1 following the acute dosing (p<0.05, unpaired t-test). During the light phase, on Days 3, 7 and 10 the vortioxetine- treated group had normal levels of NREM (not different than the control group). Yet, the paroxetine-treated group continued to exhibit an increased amount of NREM sleep compared to its control group (p<0.05, unpaired t-test). During the dark phase, paroxetine also showed an increased level of NREM sleep on Day 7 (p<0.05, unpaired t-test) and Day 10 (p=0.06, unpaired t-test), demonstrating more sleep during the period in which animals should be more active. To further investigate these observed changes in sleep, the ratio of NREM to REM was calculated by dividing the total amount of REM sleep by the total amount of NREM sleep for light phase (Figure 2(e) and (f)) and dark phase (Figure 3(e) and (f)). Normally this ratio yields approximately 25% REM and 75% NREM during sleep (i.e. during light phase) as can be seen in both control groups. The acute doses of vortioxetine and paroxetine showed a reduced REM/NREM ratio (p<0.05, unpaired t-test). This ratio was reduced for both groups throughout Day 3 (p<0.05, unpaired t-test), but for vortioxetine, it returned by Day 7, while for paroxetine, it did not return even by Day 10 (p<0.05, unpaired t-test). No changes were observed for NREM/REM during the dark phase (Figure 3(e) and (f)). Additionally, the amount of time spent in wakefulness (both active and quiet wake) was quantified for both the light (Figure 2(g) and (h)) and the dark phases (Figure 3(g) and (h)). Interestingly, vortioxetine did not seem to augment the total time spent in wake (quiet and active wake) during the light phase of Day 1, nor any other day (Days 3, 7, and 10). During the dark phase of Day 3, vortioxetine only increased total awake time. Paroxetine increased the time spent in wake during the light phase on Day 1. Yet, more importantly, paroxetine significantly decreased the time spent in wake during the dark phase on Day 7 and Day 10 (p<0.05, unpaired t-test), when the animals should be more active, which further demonstrates a disruption in the ultradian sleep-wake balance.
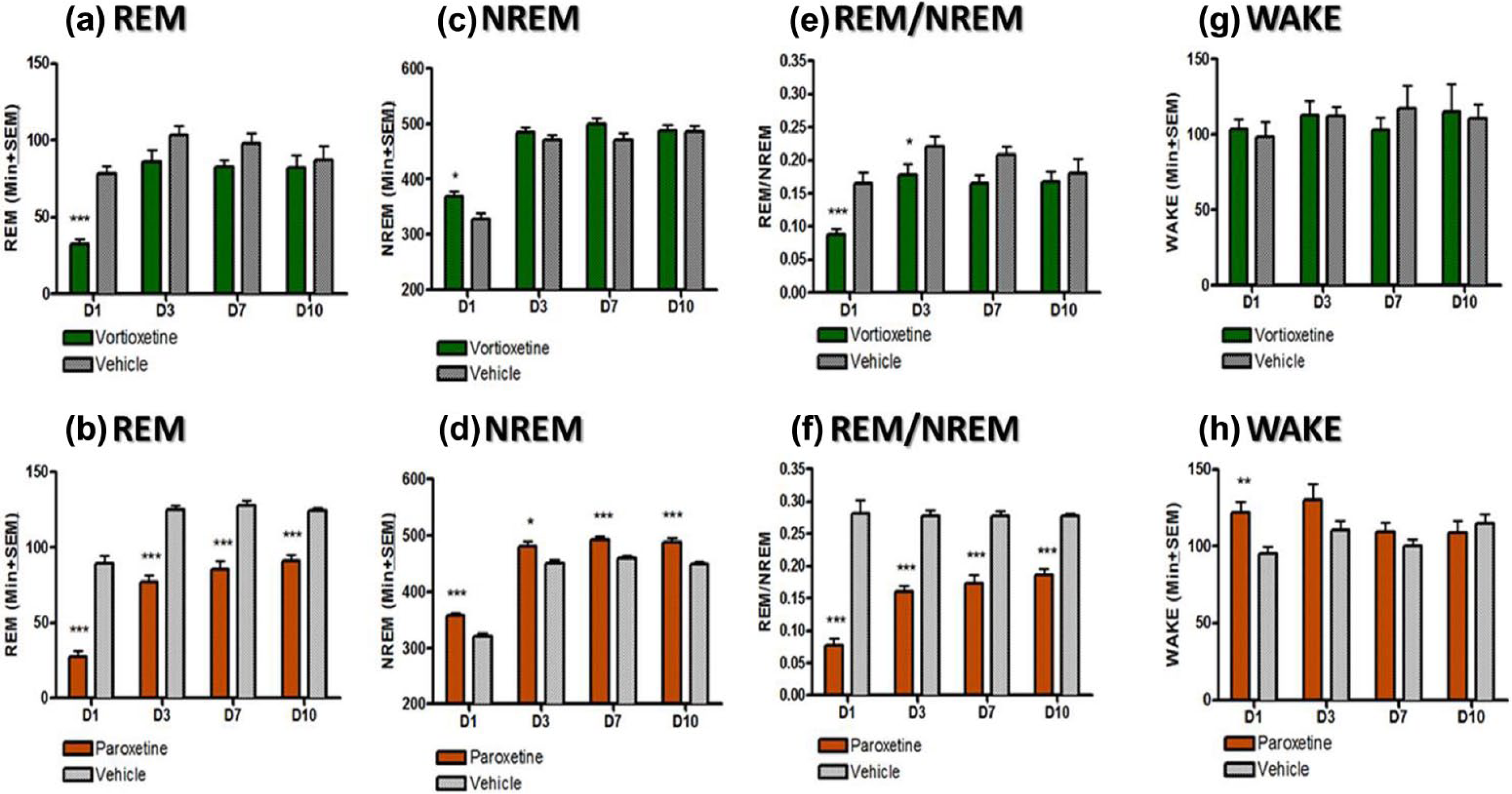
Effect of vortioxetine and paroxetine on rapid eye movement (REM), non-rapid eye movement (NREM), wake (quiet wake and active wake), and REM/NREM ratio during 12-hour light phase (06:00–18:00). Data shown reflect mean±standard error of the mean (SEM) of the sum of time spent in each stage for each 30-minute epoch after drug administration for day 1 or the whole light phase for all other days. Each treatment group was compared to its respective vehicle control for each day separately (*p<0.05; **p<0.01; ***p<0.005).
