
Abstract
Recombinant adeno-associated virus (rAAV) vectors have emerged as a promising tool for gene therapy. However, the systemic administration of rAAV vectors is not without risks, particularly for dose levels >1 × 1014 viral genome per kilogram of body weight (vg/kg). rAAV-associated toxicities can variably manifest either acutely or in a delayed manner. Acute toxicities often present shortly after administration and can include severe immune responses, hepatotoxicity, and thrombotic microangiopathy (TMA). Delayed toxicities, on the other hand, may emerge weeks to months post-treatment, potentially involving chronic liver damage or prolonged immune activation. Thrombotic microangiopathy is often associated with complement activation and endothelial damage. The activation of the complement system can additionally trigger a cascade of inflammatory responses, exacerbating systemic toxicity. While many of these toxicities are reversible with appropriate medical intervention, there have been instances where the adverse effects were severe enough to lead to fatalities. Both human and animal studies have reported these adverse effects, highlighting the critical importance of thorough preclinical testing. However, a differential toxicity profile associated with systemic AAV administration exists between humans and nonhuman primates (NHPs), in which certain toxicities reported in humans are yet to be observed in NHPs, and vice versa. This review aims to explore the recent literature on systemic rAAV toxicities, focusing on dose levels, the role of the complement activation pathway, endothelial injury, TMA, hepatotoxicity, and the bidirectional translational safety profiles from both human and animal studies.
Keywords
Introduction
Recombinant adeno-associated virus (rAAV) vectors have emerged as a promising tool for gene therapy, offering potential treatments for a variety of genetic disorders. However, the systemic administration of rAAV vectors is not without risks, as it can lead to significant toxicities, including hepatotoxicity, thrombotic microangiopathy (TMA), and immune-mediated responses.7,9,19 rAAV-associated toxicities can manifest either acutely or in a delayed manner, with the timing and severity of these adverse effects varying widely. Acute toxicities often present shortly after administration and can include severe immune responses, hepatotoxicity, and TMA or other endothelial injury syndromes such as disseminated intravascular coagulation (DIC) or acute respiratory distress syndrome (ARDS). Delayed toxicities, on the other hand, may emerge weeks to months post-treatment, potentially involving chronic liver damage or prolonged immune activation. Hepatotoxicity is commonly observed, ranging from mild liver enzyme elevations to severe liver injury and acute or chronic liver failure. Thrombotic microangiopathy is characterized by thrombocytopenia, hemolytic anemia, and acute kidney injury and is often associated with complement activation and endothelial damage. In addition, the activation of the complement system can trigger a cascade of inflammatory responses, exacerbating systemic toxicity. While many of these toxicities are reversible with appropriate medical intervention, there have been instances where the adverse effects were severe enough to lead to fatalities.7,12 Both human and animal studies have reported these adverse effects, highlighting the critical importance of thorough preclinical testing, careful dose optimization, and vigilant post-treatment monitoring to mitigate these risks and improve the safety of AAV-based gene therapies. Nonetheless, dissociative toxicity profiles between humans and animals have been observed following systemic AAV administration. Systemic AAV administration in humans has resulted in cases of ARDS and atypical hemolytic uremic syndrome (aHUS) associated with TMA that have not been observed in animal studies.7,23,27 Conversely, liver failure and death associated with acute hepatotoxicity in nonhuman primates (NHPs) and hepatocellular carcinoma associated with AAV integration in mice remain absent in humans.5,14,26 This differential pattern of certain toxicities emphasizes the continued need of mechanistic studies to better understand the mechanism of AAV-associated toxicities and improve the bidirectional translational safety approach (human clinical trials ↔ nonclinical animal studies) in AAV-based product development.
This symposium presentation reviewed the recent literature on multifaceted aspects of systemic rAAV toxicity, focusing on dose levels, the role of the complement activation pathway, endothelial injury, TMA, hepatotoxicity, cardiovascular toxicity, and findings unique or common from both human and animal studies.
Dose Levels and Systemic AAV Toxicity
The dose of rAAV vectors is a critical factor influencing the extent of systemic toxicity. Higher doses are often required for effective gene transfer rate necessary for systemic indications, such as spinal muscular atrophy and systemic muscular dystrophies. 20 However, systemic high-dose levels of rAAV have been associated with increased risks of adverse effects and fatalities in several clinical trials. 27 Studies have shown that high-dose levels, particularly those exceeding 1 × 1014 vector genomes (vg)/kg, can lead to severe toxicities, including hepatotoxicity and TMA7,12 and several cases of fatality occurred following high-dose intravenous AAV administration. 7 The balance between therapeutic efficacy and safety remains a significant challenge in the clinical application of rAAV gene therapy.
