
Abstract
Colorectal cancer (CRC) is a global cause of cancer-related mortality driven by genetic and environmental factors which influence therapeutic outcomes. The emergence of next-generation sequencing technologies enables the rapid and extensive collection and curation of genetic data for each cancer type into clinical gene expression biobanks. We report the application of bioinformatics tools for investigating the expression patterns and prognostic significance of three genes that are commonly dysregulated in colon cancer: adenomatous polyposis coli (APC); B-Raf proto-oncogene (BRAF); and Kirsten rat sarcoma viral oncogene homologue (KRAS). Through the use of bioinformatics tools, we show the patterns of APC, BRAF and KRAS genetic alterations and their role in patient prognosis. Our results show mutation types, the frequency of mutations, tumour anatomical location and differential expression patterns for APC, BRAF and KRAS for colorectal tumour and matched healthy tissue. The prognostic value of APC, BRAF and KRAS genetic alterations was investigated as a function of their expression levels in CRC. In the era of precision medicine, with significant advancements in biobanking and data curation, there is significant scope to use existing clinical data sets for evaluating the role of mutational drivers in carcinogenesis. This approach offers the potential for studying combinations of less well-known genes and the discovery of novel biomarkers, or for studying the association between various effector proteins and pathways.
Introduction
Colorectal cancer (CRC), also known as bowel cancer, is a type of cancer that occurs as a result of uncontrolled cell growth in parts of the large intestine or the appendix, and is a major cause of global morbidity and mortality. In 2020, CRC was ranked as the third most common cancer worldwide, with 1,931,590 cases (10% of global cancers) and the second highest mortality rate, with 935,173 deaths worldwide (9.4% of cases). 1 It is predicted that the number of new cases will rise to approximately 2.5 million in 2035. 2 Surgery and chemotherapy are routine standard of care interventions for the treatment of CRC. Novel interventions, such as targeted therapies against genetic mutations and the rapid development of screening techniques such as (virtual) colonoscopy, sigmoidoscopy, stool-based tests and liquid biopsies, have led to improvements in the rapid early detection of the disease and extended overall survival rates for patients with CRC. Overall, the five-year survival rate for CRC is ∼64%. 3
CRC is a heterogeneous disease driven by both genetic instability and environmental factors.4,5 Major advancements in DNA sequencing technologies have resulted in the rapid expansion of biobanks and repositories for patient tumour biopsies. This significant wealth of data offers scientists the opportunity to examine common gene mutations, the corresponding cellular pathways impacted by gene alterations, and their influence on tumour growth and development. A major challenge in the treatment of CRC is identifying the multiple biomolecular variables that impact the emergence of metastases and their prognostic implications. Various pathways mediate the initiation, progression and metastasis of CRC, and those involved in the activation of signalling cascades are ideal targets for the development of ‘targeted therapy’. Therefore, the identification of commonly-occurring mutations in patients with CRC, and the effects of these mutations on cellular pathways, are crucial for developing a deeper understanding of CRC pathogenesis, and in turn developing susceptibility biomarkers and novel drug treatments for CRC. 6
Studies related to the prevention, treatment and development of CRC use both in vitro cell cultures and in vivo preclinical models to mimic the cancer microenvironment. A review conducted in 2015 explored the use of preclinical models in CRC by investigating the current literature at that time (477 relevant articles). Overall, each reported preclinical model showed limitations arising from a lack of spontaneous CRC development, or the need for a carcinogen to induce tumorigenesis in rodent models. Moreover, murine preclinical models exhibit significant inter-animal variability in the development of intestinal tumours, which adds to the complexity of preclinical analyses. 7 Alternatively, freely accessible human clinical data sets contain a wealth of data to allow the study of expression patterns that can then be used to evaluate the prognostic significance of gene mutations in human CRC tissue. There is also a further option to use the clinical data sets to investigate the role of clinicopathologic parameters in patient prognosis, delineated by sex and age of the patient, and anatomical location and stage of the cancer.
Three genes play a significant role in CRC development and progression: the tumour suppressor gene adenomatous polyposis coli (APC), the B-Raf proto-oncogene (BRAF), and the Kirsten rat sarcoma virus gene (KRAS).6,8 The most common CRC activation pathway, which is responsible for 70–80% of all CRC diagnoses, is triggered due to inactivation of the tumour suppressor gene APC, which has an important role in the wnt signalling pathway, intercellular adhesion, cytoskeleton stabilisation, cell cycle regulation and apoptosis.9-11 Therefore, truncating mutations of APC resulting in its inactivation are thought to promote tumorigenesis and adenoma formation,
11
and subsequent KRAS and TP53 mutations propagate the development of carcinomas, commonly found in left-sided colon cancers (LCCs). However, KRAS mutations occur at a higher frequency in right-sided colon cancers (RCCs), alongside mutations in the BRAF gene.