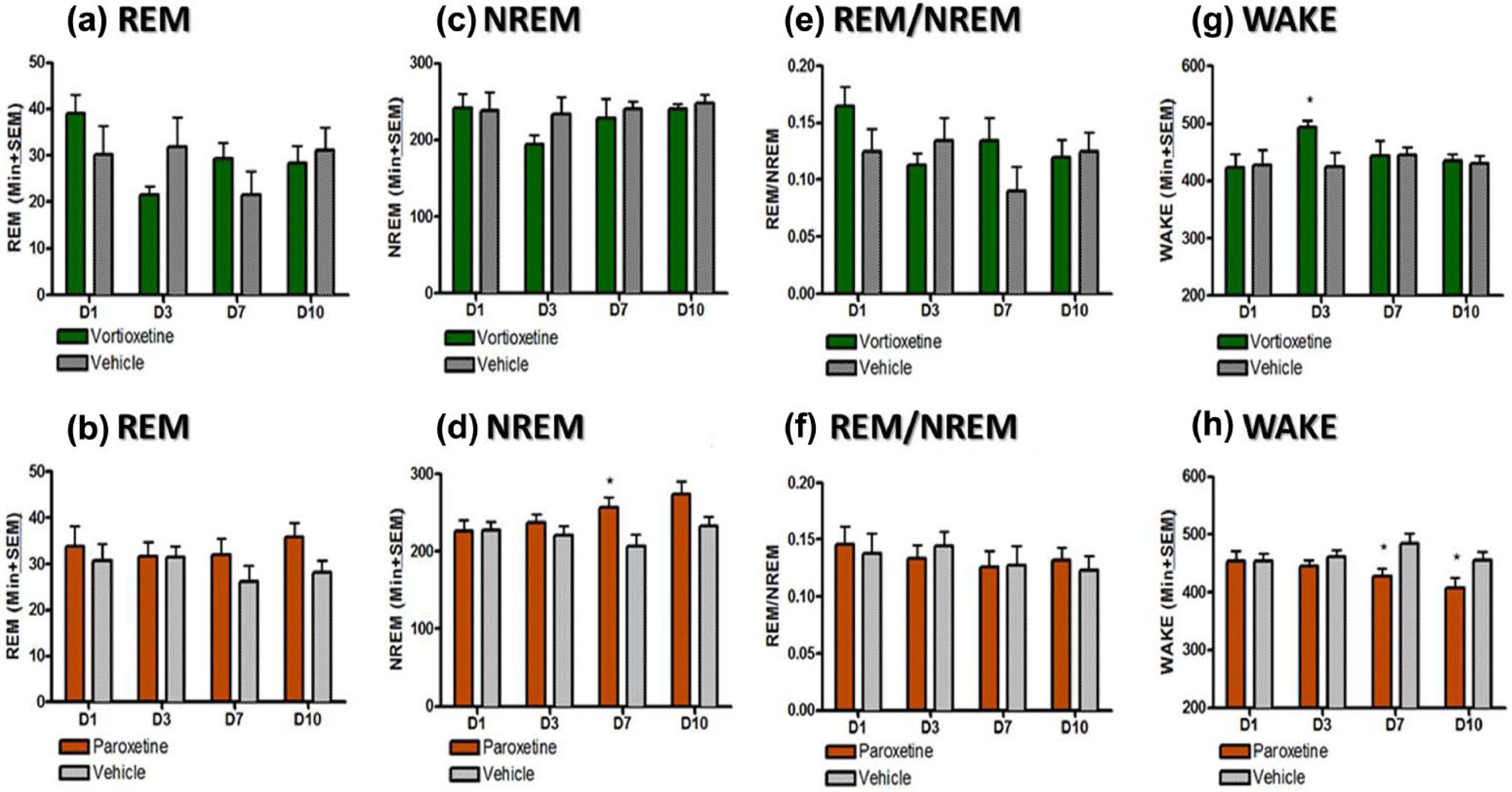
Effect of vortioxetine and paroxetine on rapid eye movement (REM), non-rapid eye movement (NREM), wake (quiet wake and active wake), and REM/NREM ratio during 12-hour dark phase (18:00–06:00). Data shown reflects mean± standard error of the mean (SEM) of the sum of time spent in each stage for each 30-minute epoch after drug administration for day 1 or the whole light phase for all other days. Each treatment group was compared to its respective vehicle control for each day separately (*p<0.05; **p<0.01; ***p<0.005).