Dose Measurements in AAV-based Gene Therapy Products
Recombinant AAV vectors consists of an icosahedral protein capsid shell composed of 60 subunits of three viral proteins VP1, VP2, and VP3 at a molar ratio of approximately 1:1:10, and a 4.7 kilobases (kb) vector DNA genome (VG) that retains the wild-type AAV inverted terminal repeats (ITR) along with the engineered transgene expression cassette placed between the ITRs. 19 Depending on the AAV serotype and amino acid sequence of the corresponding viral capsid proteins, the molecular mass of an empty AAV capsid is approximately ~3.8 MDa. 31
In nonclinical and clinical trials, systemic rAAV doses are typically expressed in terms of vector genomes (vg) per kilogram of body weight. This measurement allows for precise precise quantification of the viral DNA and dosing based on the subject’s weight, ensuring consistency across different studies and patient populations. The most common methods for quantifying rAAV vector genomes include quantitative polymerase chain reaction (qPCR) and droplet digital PCR (ddPCR) using probe typically specific to the therapeutic transgene cassette inserted in the vector genome. 29 This measurement reflects the number of viral particles containing the therapeutic transgene measured. However, the presence of defective viral particles (empty capsids that lack the vector genome or partial capsids that contain sub-genomic vector species) is not uncommon and can vary significantly depending on the manufacturing method of rAAV vectors. These defective particles are considered impurities because they do not contain the intended fully intact vector genome and can constitute up to 80% of the total AAV particles in the product formulation in certain settings.29,32 Consequently, in addition to measuring the total number of vector genomes, it is also important to assess the ratio of full to empty viral capsids (as well as assessment of partial capsids). Techniques like electron microscopy, analytical ultracentrifugation (AUC), and UV spectrophotometry can be used for this purpose.6,25 Accurate measurement of rAAV dose is crucial for optimizing therapeutic efficacy and minimizing potential side effects.
Doses of rAAV products can vary significantly depending on the therapeutic target and the specific AAV serotype used. Systemic delivery of AAV requires higher dose levels to compensate for the larger volume of distribution, and generally administered at 1014-1016 vg per patient compared with 1011-1013 vg per patient with targeted and localized AAV delivery. Dose levels for neuromuscular diseases can range from 1 × 1014 to 3 × 1014 vg/kg, with dose levels going up to 1.5 × 1017 vg per patients in certain studies. 2
A typical systemic high dose of rAAV that has a modest 50% defective virus particle (empty and partials) administered at 1 × 1014 vg/kg to an adult patient of 70 kg of body weight means the patient received a total of 5.3 × 1016 MDa, which is approximately equivalent to 1.5 mg/kg dose of capsid proteins impurities, and this number can go up to 10 mg/kg if the patient is administered 3 × 1014 vg/kg of rAAV that contains 80% defective AAV particles. As noted earlier, defective AAV particles are considered impurities and do not contribute to the therapeutic benefit intended from the administrated rAAV product, and in fact may contribute to adverse rAAV-associated toxicities as will be discussed later in the document. Nonetheless, the amount of these impurities in rAAV products, albeit as a single-dose therapeutic option, is similar and even exceeds the 1 to 15 mg/kg dose levels of therapeutic monoclonal antibodies such as bevacizumab, trastuzumab, ipilimumab, or nivolumab.
Sudden Exposure to High Dose of rAAV
As discussed earlier, high dose of rAAV administered systemically can contain high levels of foreign capsid protein impurities that do not contribute to the intended therapeutic benefit of the administered rAAV product. In addition, the high exposure to virus capsid proteins is accompanied by massive number of vector genomes that is unusual to the human subject from a natural viral infection that typically involves a significantly smaller founder virus population that replicates and increases copies of viral genome overtime. Therefore, unlike typical viral infection kinetics that starts with low viral genome copy numbers and peaks overtime, the vector genome kinetics of systemic administration of the replication defective rAAV product starts very high at >1 × 1014 vg/kg, or what’s equivalent of >7 × 1015 total vg per 70 kg patient and starts to decline overtime until it reaches a steady state of episomal vector DNA formation in transduced cells (Figure 1).