12
Recent epidemiological and clinical studies have demonstrated that the anatomical location of CRC tumours, alongside the patient sex, impacts the overall survival, as patients with RCC were correlated to a poorer prognostic outcome.12-15 Exploring the genetic and prognostic rationale of these gene mutations is important, in order to identify new biomarkers for diagnosis, determine novel potential drug targets for treatment, and explore new effective treatment combinations. The aim of the present study was to develop an integrated bioinformatics pipeline that would blend orthogonal data sets and interrogate the two main cellular pathways as drivers of CRC aggressiveness in patient populations (Figure 1). Genetic and biomolecular differences in colorectal cancer as a function of anatomical location. Right-sided colon cancers (RCCs) occur in the cecum, ascending colon and hepatic flexure. Left-sided colon cancers (LCCs) occur in the splenic flexure, descending, sigmoid and rectosigmoid colon.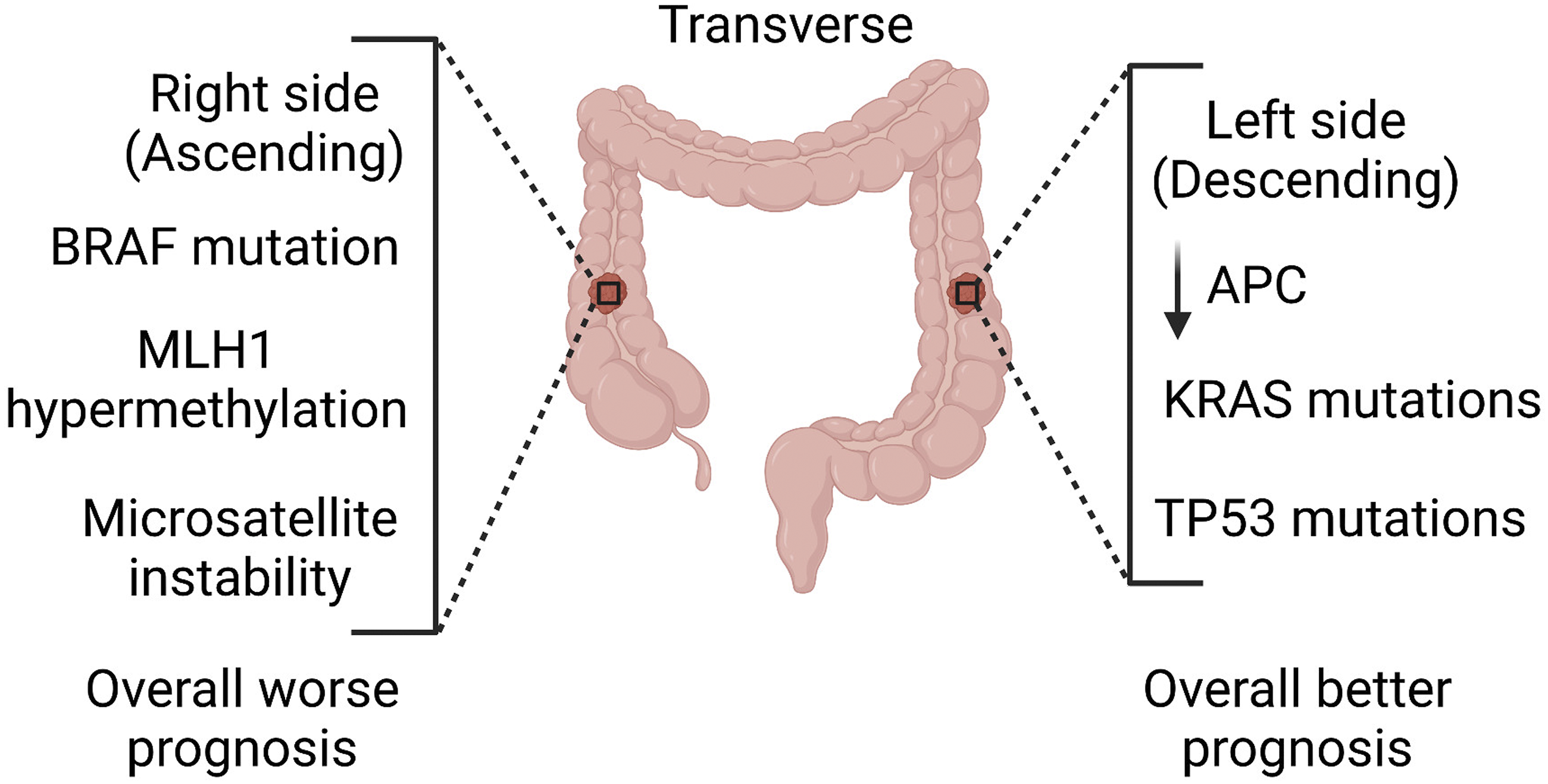
To date, several studies have applied bioinformatics tools to investigate diagnostic and prognostic data, and have implicated key pathways and genetic alterations contributing to CRC progression. Some of these studies compared the expression profiles of samples obtained from CRC patients with those obtained from healthy patients.16-20 Yang et al. used bioinformatic analyses to explore the upregulation and downregulation of microRNAs in CRC tissues, and investigated their role in tumorigenesis, invasion and migration. 21 A range of bioinformatics-based studies have reported the evaluation of actionable gene mutations (i.e. mutations likely to influence treatment options) and their role in CRC progression and metastasis.22,23
In this study, we report the analysis of driver mutations (primarily APC, BRAF and KRAS) in CRC by using existing clinical data sets that were obtained from a range of publicly available biorepositories. The objective was to develop a system to help interrogate the role of lesser-characterised genes and gene combinations from existing clinical data sets, aiding the development of novel biomarkers for CRC stratification and the identification of novel drug targets. We present the range of identified genetic alterations occurring in APC, BRAF and KRAS in CRC, as well as their association with patient prognostic outcomes. To our knowledge, this study is the first reporting the use of multiple bioinformatics-based resources for research on CRC.
Methods
cBioPortal
cBioPortal (https://www.cbioportal.org/) is an open access resource for cancer genomics that was originally developed by the Memorial Sloan Kettering Cancer Center. 24 This resource was used to investigate common gene mutations found in patients with CRC. A total of seven studies (n = 2575) covering colorectal adenocarcinoma, metastatic colorectal cancer, colon adenocarcinoma and colon cancer were explored, and a list of frequent mutated genes was generated.25–31 Additionally, using the same seven studies, the APC/BRAF/KRAS gene pairing was selected. Under the ‘cancer type summary’ tab, ‘cancer type’ was selected to generate a bar chart showing the alteration frequency in each cancer type.
PrognoScan
PrognoScan (http://dna00.bio.kyutech.ac.jp/PrognoScan-cgi/PrognoScan.cgi) is a database for performing a meta-analysis of the prognostic value of a certain mutation occurring in cancer through the incorporation of results from gene expression studies from multiple sources, such as the Gene Expression Omnibus (GEO) as well as reports from individual laboratories. 32 It relates gene expression data to prognostic outcomes, which enables the evaluation of potential biomarkers and their role in cancer prognosis. In this study, PrognoScan was used to assess the correlation between APC, BRAF and KRAS mRNA expression levels, and patient overall survival (OS), disease-free survival (DFS) and disease-specific survival (DSS). The output generated from PrognoScan includes P values (Cox), hazard ratios and confidence intervals. APC (Gene ID: 324), BRAF (Gene ID: 673) and KRAS (Gene ID: 3845) were independently entered in the PrognoScan website and the P values (Cox), hazard ratios and confidence intervals were obtained for colorectal cancer studies. R Studio software, in combination with the forestplot package, was used to generate a forest plot from the values obtained via PrognoScan.