Furthermore, the transitions of NREM to wake were investigated (Figure 4). Typically during normal sleep, there are very few NREM sleep-to-wake transitions given that most NREM sleep bouts are followed by REM prior to wakefulness. Vortioxetine had normal NREM to wake transitions for the duration of the study (Figure 4(a)). However, paroxetine (Figure 4(b)) showed an increased number of these transitions on day 1, 7 and 10 (p<0.05, unpaired t-tests). Taken together the decreased time spent in wake during the dark phase and the increased number of NREM to wake transitions suggest that with the paroxetine treatment rats were waking up more when they are supposed to be asleep and had poor sleep efficiency since they had fewer NREM to REM transitions. Seemingly, animals are sleeping more (e.g. in NREM or REM more) during the subjective night (i.e. dark phase) when they are supposed to be in wake. This may correspond to a commonly reported adverse effect of SSRIs – that is, day-time drowsiness.
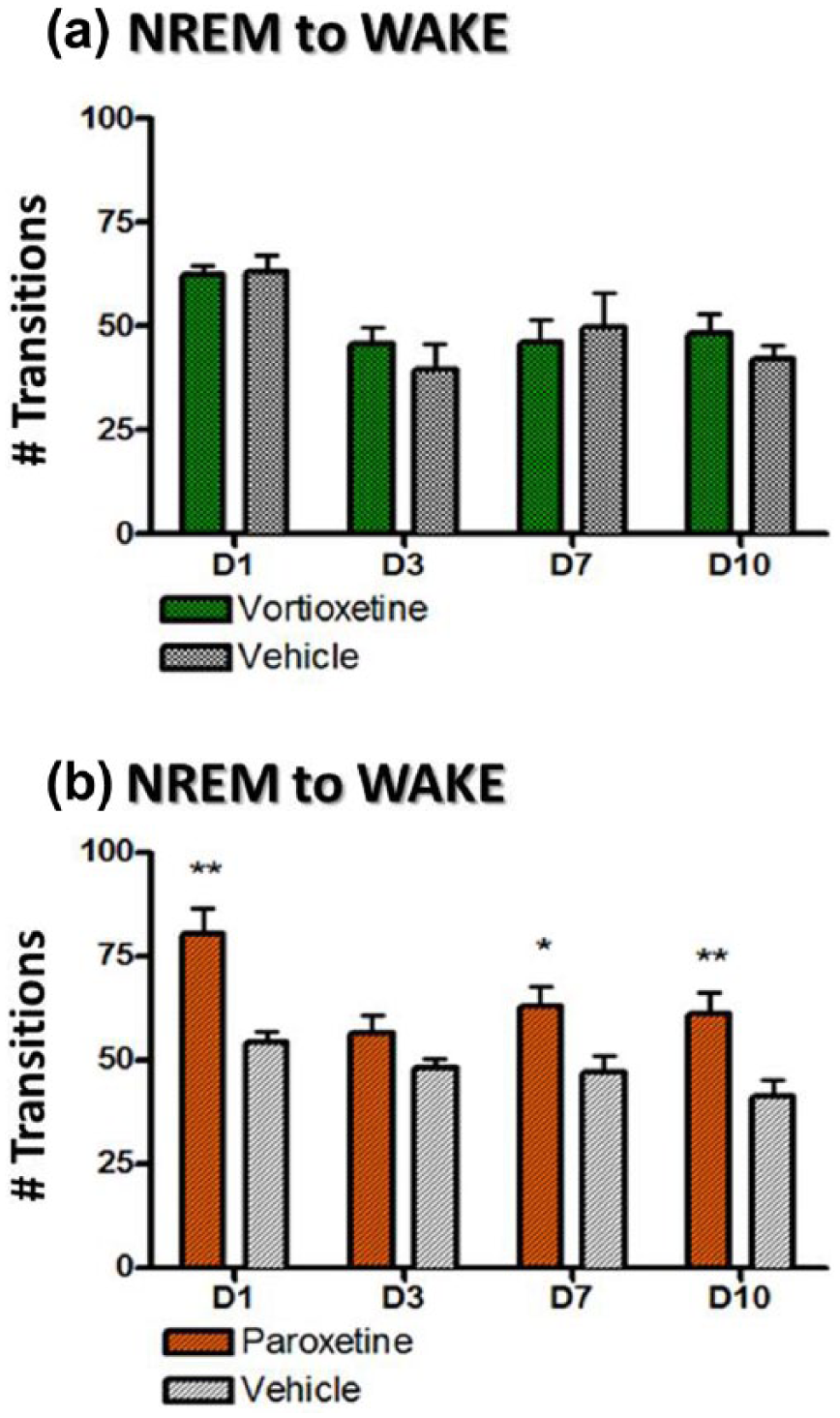
The mean (±standard error of the mean (SEM)) number of transitions from non-rapid eye movement (NREM) to wake during days 1, 3, 7 and 10 and calculated for 24 h after drug administration for day 1 and the time-matched 24-hour period for all other days. Each treatment group was compared to its respective vehicle control for each day separately (*p<0.05; **p<0.01; ***p<0.005).