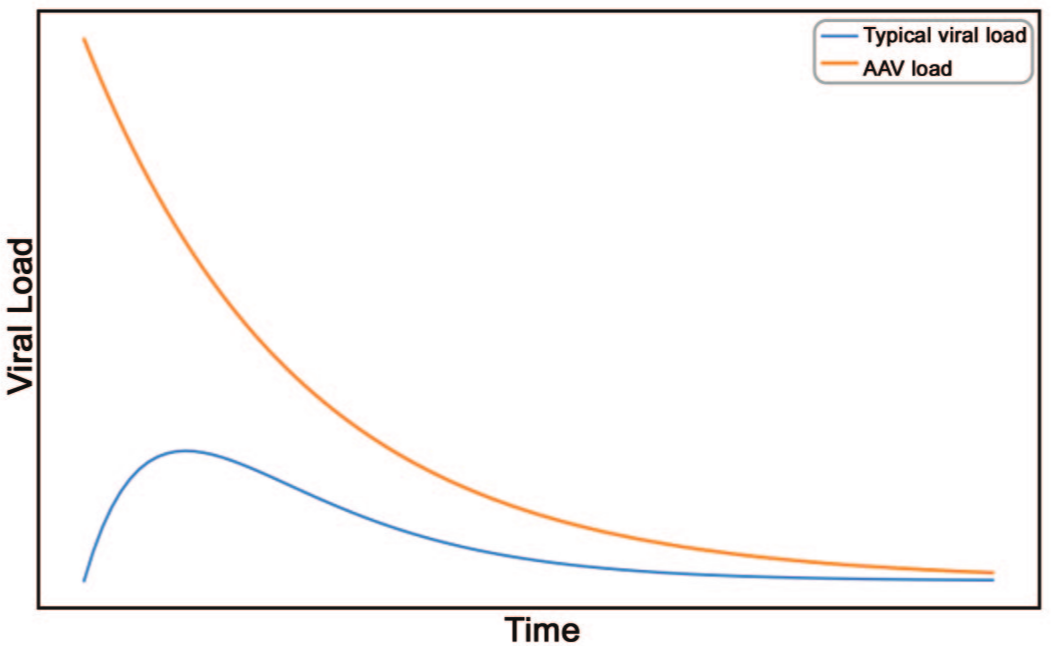
Schematic comparison of viral load kinetics between typical viral infections and rAAV systemic administration. The blue curve represents the typical progression of viral load in an infected individual, starting with a small founder virus population, peaking around day 3 and gradually declining to baseline by day 14. The orange curve shows the kinetics of systemic rAAV administration, which starts high on day 0 following intravenous vector administration and gradually drops to persistent low episomal vector genome copies in the transduced cells (figure generated by GPT-4).
Immunogenicity of rAAV Products
The immunogenicity of rAAV vectors, particularly at systemic high doses, is a significant challenge in gene therapy. Nearly all components of rAAV (capsid, vector genome, and potentially transgene product) are considered immunogenic. Toll-like receptors (TLRs) play a crucial role in the innate immune response to AAV. Specifically, TLR2 and TLR9 are activated upon recognition of AAV capsid and vector DNA, respectively. TLR2, located on the cell surface, detects the viral capsid proteins, while TLR9, found in endosomes, senses unmethylated CpG motifs in the AAV genome. This activation leads to the production of pro-inflammatory cytokines and type I interferons, which can enhance the adaptive immune response and contribute to immunotoxicity. 1 However, the intensity of the response is lower with rAAV compared with other viral vectors such as adenovirus or natural infectious viremia.
The complement system, another component of the innate immune response, is also activated by rAAV vectors either directly in the systemic circulation or indirectly following tissue transduction and tissue damage. Both the classical and alternative pathways can be triggered by AAV capsids and the immune complexes that they form with anti-capsid antibodies, leading to the formation of the membrane attack complex (MAC) or opsonization of the virus by reticuloendothelial cells. 11 This activation not only facilitates the clearance of AAV particles but also recruits immune cells to the site of infection, exacerbating inflammation. The complement activation can result in the production of anaphylatoxins, which further amplify the immune response and contribute to the adverse effects observed with high-dose systemic AAV administration. 11
Adaptive immune responses against AAV involve both B cells and cytotoxic T lymphocytes (CTLs). B cells produce neutralizing antibodies against AAV capsids, which can prevent subsequent transduction and reduce the efficacy of gene therapy. 1 In addition, CTLs can recognize and destroy transduced cells presenting AAV-derived peptides on major histocompatibility complex (MHC) class I molecules. This cytotoxic response is particularly problematic in high-dose systemic administration, as it can lead to significant tissue damage in addition to the loss of therapeutic gene expression potentially observed at lower dose level. The combined effect of these immune responses underscores the need for strategies to mitigate immunogenicity and improve the safety and efficacy of AAV-based gene therapies.