Oncomine
Oncomine (https://www.oncomine.org/resource/login.html) is a bioinformatics platform with an extensive collection of data sets. 33 Analysis of the APC/BRAF mRNA expression pattern was performed by selecting the following parameters: gene, APC/BRAF; differential analysis, cancer vs normal analysis; cancer type, colorectal cancer; sample type, clinical specimen; and data type, mRNA.
It was ordered by: Under-expression, gene rank (APC); and Overexpression, gene rank (BRAF). A two-fold change, P value corresponding to 1 × 10–4, and top 10% gene rank, were selected as the threshold for this analysis. All statistical analyses containing mRNA reporters 215310_at (APC), 203525_s_at (APC) and 243829_at (BRAF) were directly exported from Oncomine.
KMplot
KMplot (http://kmplot.com/analysis/index.php?p=service) is an online survival evaluation platform to enable the meta-analysis of a number of different data sets comprised of gene expression data. 34 The correlation between overall survival and APC/BRAF mRNA expression was assessed by using: mRNA [RNA-seq], Start KM Plotter for pan-cancer; Gene symbol, APC/BRAF; Select all; select Draw Kaplan–Meier plot.
Statistical analysis
A comparison of APC/BRAF mRNA expression levels was directly obtained from Oncomine with a Student’s paired t-test. The prognostic data obtained from PrognoScan was selected according to the calculated Cox p values and corresponding hazard ratios (95% confidence interval) for various endpoints (OS, DFS, DSS), which were visualised with a forest plot. For comparison of patient survival endpoints, patients were categorised according to survival, and survival difference between high and low expression groups was determined by a log-rank test in PrognoScan. False discovery rates below 5% and p < 0.05 were considered as statistically significant for all comparisons.
Results
Results were obtained for the APC, BRAF and KRAS genes in all analyses.
Genetic alterations and their frequency in CRC, by using CBioportal
The frequency and type of APC, BRAF and KRAS genetic alterations were examined as a function of CRC clinicopathologic parameters by using data from seven clinical CRC studies (2575 samples) in cBioportal.25–31