Chronic vortioxetine/paroxetine occupancy data
Prior to conducting the subchronic paroxetine sleep-EEG study, a satellite group of animals was treated with paroxetine at 1, 3, and 5 mg/kg/day p.o. (via drinking water) for 14 days (n=3 rats/dose). SERT occupancy was determined as described previously and all doses yielded greater than 80% SERT occupancy (Table 2). Additionally, animals treated with 2.5 mg/kg/day paroxetine from the sleep-EEG portion of this study were assessed in terms of SERT occupancy, and these animals also had greater than 80% SERT occupancy (Table 2). Importantly, previous data demonstrates that the subchronic dose of vortioxetine infused in chow used in this study also yields greater than 80% SERT occupancy (Wallace et al., 2014). Previous reports have shown that ondansetron at 0.3 mg/kg yields 5-HT3 receptor occupancy in the forebrain only in the range of 30–35% (Du Jardin et al., 2014; Leiser et al., 2014b).
Effects of subchronic paroxetine dose on serotonin transporter occupancy, as assessed with ex vivo autoradiography.
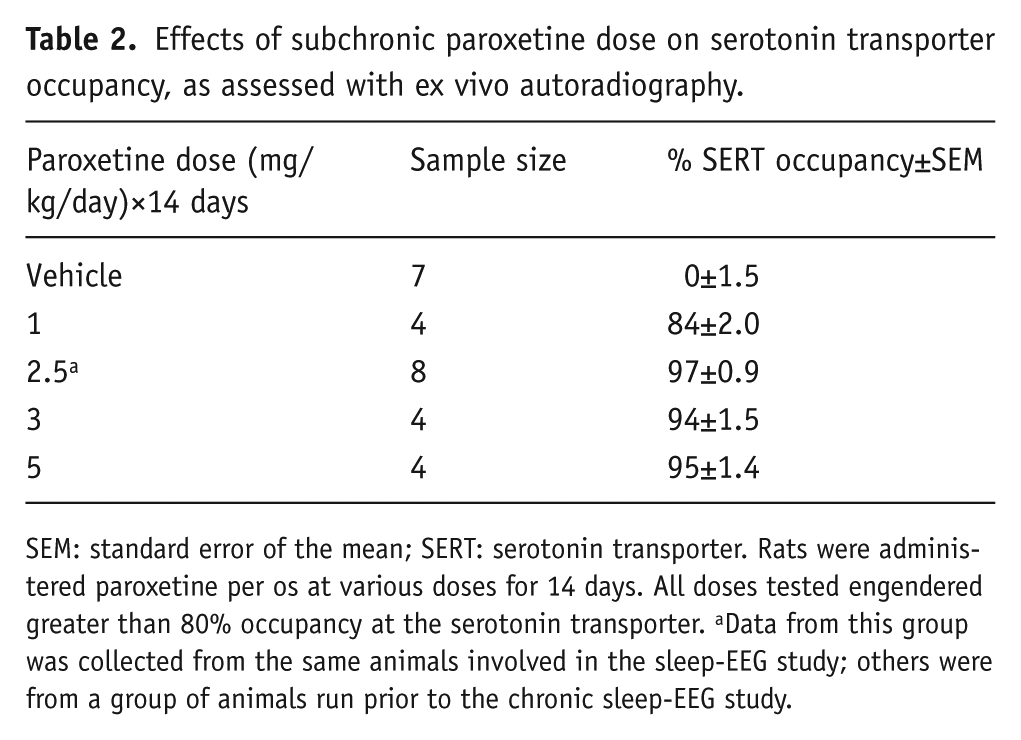
SEM: standard error of the mean; SERT: serotonin transporter. Rats were administered paroxetine per os at various doses for 14 days. All doses tested engendered greater than 80% occupancy at the serotonin transporter. aData from this group was collected from the same animals involved in the sleep-EEG study; others were from a group of animals run prior to the chronic sleep-EEG study.