Complement Activation Pathway
High-dose systemic administration of rAAV vectors can significantly activate the complement system, leading to various immunotoxic effects. The classical pathway is primarily triggered by the binding of pre-existing or newly formed anti-AAV antibodies to the viral capsid. This interaction activates C1q, initiating a cascade that results in the formation of C3 convertase and subsequent cleavage of C3 into C3a and C3b. C3b acts as an opsonin, marking AAV particles for phagocytosis by immune cells, while C3a functions as an anaphylatoxin, promoting inflammation by recruiting and activating leukocytes. 27 The classical pathway’s activation is a critical factor in the rapid clearance of AAV vectors from the bloodstream, reducing the efficiency of gene delivery. 11
Simultaneously, the alternative pathway can be activated independently of antibodies through the direct interaction of AAV capsids with complement proteins. This pathway is continuously active at a low level, providing a rapid response to pathogens. When AAV capsids are present in high doses, they can stabilize the alternative pathway C3 convertase (C3bBb), leading to an amplification loop that generates large amounts of C3b. This process further enhances opsonization and the formation of the MAC, which can lyse cells and contribute to tissue damage. 9 The alternative pathway’s amplification can exacerbate the inflammatory response, leading to adverse effects such as TMA and acute kidney injury. 22 It should be noted that the alternative complement pathway role in triggering TMA following rAAV administration continues to require further exploration.9,30 In addition, the lectin pathway can be activated secondary to tissue damage by damage-associated molecular patterns exposed on necrotic cells or apoptotic bodies initiating complement activation such that it may be difficult to determine complement activation as causative or secondary in some instances. 9
The terminal pathway of the complement system, involving the formation of the MAC, is a common endpoint for the classical, lectin, and alternative pathways. The MAC creates pores in the membranes of target cells, leading to cell lysis and death. In the context of high-dose systemic AAV administration, the extensive formation of MAC can result in significant tissue damage, particularly in the liver and kidneys, where AAV vectors tend to accumulate. 11 In addition, the release of anaphylatoxins (C3a, C4a, and C5a) during complement activation can lead to systemic inflammatory responses, including cytokine release syndrome (CRS), which poses a severe risk to patients. Understanding these mechanisms is crucial for developing strategies to mitigate complement activation and improve the safety of AAV-based gene therapies.
Clinical Manifestation of rAAV-Mediated Complement Activation
Recent clinical investigations compiled from several clinical trials involving systemic administration of rAAV serotype 9 (AAV9) vectors at dose levels ranging from low (1.5 × 1013 vg/kg) to high (1.1 × 1014 vg/kg) demonstrated the immune responses triggered by systemic administration of high-dose rAAV vectors and the potential associated mechanism of TMA. 27 The study involved 38 individuals who received AAV9 and were divided into two groups based on their prophylactic immunomodulation regimens. Group 1 received corticosteroids, while Group 2 received a combination of rituximab, sirolimus, and steroids to prevent anti-AAV antibody formation. The results showed that participants in Group 1 experienced a rapid increase in immunoglobulin M (IgM) and IgG levels, along with elevated D-dimer and decreased platelet counts (thrombocytopenia), and the presence of schistocytes on blood smears (suggesting endothelial damage), indicative of TMA. Complement activation was evident in these participants, with both the classical and alternative pathways being activated, as shown by depleted C4 and elevated levels of soluble C5b-9, Ba, and Bb antigens. In contrast, group 2 participants did not exhibit significant changes in IgM or IgG levels and had minimal complement activation. This suggests that the immunomodulation regimen in group 2 was mitigating the immune response against AAV and the occurrence of acute severe adverse events. However, it is worth noting that dose level may be another variable as the average dose level in group 2 immunosuppressed patients was mainly in the low to moderate range (3.6 × 1013 vg/kg on average) compared with group 1 corticosteroids only patients comprised in majority of high-dose levels (1.18 × 1014 vg/kg on average). Hence, dose level may have also contributed to the study outcome. To mitigate the immunotoxicity associated with high-dose systemic AAV administration, the study suggests a comprehensive immunosuppression regimen. The use of corticosteroids alone, as seen in group 1, was insufficient to prevent the formation of anti-AAV antibodies and subsequent complement activation. The combination of rituximab, sirolimus, and steroids in group 2 effectively suppressed the immune response, albeit with the potential role of lower dose levels in this group compared with group 1 patients. Rituximab, an anti-CD20 monoclonal antibody, depletes B cells, thereby reducing the production of anti-AAV antibodies. Sirolimus, an mTOR inhibitor, further modulates the immune response by inhibiting T-cell activation and proliferation.