Across the seven studies surveyed in cBioportal, the percentage of samples in which somatic mutations occurred in APC was 69.6%, with a corresponding frequency of 13.7% and 38.2% for BRAF and KRAS mutations, respectively. In the case of APC mutations, this corresponds to 2814 driver mutations (2747 of these were truncating mutations, 213 missense, 45 splice and 12 fusion); for BRAF, there were 327 driver mutations (319 missense, 3 in-frame, and 5 fusion); and for KRAS, there were 525 driver mutations (525 missense). Most of the mutations located in the APC, BRAF and KRAS sequences (Figure 2a), were annotated as oncogenic in cBioportal. The bar chart obtained from cBioPortal (Figure 2b) shows that the mutation frequency varied in range across studies — for APC, the range was 40–90% across the seven CRC studies selected while, for BRAF, the range was 4–20% and for KRAS it was 28–57%. The distribution, frequency and types of APC, BRAF and KRAS mutations occurring in their protein sequences, across seven CRC studies. (a) A lollipop diagram featuring data from CRC studies selected in cBioportal, illustrating the distribution, frequency and types of changes occurring for APC, BRAF and KRAS mutations. The circles indicate sites in which the mutations occur, and the length of each lollipop represents the number of patients with the specific mutation. (b) The frequency and types of genetic alteration in APC, BRAF and KRAS, according to the different cancer types analysed in seven studies. CRA = colorectal adenocarcinoma; CRC = colorectal cancer; MCRC = metastatic colorectal cancer; CA = colon adenocarcinoma; CC = colon cancer; CRAT = colorectal adenocarcinoma triplets.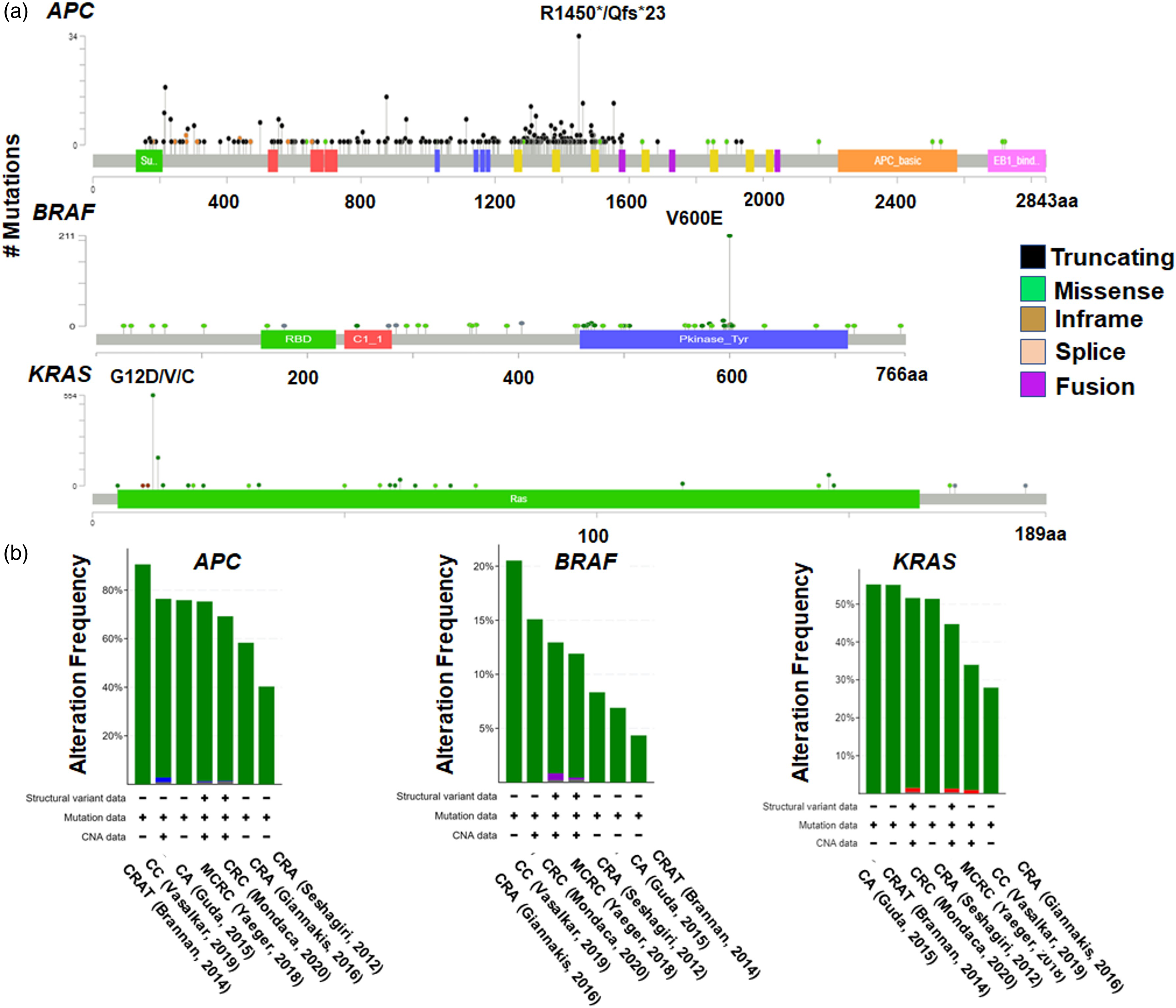
Significant variation was observed in the types of genetic alterations occurring for APC, BRAF and KRAS (Figure 3a–c). Moreover, the range and types of genetic alterations were found to significantly differ as a function of the anatomical location of the tumour in the colon (Figure 3d–f). mRNA expression levels and details of the mutations for the three genes analysed. The mRNA expression levels for (a) APC, (b) BRAF and (c) KRAS, selected from seven studies in cBioportal are shown. The frequency of mutations for the three genes and the proportion of gene alterations as a function of anatomical site are shown in (d) for APC, (e) for BRAF and (f) for KRAS. AC = ascending colon; C = cecum; DC = descending colon; HF = hepatic flexure; SC = sigmoid colon; SF = splenic flexure; TC = transverse colon.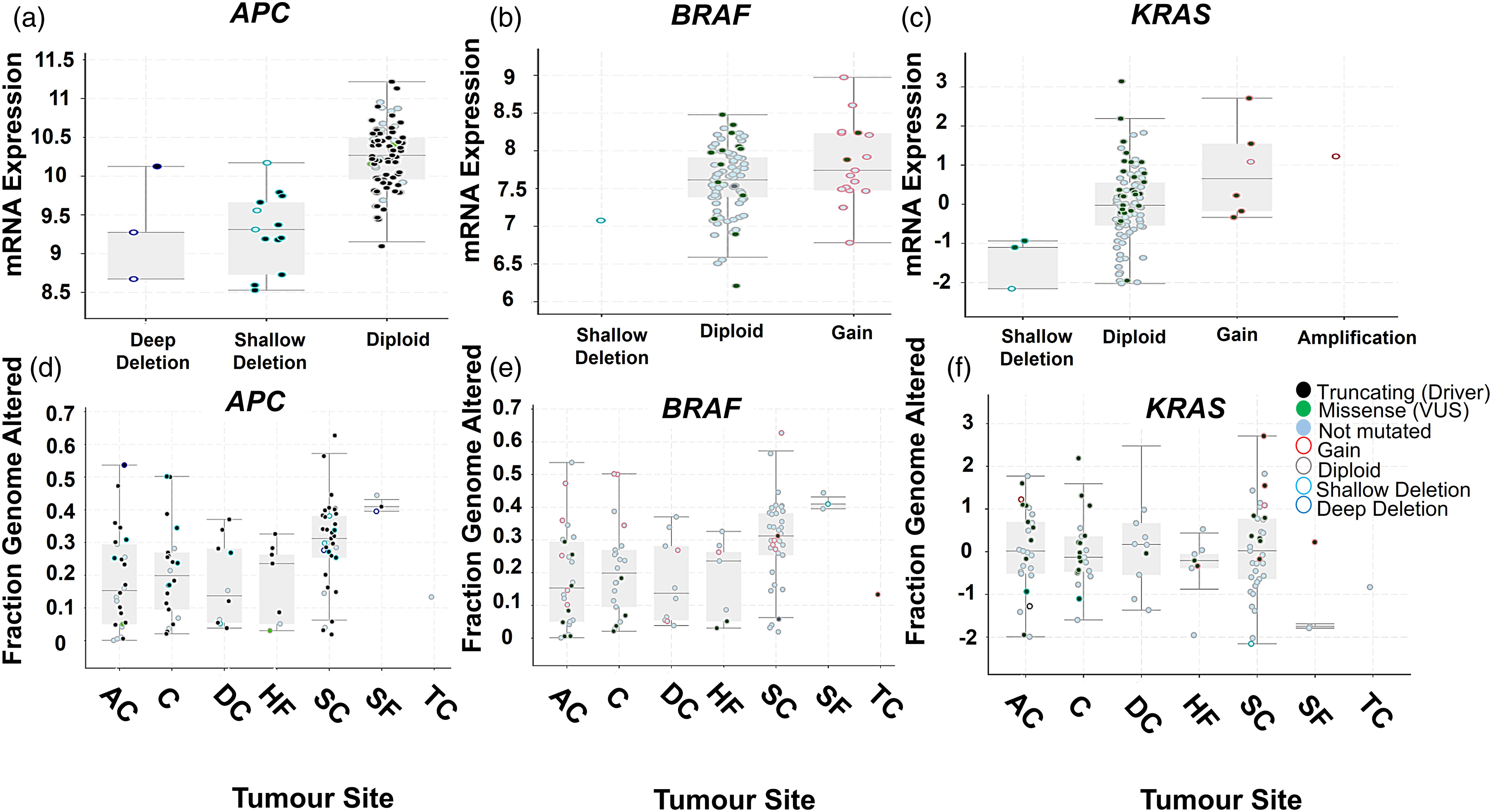
mRNA expression in healthy colon tissue and distinct types of CRC tissue