Acute paroxetine vs paroxetine with ondansetron
In a separate experiment, paroxetine when dosed acutely (0.5 mg/kg, s.c., n=12) significantly suppressed REM with a REM onset latency of 145±8 min post dose (Figure 5(a)). However, when paroxetine was dosed in combination with ondansetron (0.3 mg/kg, s.c., n=8), REM onset latency was significantly shorter at 107±8 min post dose (p=0.004, unpaired t-test). For comparison, after vehicle (n=10) administration alone, REM onset latency was 72±8 min and ondansetron (n=7) was 98±15 min post dose (Figure 5(b)). There was no difference between the vehicle and ondansetron groups (p=0.331, unpaired t-test).
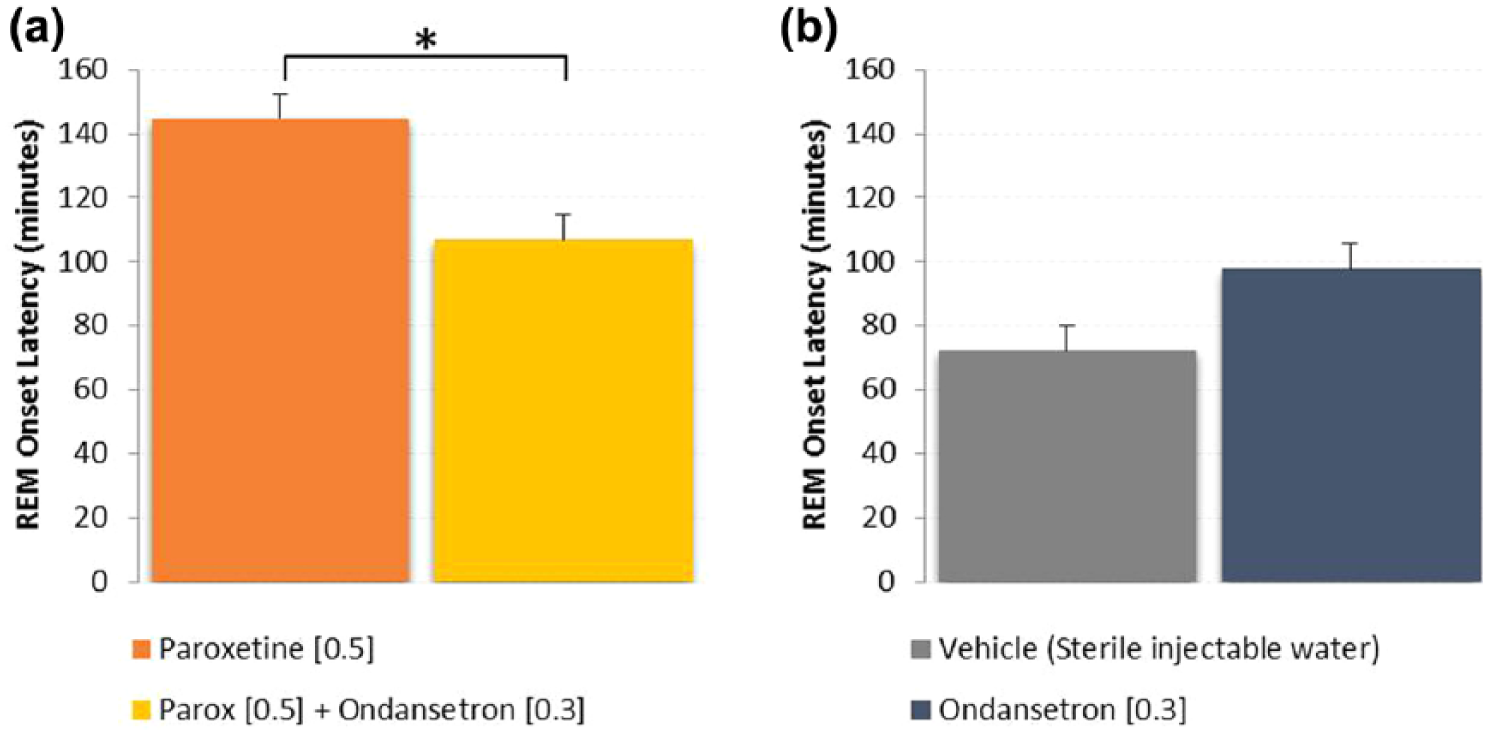
Rapid eye movement (REM) onset latency following acute administration of paroxetine alone and in combination with ondansetron shows that the 5-HT3 antagonist reduced REM onset latency (*p<0.05, unpaired t-test). Ondansetron alone was not different than vehicle (p=0.331). Paroxetine was significantly greater than vehicle (p<0.001). Ondansetron in combination with paroxetine did not reach vehicle control levels (p<0.01, unpaired t-test), but did significantly reduce REM onset from paroxetine alone (p=0.004, unpaired t-test).