The study concludes that TMA in the context of high-dose AAV gene therapy is primarily driven by anti-capsid antibodies, which activate the classical complement pathway, and is further amplified by the alternative pathway. The findings highlight critical time points and interventions that can help manage immune-mediated events, thereby improving the safety and efficacy of systemic rAAV gene therapy. More details on clinical manifestation of systemic rAAV gene therapy are reviewed elsewhere. 7
Nonclinical Manifestation of AAV-Mediated Complement Activation
Severe hepatotoxicity and occasionally fatality has been observed in NHPs receiving high-dose systemic AAV gene therapy.16,26 While these studies led to significant acute liver injury characterized by elevated liver enzymes, alternative complement activation and not TMA, the acute molecular manifestations of hepatotoxicity in monkeys within days post vector administration overlapped with those associated with the acute toxicity profile seen after high doses of systemic rAAV in humans and was characterized by thrombocytopenia and activation of the complement pathway (primarily the alternative pathway).21,26
Liver findings in cynomolgus monkeys treated with AAV-PHP.B-CBh-SMN1 showed a dose-dependent acute hepatocellular damage, particularly in animals receiving a dose of 1 × 1014 vg/kg. Four days after a single intravenous bolus injection, the liver exhibited moderate hepatocellular degeneration and necrosis, characterized by diffuse swelling and multifocal to coalescing necrosis of hepatocytes. At higher magnification, hepatocytic vacuolation, cellular disintegration, and intracytoplasmic eosinophilic inclusions of various sizes were observed. Immunohistochemical staining for albumin in treated animals showed increased cytoplasmic staining of hepatocytes with a mosaic pattern, indicating altered protein expression. In addition, Oil Red O staining of neutral lipids revealed a dose-related increase in lipid droplets, suggesting hepatocellular malfunction. 26
More recently, high-dose intravenous clade F rAAV vectors (e.g., AAV9 and AAVhu68) encoding enhanced green fluorescent protein (eGFP) or therapeutic proteins highlighted significant sinusoidal endothelial and platelet-related changes in the livers of rhesus or cynomolgus macaques administered rAAV. 16 Platelet levels were monitored in macaques receiving high doses of AAV, defined as >5 × 1013 vg/kg. The study observed a reduction in platelet levels within days post-administration, indicating potential platelet consumption or sequestration, along with activation of the alternative complement pathway and lack of activation of the classical pathway. Immunostaining of liver sections showed the presence of fibrin thrombi, as indicated by fibrinogen staining, platelet