Three studies (Skrzypczak et al., 35 Kaiser et al. 36 and Hong et al. 37 ) were analysed by using Oncomine, to compare the relative expression patterns of APC, BRAF and KRAS mRNA expression levels in healthy colon tissue and CRC tumours defined by their anatomical location.35-37
Boxplots of APC mRNA expression levels (Figure 4) obtained from Oncomine show a significant reduction of APC expression levels in distinct types of CRC tissue when compared to healthy colon tissue. Analysis of APC mRNA expression level patterns across the three studies showed that results varied between the different CRC tissue subtypes used in the respective studies. In the case of BRAF (Figure 4f–g), it was evident that mRNA expression levels were significantly elevated in CRC tissue, when compared to healthy colon tissue. Corresponding fold-changes in expression levels and other study information derived from Oncomine are summarised in Table 1. APC and BRAF mRNA expression levels in normal colon and CRC tissue. The box plots correspond to the mRNA expression levels of: APC (203525_s_at APC reporter) in plots (a) to (e); and BRAF (243829_at BRAF reporter) in plots (f) and (g). All box plots were generated from Oncomine searches. Corresponding data sets include: in (a) to (c), Skrzypczak Colorectal 235 (n = 40); in (d), Kaiser Colon
36
(n = 105); in (e), Hong Colorectal
37
(n = 82). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, as determined by using the Student’s t-test. Combined APC and BRAF expression levels are frequently altered in CRC compared with healthy colon tissue. Fold-changes in APC (reporter 203525_s_at) and BRAF (reporter 243829_at) mRNA expression levels, relative to matched healthy colon tissue as a function of CRC tumour subtypes, according to Oncomine. n = number of patients. No statistically significant gene alterations were found for KRAS on Oncomine.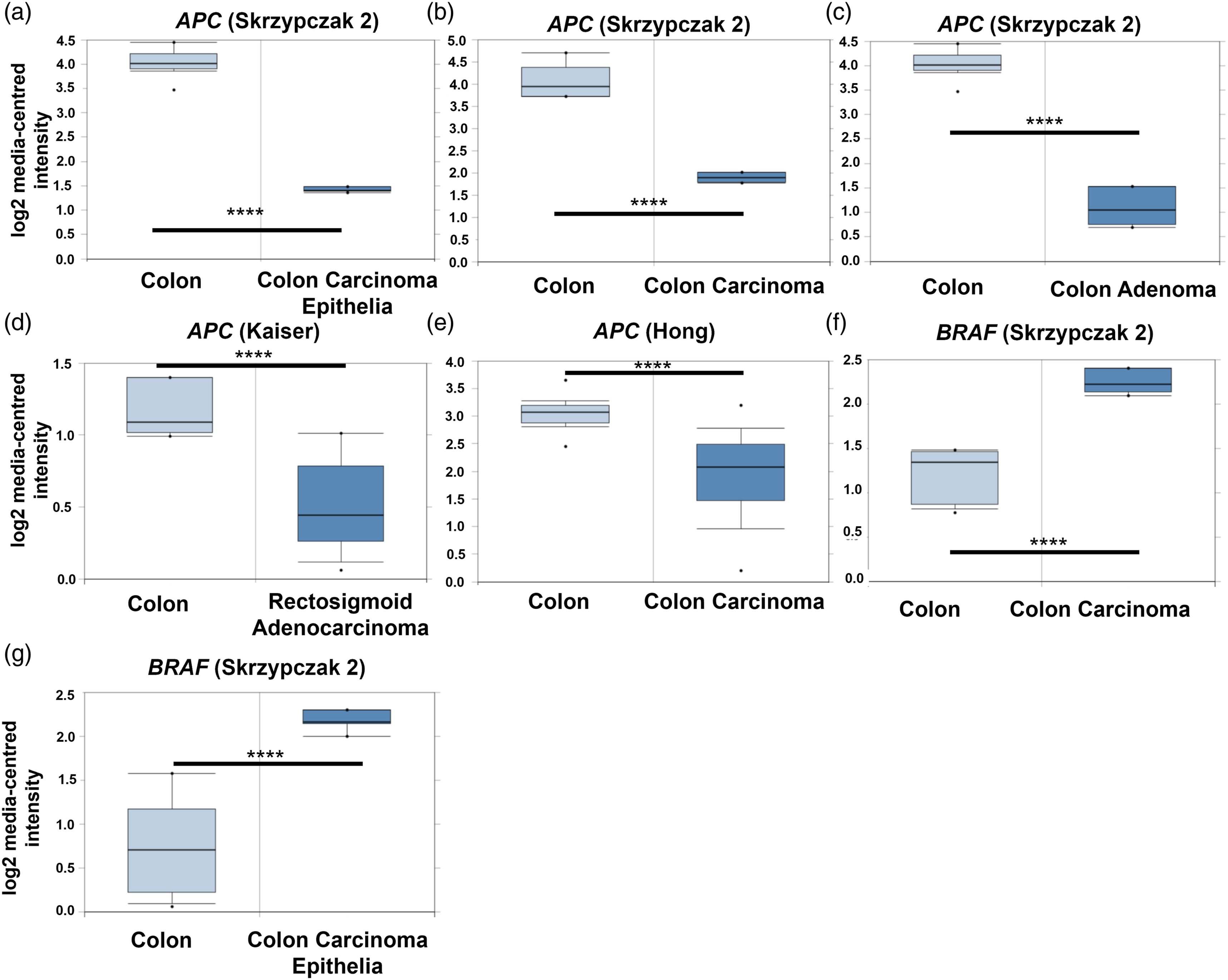
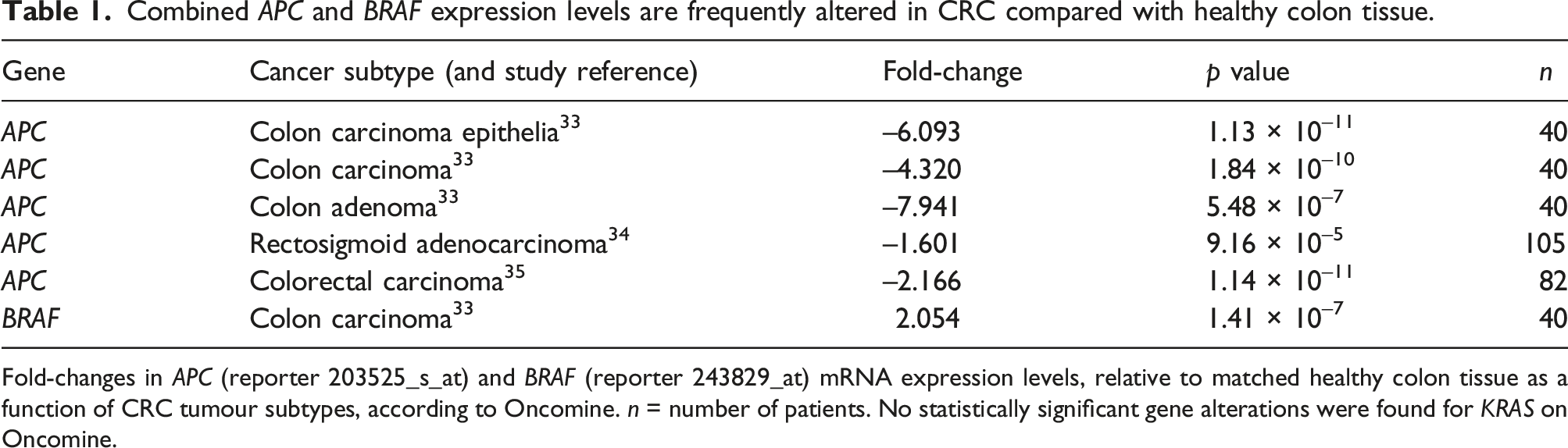
The negative fold-change values > 2, together with P values < 0.05, show that APC gene expression is significantly reduced in CRC tumour tissue. In cases of colon carcinoma epithelia, colon carcinoma and colon adenoma epithelia, high negative fold-changes were observed, confirming a reduction in APC mRNA expression levels in these cancer types, in comparison with rectosigmoid adenocarcinoma. The P values of reported fold-changes were found to be statistically significant (P < 0.05), except for two cases (the