Acute vortioxetine with SR57227A
In the final experiment vortioxetine was combined with the 5-HT3 receptor agonist SR57227A (Figure 6). When dosed alone vortioxetine (1 mg/kg, s.c.; n=10) produced a REM onset latency of 75±9 min post dose compared to vehicle (n=10), which produced a REM onset latency of 62±5 min (Figure 6(a) and (b)). In combination with SR57227A, vortioxetine (n=10) produced a significantly greater REM onset latency of 116±8 min (p=0.003, unpaired t-test) (Figure 6(a)). SR57227A treatment alone (n=10) produced a REM onset of 117±10 min, which was significantly greater than vehicle (p=0.0001, unpaired t-test) (Figure 6(b)).
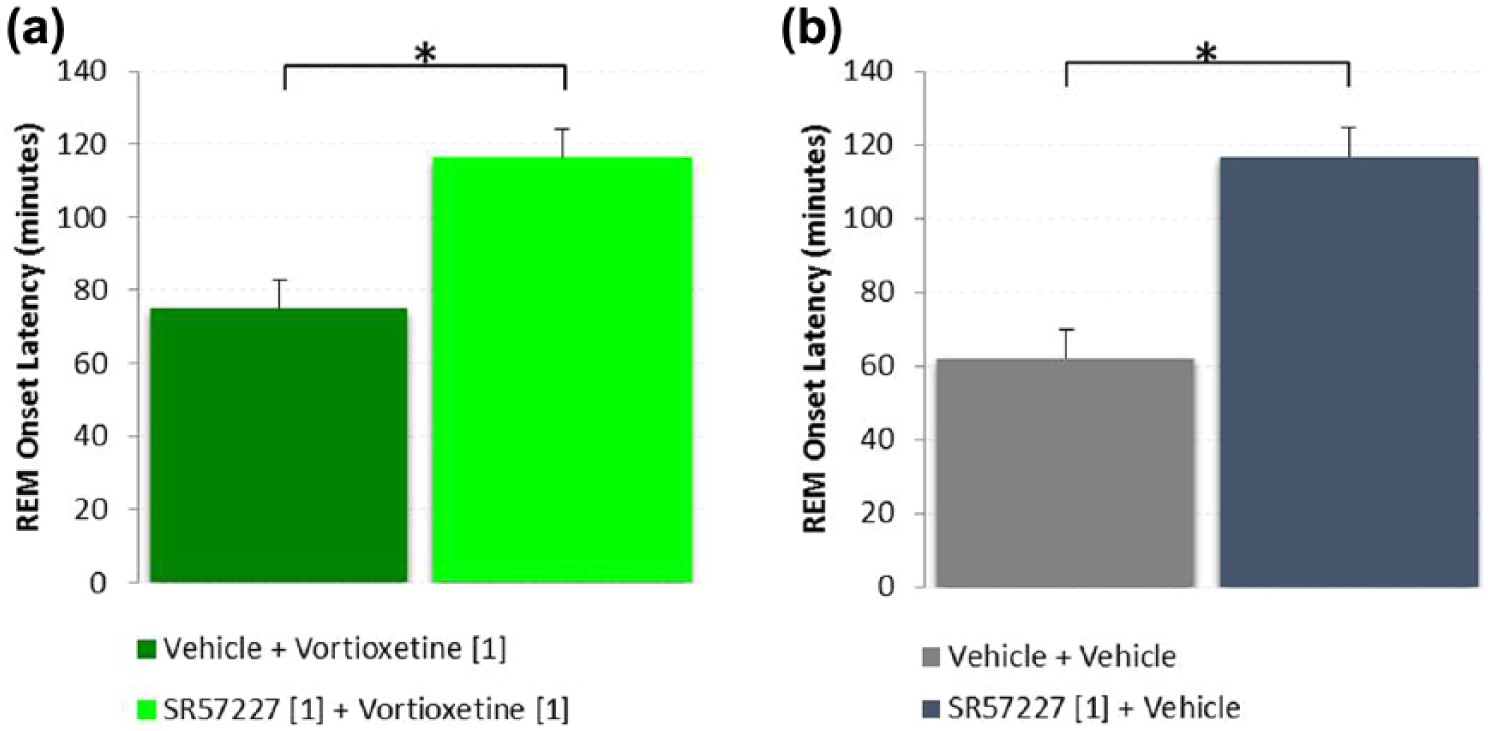
Rapid eye movement (REM) onset latency following acute administration of vortioxetine alone and in combination with SR57227A shows that the 5-HT3 agonist increased REM onset latency (*p<0.05, unpaired t-test). SR57227A alone induced a significant delay in REM onset compared to vehicle (p<0.001). Vortioxetine alone was not different than vehicle (p=0.228), however SR57227A in combination with vortioxetine significantly delayed (increased) REM onset compared to vortioxetine alone (p=0.003, unpaired t-test).
Discussion
In our comparative EEG study in rats of acute and subchronic effects of paroxetine and vortioxetine, we have shown that vortioxetine impacted the sleep architecture much less than paroxetine, in particular on measures of REM sleep and NREM to wake transitions which are considered as measures of sleep fragmentation.