sinusoidal accumulation, marked by CD41, and liver sinusoidal endothelial cells (LSECs) injury with loss of expression of LSECtin and VEGFR2 (normal LSECs surface markers) (Figure 2A-C). Interestingly, this study identified serum hyaluronic acid (HA) levels as a potential biomarker of AAV-associated LSEC injury (Figure 2D and E), as it is a metabolite scavenged by LSEC via a receptor specific uptake and this receptor was shown to be severely downregulated on single-nuclei RNAseq analysis of livers from NHP that died of acute toxicity. As predicted by the histology and molecular pathology analyses, HA was elevated in macaques receiving high-dose AAV vectors and confirmed the LSEC injury seen histologically and by snRNAseq. In addition, there was a significant correlation between increased serum HA levels and reduced platelet counts on day 3 post-administration, indicating that LSEC injury and platelet reduction are linked. This correlation underscores the impact of high-dose AAV administration on liver endothelial cell integrity and platelet dynamics. 16
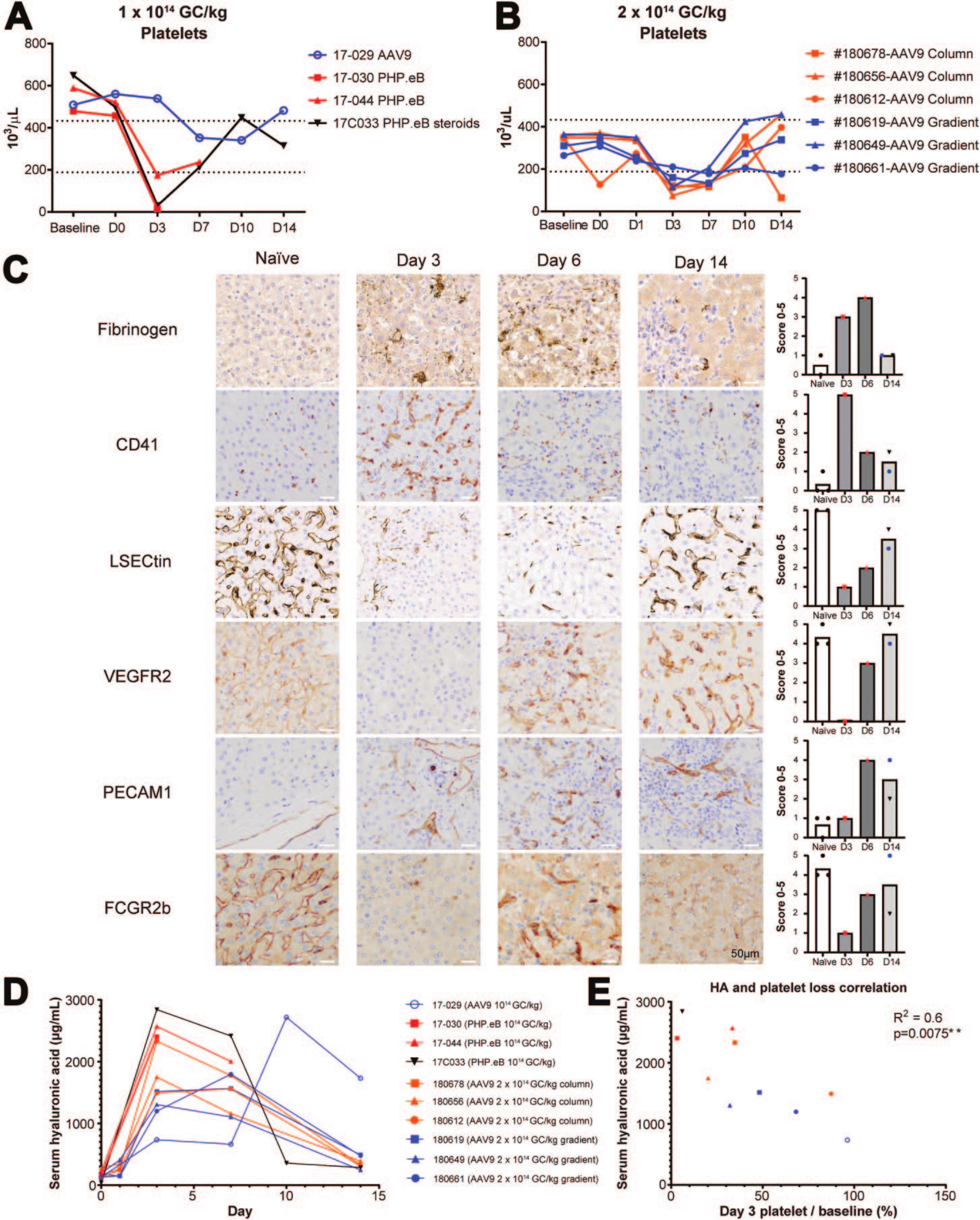
Acute thrombocytopenia and LSEC injury following high-dose systemic intravenous administration of AAV.eGFP in rhesus macaques. (A) Acute thrombocytopenia starting on day 3 following the administration of AAV-PHP.eB or AAV9 at 1 × 1014 vg/kg or (B) 2 × 1014 vg/kg (C) Representative images and semi-quantitative scoring of liver section immunostaining of fibrinogen (fibrin thrombi marker), CD41 (platelet marker), LSECtin (liver sinusoidal endothelial cell lectin, a sinusoidal endothelium marker), VEGFR2 (sinusoidal endothelium marker), PECAM1 (CD31, non-sinusoidal endothelial marker), and FCGR2b (sinusoidal-specific FC γ receptor 2b). Scale bar, 50 μm. (D) Serum hyaluronic acid (HA) levels (biomarker of LSEC integrity) in monkeys that received AAV-PHP.eB or AAV9 vectors encoding eGFP at 1 × 1014 vg/kg or 2 × 1014 vg/kg. (E) Correlation between serum HA level and thrombocytopenia on day 3 post high-dose AAV. Pearson correlation R2 0.6, 2-tailed P value .0075. 16
The low frequency of animals developing such severe toxicity in this and another study highlights the importance of enrolling adequate number of animals per dose group in the study design, and the need to include several blood collection time points during the first week post-AAV dosing, especially a baseline at pre-vector administration, day 3, and day 7 post-dose.14,16 In addition, for animals that received high doses of AAV expressing therapeutic transgenes, steroid treatment helped mitigate the acute liver enzyme elevations but did not alleviate the acute thrombocytopenia. 16 Hence, the use of prophylactic steroids should be discussed on a case-by-case basis but is generally recommended if planned for use in clinical trials.