colon carcinomas in Skrzypczak et al. 35 and Hong et al. 37 ), which both applied to the 215310_at reporter (see Online Supplementary Figure S1, D). Positive fold-changes (> 2), coupled with P values < 0.05, confirm that BRAF gene expression is significantly increased in colon carcinoma relative to healthy tissue. No statistically significant gene alterations were retrieved from Oncomine for KRAS mutations.
Next, cBioportal was used to analyse the co-expression patterns of genes that are frequently altered and overexpressed alongside APC, BRAF or KRAS. Figure 5 shows the top ten of these frequently altered genes, as well as the mutual exclusivity of APC, BRAF and KRAS expression in CRC. Our results showed that TP53 and TTN are frequently altered in the presence of APC, BRAF or KRAS mutations. Also, it was evident that KRAS and APC mutations frequently co-occur, while there is a tendency for KRAS and BRAF, and APC and BRAF genetic alterations to be mutually exclusive. Genes frequently co-expressed with APC, BRAF and KRAS, according to cBioportal data. cBioportal was used to identify genes that are frequently co-expressed with (a) APC, (b) BRAF and (c) KRAS. The mutual exclusivity of APC, KRAS and BRAF expression levels, and the corresponding log odds ratio, determined by using cBioportal, are shown in (d). ‘Unaltered group’ and ‘Altered group’ refer to wild-type and mutated genes, respectively.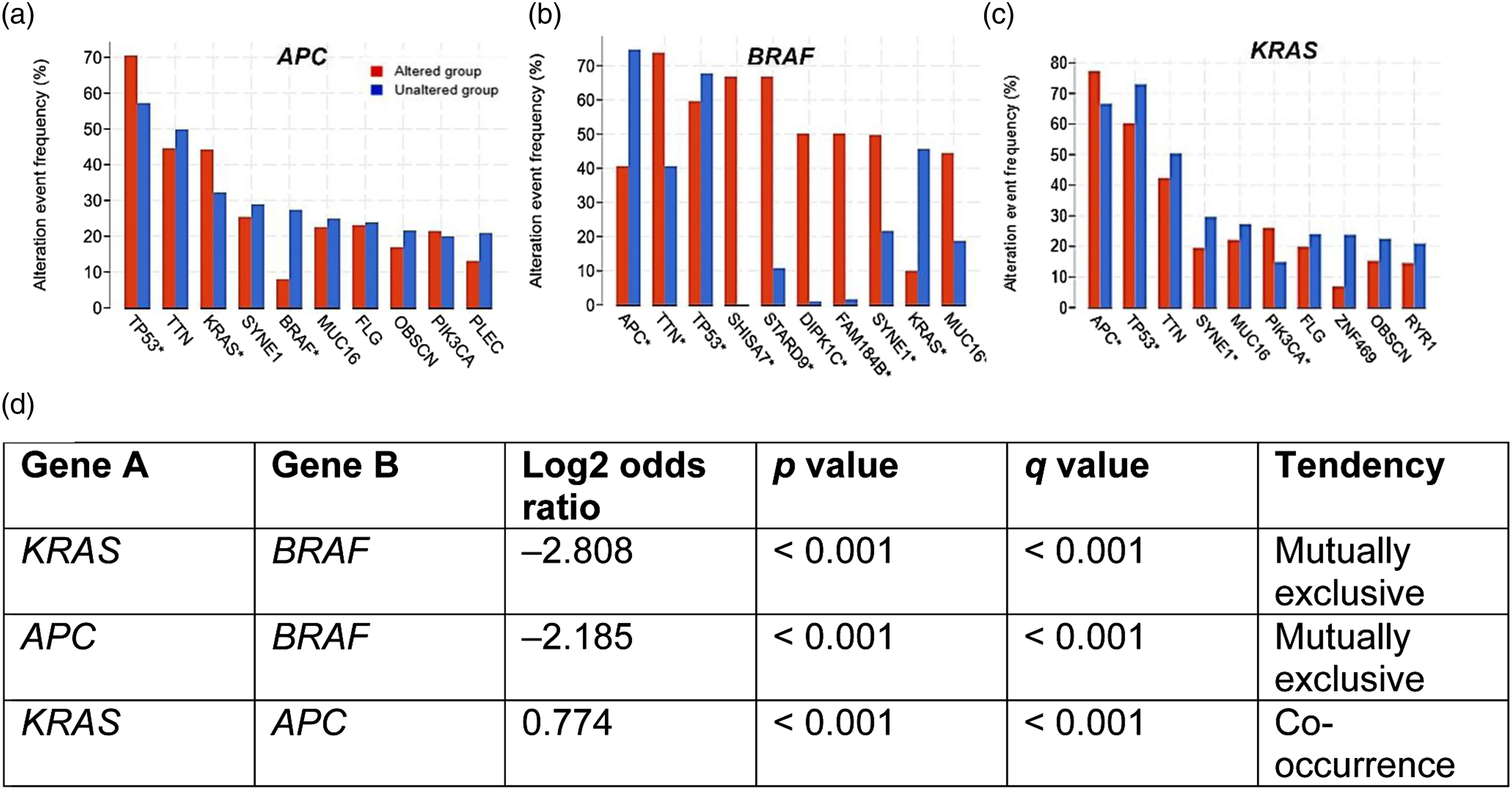
The prognostic value of genetic alterations in CRC
PrognoScan was used to assess the prognostic value of APC, BRAF and KRAS expression in CRC. Four data sets were located by using PrognoScan, and meta-analyses of APC mRNA expression levels and their impact on specific endpoints — including disease-specific survival (DSS), disease-free survival (DFS) and overall survival (OS) — were represented in forest plots (Figure 6). Except for the GSE17537 data set (OS, DFS, DSS), no statistically significant associations were observed between APC expression levels and patient survival endpoints. Analysis of the forest plot indicates that in the case of GSE17537, APC expression favours better patient survival outcomes. BRAF overexpression in the GSE17536 data set was associated with unfavourable DFS endpoints. The remainder of the studies examined, yielded no statistically significant observations regarding the impact of BRAF on patient prognosis. In the case of KRAS, statistically significant