Whereas paroxetine suppressed REM sleep both acutely and continually during the sub-chronic treatment, with vortioxetine treatment only the acute dose had an effect. The effect of paroxetine is in agreement with the effects of SSRIs described in the literature, where chronic treatment with the SSRIs citalopram and escitalopram demonstrated long-term REM suppression in rats. Specifically, after five weeks of chronic treatment with citalopram in rats there was still a 21% reduction in total REM sleep (Neckelmann et al., 1996) and after three weeks of treatment with escitalopram in rats there was a reduction in REM sleep and onset latency (Vas et al., 2013). Moreover, our data in rats are consistent with a previous report in healthy subjects showing that REM sleep was significantly suppressed by both citalopram and paroxetine (Wilson et al., 2004). That is, in our study the total number of NREM to wake transitions in 24 h was significantly increased in paroxetine-treated animals on days 1, 7 and 10. Also, looking beyond the acute effects, in the dark phase paroxetine, but not vortioxetine, reduced the total time in wake and increased the total time in NREM on days 7 and 10. In the light phase paroxetine, but not vortioxetine, decreased REM but also increased NREM on days 3, 7, and 10. These analyses demonstrate that paroxetine altered ultradian sleep patterns and thus we may relate to the metric of sleep fragmentation used clinically: in healthy subjects sleep fragmentation was significantly increased by both citalopram and paroxetine (Wilson et al., 2004).
Because vortioxetine, at a given SERT occupancy, seemed to affect REM sleep differently than paroxetine in both our study in rats and Wilson et al.’s study in healthy subjects (this issue), we sought to experimentally address the hypothesis we have previously suggested that the 5-HT3 receptor antagonistic properties of vortioxetine may lessen its effect on REM (Bétry et al., 2011; Leiser et al., 2014a; Sanchez et al., 2015). This hypothesis is derived in part from published studies of selective 5-HT3 receptor ligands studied in animals with a normal 5-HT tone, i.e. without the elevated tone produced by SERT inhibition (see introduction and Table 1). To test the hypothesis we first measured the acute REM-suppressing effect of paroxetine alone and with pretreatment with the selective 5-HT3 receptor antagonist ondansetron. As hypothesized, the combination significantly attenuated the REM-suppressing effects of paroxetine. Next, to further establish the role of 5-HT3 receptors in mediating these effects, we tested whether adding a selective 5-HT3 receptor agonist to vortioxetine would counteract the effects of its 5-HT3 antagonism and yield greater REM suppression. To test this hypothesis we co-administered SR57227A with vortioxetine. SR57227A is a selective 5-HT3 receptor agonist (Delvaux, 2002). This combination elicited a longer delayed REM onset latency than vortioxetine alone. This effect was similar to that reported in healthy subjects where SR57227A produced a dose-dependent shift of REM toward the end of the night (Staner et al., 2001). In summary, acutely, vortioxetine (1 mg/kg) elicited a mean REM onset latency of approximately 75 min, while paroxetine (0.5 mg/kg) elicited a much greater mean REM onset latency of approximately 145 min, but when the 5-HT3 receptor agonist SR57227A was combined with vortioxetine the combination elicited a mean REM onset latency closer to paroxetine, suggesting that the reduced effect on REM suppression elicited by vortioxetine alone is, at least in part, due to its 5-HT3 receptor antagonism. Vortioxetine is also a SERT inhibitor and thus it does have REM-suppressing properties, however the effects contributed by SERT seem to be offset by vortioxetine’s other actions. The mechanism by which these effects happen is currently not well understood but is discussed below in the context of all findings. However, the interaction of SR57227A with vortioxetine likely takes place at 5-HT3 receptors since vortioxetine at a dose of 1.0 mg/kg practically only occupies SERT and 5-HT3 receptors (Sanchez et al., 2015).
5-HT3 receptors are expressed on inhibitory GABAergic interneurons, where they are thought to provide a serotonin-mediated fast excitatory drive (Kawa, 1994; Leiser et al., 2015; Pehrson and Sanchez 2015). 5-HT3 receptor antagonists suppress the firing of GABAergic interneurons as well as elicit changes to neuronal firing downstream of GABAergic interneurons (Ashby et al., 1991; Puig et al., 2004; Reznic and Staubli, 1997). Additionally, 5-HT3 receptor antagonists increase ACh levels as measured by microdialysis (Barnes et al., 1989; Giovannini et al., 1998) and increase cortical theta rhythms as measured by EEG (Staubli and Xu, 1995). Notably, vortioxetine has been shown to both increase ACh measured at acute timepoints (Mørk et al., 2013) and increase theta oscillations in the frontal cortex of rats (Leiser et al., 2014b) after acute administration. These effects are likely driven by its 5-HT3 receptor antagonism. Therefore, since REM sleep only occurs when the aminergic system suspends its inhibitory effect on cholinergic activity (Aloe et al., 2005) and, given that 5-HT3 receptor antagonism can increase ACh, this may stimulate REM onset.