Hepatoxicity Associated With Systemic AAV
As noted earlier, high-dose systemic rAAV administered results in acute hepatoxicity mediated by complement activation of innate immune system. In addition, cytotoxic T lymphocytes (CTLs) play a pivotal role in the adaptive immune response against AAV vectors. This immune-mediated cytotoxicity was first recognized to lead to the elimination of transgene-expressing cells, resulting in a loss of therapeutic gene expression and potential tissue damage.21,24 Cytotoxic T lymphocyte responses to rAAV can cause significant hepatotoxicity. The destruction of hepatocytes by CTLs can lead to elevated liver enzymes, indicative of liver injury, and can exacerbate underlying liver conditions. 9 This hepatotoxicity is a major concern in high-dose systemic AAV administration, where the immune response is more pronounced as recently reported by the subacute liver failure in two pediatric patients who received Zolgensma (also known as onasemnogene abeparvovec-xioi or AVXS-101), an FDA-approved rAAV for spinal muscular atrophy (SMA). Both cases involved elevated liver enzymes, coagulopathy, and encephalopathy, meeting the criteria for acute liver failure. The liver injury was transient and managed with supportive care and corticosteroids, but these cases raise concerns about the potential for severe hepatotoxicity associated with high-dose systemic AAV administration and emphasize the need for careful monitoring of liver function in patients receiving AAV-based gene therapies, particularly those with pre-existing liver conditions. 10 The significance of hepatoxicity associated with high-dose systemic rAAV administered cannot be further emphasized by the trial pause of resamirigene bilparvovec developed for the treatment of X-linked myotubular myopathy (XLMTM). The trial was paused due to severe adverse events observed at the higher dose of 3.5 × 1014 vg/kg as three unexpected deaths occurred among participants receiving this higher dose. These adverse events included acute liver failure, sepsis, and multi-organ failure. The severity and nature of these events prompted an immediate halt to the higher dose administration and a thorough investigation to understand the underlying causes. 28
Cardiovascular Toxicity Associated With Systemic AAV
A high number of AAV-based cardiovascular gene therapies are currently under development for treating genetic cardiomyopathies and non-genetic heart failure. This is due to the robust tropism of several AAV serotypes for cardiomyocytes after systemic administration across species. However, the heart being both the target organ for genetic medicine delivery and a common target for toxicities makes the drug development journey challenging for cardiovascular gene therapy. This challenge is exemplified by cardiac toxicity due to frataxin overexpression,3,18 which halted the development of several Friedrich ataxia gene therapy programs, the high susceptibility of the heart to immune-mediated myocarditis in gene therapy programs for Pompe or Duchenne muscular dystrophy,17,23 and the troponin I elevations observed following Zolgensma administration in both nonclinical and clinical studies. 4
AAV-related cardiovascular safety findings in humans following systemic administration of high doses of AAV generally fall into two categories. The first category includes acute toxicities within the first two weeks post-dosing, where innate immune response activation and high levels of transduction can cause thrombocytopenia, TMA, DIC, CRS, generalized edema, ARDS, or acute cardiac events. 23 The second category involves delayed myocarditis, either caused by an adaptive immune response to non-self portions of the transgene or transgene-specific overexpression-related toxicities.3,17,18 The acute toxicity responses are mainly variations of endothelial injury syndromes targeting different vascular beds, while the subacute and chronic toxicities are caused by T-cell-mediated inflammation in the heart tissue.
Subacute to chronic cardiac toxicity can develop in NHPs. In one example, rhesus macaques receiving an AAVhu68 vector expressing GAA, the enzyme deficient in Pompe disease, 17 developed cardiotoxicity with unusual high inter-individual variability and no apparent dose response. Cardiac toxicity manifested as troponin I elevations from day 40 to day 60, and histological analysis revealed a cytotoxic T-cell-rich infiltrate causing cardiomyocyte degeneration. Marked myocarditis in some animals uniquely exhibited strong interferon-gamma ELISpot responses to the transgene peptide pool D (C-terminal region of human GAA). Genotyping of MHC class I alleles revealed that cardiac toxicity was linked to an uncommon MHC class I haplotype in this cohort of rhesus macaques. Epitope mapping narrowed down the immunodominant epitope to a limited C-terminal region of the human GAA transgene. 17 Similar findings were reported by another group in which cardiotoxicity associated with elevated cardiac troponin I (cTnI) and N-terminal pro-b-type natriuretic peptide (NT-proBNP) and mixed inflammation in the heart was observed in cynomolgus monkeys receiving high doses (2 × 1014 vg/kg and 5 × 1014 vg/kg) of an AAV8 vector expressing the human but not the cynomolgus GAA transgene protein. Interestingly, the highest dose (5 × 1014 vg/kg) resulted in two unscheduled deaths; one of which due to echocardiographic findings without concerning clinical signs. 8 The in-depth characterization in both studies helped assess the risk of translation to humans, as the immune response is directed to a non-self human epitope in macaques, which humans would typically tolerate unless this region is deleted in some patients by their disease-causing mutation.