associations were found in the GSE17537 data set, where mutations leading to upregulated KRAS levels correspond to unfavourable patient survival endpoints. Forest plots of the prognostic significance of APC, BRAF and KRAS for several disease-related endpoints. The prognostic endpoints studied were overall survival (OS), disease-free survival (DFS) and disease-specific survival (DSS) for patients with CRC, according to PrognoScan. ‘P value’ denotes the Cox’s p value, with a P value of < 0.05 deemed as statistically significant. ‘HR’ corresponds to hazard ratio (represented by the grey square) and ‘95% CI’ to the 95% confidence interval (represented by the solid black line), N corresponds to the number of patients.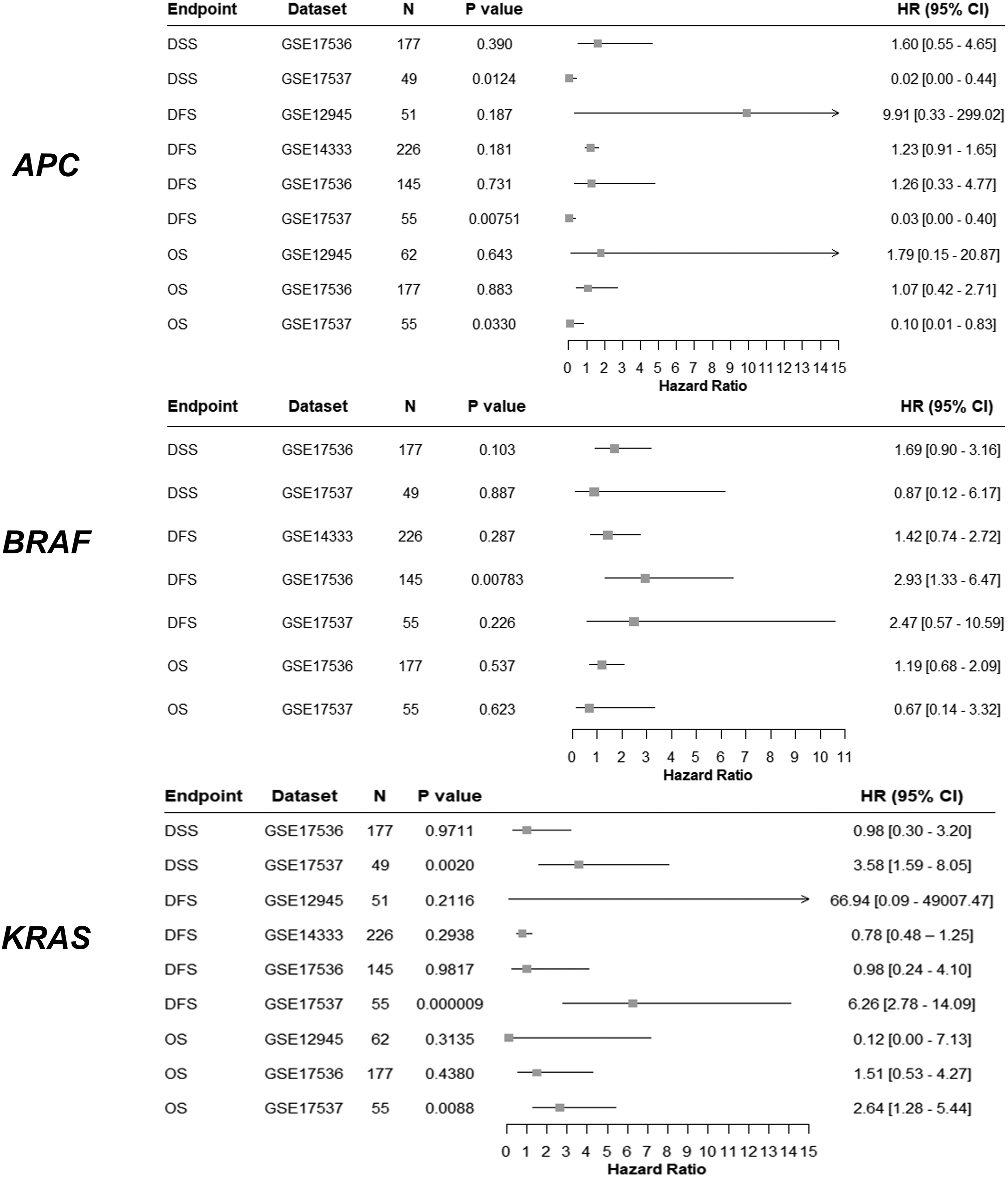
Discussion
In this study, we investigated the evidence base for the prognostic role of APC, BRAF and KRAS expression patterns and the frequency of mutations, by using a combination of open-source bioinformatics tools and clinical data sets. Our rationale for selecting these genes for the present study was informed by the established link between their genetic alteration and CRC tumour aggressiveness.38-41
Here, we report the application of an accessible bioinformatics pipeline for performing a meta-analysis of the prognostic role of gene mutations by employing freely available online bioinformatics resources (cBioPortal, PrognoScan, Oncomine and KMPlot) to interrogate existing clinical data on APC, BRAF and KRAS as actionable drivers of cancer aggressiveness in patients with CRC. The implementation of this approach has several advantages, such as contributing to the refinement and replacement of preclinical in vivo models for studying the role and significance of these mutations in cancer progression. Developing a deeper understanding of the frequency of mutations, their clinical significance and their impact on patient prognosis, provides a strong foundation for the careful design of preclinical studies examining the role of these mutations in any responses to novel experimental therapeutics. Moreover, the analysis of large patient clinical data sets increases the translational relevance of findings from these studies to humans through analysis of the pre-existing clinical data. To our knowledge, this study is the first to combine this range of bioinformatic tools for the evaluation of APC, BRAF and KRAS genetic alterations and prognostic value in CRC.