To understand how 5-HT3 receptors could modulate sleep, we must first discuss some basic premises of how REM sleep is initiated (see Brown et al., 2012; Datta and Maclean, 2007; Staner et al., 2008 for more in depth review of this process). Briefly, the two brain regions predominantly involved in generation of REM are the PPT nucleus, which is maximally active immediately before and during REM sleep, and the LDT. The output of these brain regions have been coined the cholinergic REM-ON neurons. Interestingly, local injections of cholinergic agonists into the PPT triggers REM sleep (Jones, 1991). The DRN projects its 5-HT neurons to the LDT/PPT complex and when the DRN is tonically active, such as in wake, it inhibits the LDT/PPT thereby suppressing its REM-ON neurons. This inhibitory drive supplied by the DRN will be active as long as the DRN is being activated with wake-active neurons (e.g. NE, DA, HA, OX, ACh). However, as the diurnal switch draws near and less wake-active neurons are supplying input to the DRN, the sleep-active neurons (e.g. GABA and melanin-concentrating hormone/melanin) inhibit the DRN, withdrawing the DRN’s inhibitory drive on the LDT/PPT, thereby activating REM-ON neurons. The LDT/PPT sends glutamatergic neurons to the medial pontine reticular formation (mPRF), which in turn sends glutamatergic neurons to stimulate the cholinergic REM-ON neurons. As mentioned above, when active the DRN inhibits the mPRF, but during sleep, this inhibition is lost, further reinstating REM. Once the cholinergic REM-ON neurons are activated in the LDT/PPT, the locus coeruleus and the DRN are in turn inhibited explaining why these structures exhibit no firing rate during REM. Recall from the introduction that during wakefulness serotoninergic tone gives rise to enhanced cortical activity and arousal, while during sleep the awake-related neurons slow down, thereby withdrawing their effects on REM sleep-related neurons. As 5-HT levels drop, this triggers an increase in the acetylcholinergic REM-ON neuronal activity, which in turn excites GABAergic neurons to inhibit the REM-OFF neurons (5-HT neurons of the DRN and NE neurons of the LC), resulting in initiation of REM sleep (Aloe et al., 2005; Sutcliffe and de Lecea, 2002).
Another possibility is that the 5-HT3 receptor antagonism-induced increase in REM is mediated by glutamate. 5-HT3 receptors are also located on glutamatergic interneurons in the DRN where they elicit glutamate release (Monti and Monti, 2000, Monti et al., 2011). Activation of glutamatergic interneurons that express 5-HT3 receptors in DRN facilitates the release of glutamate, which, in turn, acts on postsynaptic N-Methyl-D-aspartate (NMDA) and non-NMDA receptors expressed by serotonergic neurons of the DRN and increases the release of 5-HT at postsynaptic sites (Monti, 2011). Presumably, 5-HT3 antagonists would inhibit these glutamatergic interneurons and in turn decrease 5-HT release at postsynaptic sites (Monti and Jantos, 2008). If so, as 5-HT levels drop, this triggers an increase in the acetylcholinergic REM-ON neuronal activity, and the cascade occurs as described above until REM sleep is initiated.
Finally, vortioxetine may act via other mechanisms as well to overcome the typical SSRI-induced sleep disruption. For example, vortioxetine may mediate its effect on sleep via its 5-HT1B partial agonist properties (El Mansari and Blier, 2015) to contribute to its overall lesser effect on REM. That is, under elevated 5-HT tonus (e.g. SERT inhibition), the 5-HT1B receptor partial agonism could in fact function to reduce the overstimulation of receptors thereby acting similar to an antagonist (Zhu, 2005) and we know from the literature that 5-HT1B receptor antagonism induces REM sleep (Boutrel et al., 1999; Monti et al., 2010). Moreover, a preclinical sleep EEG study in rats by Bonaventure et al. (2007) reported an increase in the number of micro-arousals in citalopram-treated rats and showed that the 5-HT7 receptor antagonist SB-269970 counteracted this effect. Ex vivo autoradiography studies of vortioxetine’s target to dose relation would suggest that vortioxetine occupies 5-HT7 receptors in our chronic sleep EEG study. It may therefore be hypothesized that vortioxetine’s neutral effect of NREM to wake transitions involves its 5-HT7 receptor antagonistic properties. In fact, it has been demonstrated that 5-HT7 receptors can mediate phase advances of the circadian biological clock through increases in cAMP production (Sprouse et al., 2004) and that the combination of an SSRI and a 5-HT7 receptor antagonist has a greater impact on circadian rhythms than that observed with either agent alone (Westrich et al., 2013, 2015). Nevertheless, it is clear that more work is needed to fully understand the mechanisms involved in vortioxetine’s effects on REM sleep and its different clinical pharmacological profile than a SSRI.
Overall, our preclinical findings support that these two antidepressants at clinically relevant levels of SERT occupancy have different modes of action on sleep-wake rhythms. This is in line with the overall conclusion of the clinical study in healthy subjects reported by Wilson et al., and previous reports (Baldwin et al., 2012; Wilson et al., 2013). Whether these differences have an impact on sleep in depressed patients or contribute to vortioxetine’s efficacy as an antidepressant is worthy of study.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.