AAV-associated cardiotoxicity can be directly mediated by the transgene and independent of the immune system. One such case example is the mitochondrial toxicity observed secondary to frataxin overexpression in rodents and NHPs.3,18 Frataxin cardiac overexpression was tolerated up to nine-fold the normal endogenous levels in mice but led to significant mitochondrial toxicity and heart damage above twenty-fold. 3 In this case, there is no immune response implication, and the transgene function itself impaired the mitochondrial respiratory chain and ultrastructure, leading to cell death and secondary fibrosis. As predicted in rodent models, AAV expressing human frataxin administered intravenously to macaques at dose levels of 3 × 1013 vg/kg or greater resulted in troponin I elevations by 90 days post-dose, cardiomyocyte degeneration on histopathological examination, and fatal arrhythmia in one study animal 119 days post vector administration. 13
A recent meta-analysis presented at the American Society of Gene and Cell Therapy (ASGCT) 27th Annual Meeting analyzed the toxicity profile in NHPs that received an intravenous administration of AAV at the Gene Therapy Program at the University of Pennsylvania for which electronic records were available. 15 This cohort included 448 NHPs representing a variety of capsids, promoters, and transgenes. Serious outcomes defined by severe toxicity warranting unscheduled necropsy were generally uncommon following systemic AAV administration in 13 out of total 448 animals surveyed (3%) or 13 out of 85 (15%) animals that received a dose of ≥5 × 1013 vg/kg. Most severe events warranting unscheduled euthanasia occurred within the first two weeks post-dose (average time point approximately 5 days post-dose) and was characterized by elevated liver enzymes and thrombocytopenia that was often followed by generalized edema due to capillary leakage, hyberbilirubinemia, and/or coagulopathy. Clade F capsids or engineered clade F capsids were most frequently associated with serious outcomes, likely because these capsids were used at very high doses. Finally, there was no clear transgene effect, as serious outcomes were seen with GFP, therapeutic transgenes, or capsid libraries. 15
Conclusions
High-dose systemic administration of rAAV vectors presents significant challenges due to the complex interplay of innate and adaptive immune responses, tissue biodistribution profile, transgene expression, systemic and local complement activation, and resultant toxicities. Direct vector effect and the innate immune response can lead to acute toxicities such as thrombocytopenia, TMA, and DIC. These responses are often accompanied by hepatotoxicity, characterized by elevated liver enzymes and liver sinusoidal endothelial cell injury, leading to platelet sequestration and aggregation. Adaptive immune responses, particularly the production of neutralizing antibodies and cytotoxic T lymphocytes, further exacerbate these effects, contributing to chronic toxicities such as hepatoxicity, myocarditis, and cardiomyocyte degeneration. The potential for chronic cardiotoxicity, marked by troponin I elevations and T-cell-mediated inflammation, and for acute endothelial injury syndromes underscore the need for careful dose management and monitoring. Understanding these mechanisms is crucial for developing strategies to mitigate these adverse effects and improve the safety and efficacy of AAV-based gene therapies.
Footnotes
Acknowledgements
The author thanks Dr Juliette Hordeaux for the critical review of the manuscript.
Correction (December 2024):
Article updated to correct the reference citation in the legend of Figure 2 and to correct the spelling of hepatotoxicity under the “Nonclinical Manifestation of AAV-Mediated Complement Activation” section.
Declaration of Conflicting Interests
The author declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author received no financial support for the research, authorship, and/or publication of this article.
Disclosure
During the preparation of this work, the authors used the assistance of an AI language model (GPT-4) developed by OpenAI to draft or edit sections of the manuscript as well as to generate ![]() in the manuscript. After using this tool, the author reviewed and edited the content as needed and takes full responsibility for the content of the publication.
in the manuscript. After using this tool, the author reviewed and edited the content as needed and takes full responsibility for the content of the publication.