The model genes APC, BRAF and KRAS were selected to demonstrate the utility of this presented bioinformatics pipeline. APC, BRAF and KRAS mutation assessments have been useful in the expedient prognosis of CRC, and their relative expression levels and mutational status have been implicated in treatment responses.6,8
In the present study, our analysis of existing clinical data sets shows a significant reduction in APC mRNA expression levels in distinct anatomical regions of colon tumour tissue, when compared to healthy colon tissue (Figure 4). APC is a well-recognised tumour suppressor gene which is highly mutated in CRC. Our findings of frequent mutations and downregulation of APC expression levels are consistent with previous reports that show the role of APC loss or gain of function contributing to disease progression in CRC tumorigenesis. 38 The loss of APC tumour suppressive functions and gain of function from truncated APC are widely recognised to contribute to the initiation, progression and maintenance of CRC.6,8,29
Our analysis of the expression of BRAF demonstrated a significant increase in BRAF expression in CRC tissues relative to healthy tissue. Also, BRAF mutations function as strong negative prognostic markers, relating to the V600E (valine to glutamate at codon 600) mutation that leads to gain of function. These mutations have previously been observed in mismatch repair deficient tumours and right-sided colon tumours in older women.39,42,43
KRAS is the most frequently altered protein in cancer, with the G12D mutation being the most prevalent form, leading to aberrant function and poor cancer prognosis. 44 The presence of mutations in BRAF (occurring as a downstream biomolecular effector of KRAS), and in KRAS itself, has frequently been implicated in poor prognosis for CRC patients and a marker of resistance to chemotherapy.8,45 Moreover, genetic alterations in APC, BRAF, and KRAS frequently occur in the presence of other genes. For example, in the case of each of the genes examined here, our cBioportal analyses showed that TP53 and TTN were frequently mutated, with the presence of mutations also noted in APC, BRAF and KRAS in each case. Understanding the patterns and clusters of genetic alterations in CRC, in particular their prevalence and co-occurrence, can serve as a prognostic tool in CRC patient stratification.
Our analysis of pan-cancer prognostic data (see Online Supplemental Material) indicates that the prognostic value of BRAF mutations is not unique to CRC, but has also been observed in renal cell carcinoma and other cancer types. However, previous research has shown that patients with renal cell carcinoma lack BRAF mutations, in comparison to patients with CRC.46,47 According to the literature, BRAF mutations occur frequently in melanoma, ovarian and thyroid cancer.48,49
Following our analyses of gene mutation signatures and expression levels for APC and BRAF, we examined the prognostic value of these genes. Our results show that loss of APC and gain of BRAF both contribute to poorer survival endpoints in patients with CRC. All our findings are consistent with previous preclinical and clinical studies profiling the prevalence of mutations in APC, BRAF and KRAS, and their correlation with patient prognosis. 50 Differences in the prognostic outcomes observed between data sets (Figure 6) highlights the importance of meta-analyses in studying the prognostic significance of genes comparing outcomes from different clinical studies.
A limitation of the present study is a lack of stratification of data sets according to patient metadata, clinicopathologic parameters and anatomical location of CRC tumours. The biology underlying left-sided and right-sided CRC has been associated with differences in tumour pathology, which vary according to patient parameters (e.g. age, sex, lifestyle factors).50,51 The prognostic value of APC, BRAF and KRAS expression, and the role of these genes in CRC tumorigenesis, has previously been associated with patient sex and location of the CRC tumour, which has implications for establishing the clinical significance of these actionable mutations according to tumour location. For example, BRAF mutations are typically associated with right-sided colon cancer.
Further investigation of the genetic and phenotypic role of the APC, BRAF and KRAS genes by using this bioinformatics approach can contribute to the identification of new biomarkers, or to the development of novel potential drug targets or combination strategies for overcoming chemoresistance, without needing to use animal models. The rapid global rise in the number of biobanks being established has led to a concomitant increase in the level of available patient data associated with biobanked human tissue specimens, and these data often cover a wide range of parameters. In conjunction with the increasing availability of freely-accessible online bioinformatic tools and software, this wealth of data can be used to perform the functional analysis of genes and reduce the need for using pre-clinical animal models. The bioinformatics pipeline developed in this study can be directly applied to the evaluation of the expression and prognostic value of other actionable genes in various types of cancer.
Conclusion
In this study, we analysed APC, BRAF and KRAS mutations, their expression profiles and prognostic roles as actionable genes in CRC. Detailed analysis of genomic alterations in CRC can provide novel insights into disease progression, supporting the development of novel drug treatment combinations that will ultimately improve patient outcomes. Findings from this study demonstrate the importance of widespread accessibility to clinical data sets and the need for a bioinformatics pipeline for investigating the role of genes in the diagnosis and therapy of CRC.
Supplemental Material
Supplemental Material - Development of an Accessible Gene Expression Bioinformatics Pipeline to Study Driver Mutations of Colorectal Cancer
Supplemental Material for Development of an Accessible Gene Expression Bioinformatics Pipeline to Study Driver Mutations of Colorectal Cancer by Lisa van den Driest, Caroline H. Johnson, Nicholas J.W. Rattray and Zahra Rattray in Alternatives to Laboratory Animals
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
Funding
The authors acknowledge funding of this research by Tenovus Scotland, RSE Research Reboot, and the FRAME internship for Lisa Van Den Driest.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.