
Abstract
Nonclinical studies of test articles (TAs) in nonhuman primates are often designed to assess both biodistribution and toxicity. For this purpose, studies commonly use intravenous perfusion of ice-cold (2°C-8°C) saline to facilitate measurements of TA-associated nucleic acids and proteins, after which tissues undergo later fixation by immersion for histological processing and microscopic evaluation. Intriguingly, minimal apoptosis/single cell necrosis (A/SCN) of randomly distributed neural cells is evident in the cerebral cortex and less often the hippocampus in animals from all groups, including vehicle-treated controls. Affected cells exhibit end-stage features such as cytoplasmic hypereosinophilia, nuclear condensation or fragmentation, and shape distortions, so their lineage(s) generally cannot be defined; classical apoptotic bodies are exceedingly rare. In addition, A/SCN is not accompanied by glial reactions, leukocyte infiltration/inflammation, or other parenchymal changes. The severity is minimal in controls but may be slightly exacerbated (to mild) by TA that accumulate in neural cells. One plausible hypothesis explaining this A/SCN exacerbation is that cold shock (perhaps complicated by concurrent tissue acidity and hypoxia) drives still-viable but TA-stressed cells to launch a self-directed death program. Taken together, these observations indicate that A/SCN in brain processed by cold saline perfusion with delayed immersion fixation represents a procedural artifact and not a TA-related lesion.
This article is an opinion piece submitted to the Toxicologic Pathology Forum (TPF). This perspective is the particular view of the authors and does not represent an official position of the Society of Toxicologic Pathology (STP), British Society of Toxicological Pathology (BSTP), or European Society of Toxicologic Pathology (ESTP), nor should it be considered to reflect the opinions, policies, or positions of the authors’ employers or regulatory agencies. The TPF is designed to stimulate discussion of topics relevant to regulatory issues in toxicologic pathology. Readers of Toxicologic Pathology are encouraged to send their thoughts on TPF opinion articles or ideas for new discussion topics to
Introduction
Development of new products to treat neurological diseases is a major global health need. In recent decades, increased attention has been given to devising innovative modalities, such as biologics and gene therapies, to specifically engage distinct cell populations within the central nervous system (CNS). The importance of neurotoxicity as a key concern in nonclinical safety testing is reflected in the broad range of regulatory guidance,5,6,15,21 industrial “best practice” recommendations,3,4,8,9,19,27-30,32,33 and globally accepted diagnostic terminology10-13,23,38,43 for pathologic findings in neural tissues of common laboratory animal species. Accordingly, toxicologic pathologists must stand ready to identify and characterize microscopic findings in a wide spectrum of CNS and peripheral nervous system (PNS) organs, and interpret their implications (ie, test article [TA]-related vs procedure-associated vs incidental background vs artifactual changes vs normal microanatomic features) with respect to assessing risk posed to human patients.17,18,20,31
Historically, the pivot toward biologics and gene therapies often necessitated the choice of nonhuman primates (NHPs) as the nonrodent test species, or even the only nonclinical species, because human-based biomolecules are often pharmacologically active in animals only when interacting with primate molecular pathways. In recent years, the scarcity of NHPs has led to a pivot in nonclinical study design to assess multiple endpoints using limited number of animals. When the CNS is the intended site for therapeutic action, a common approach for investigating biologics and gene therapies is to investigate TA biodistribution in tandem with toxicity testing. Such study designs require adjustments in the tissue sampling and handling practices at necropsy to balance molecular quality (eg, RNA and/or protein expression in target cell populations) against anatomic preservation.
This article describes apoptosis/single cell necrosis (A/SCN) in the brain as a subtle but frequent microscopic finding in NHPs from combined biodistribution/toxicity studies that share a particular study design. Because it occurs in vehicle-treated control animals, the change is interpreted as a likely procedural artifact associated with terminal intravascular whole-body perfusion with ice-cold saline followed by delayed immersion fixation, although experimental investigation will be needed to confirm this possible mechanism. The intent of this article is to raise awareness of this finding and specifically to ensure that it will neither be attributed to TA exposure nor interpreted as adverse without due consideration of the study design as a potential contributing factor.
Anatomic Appearance of Apoptosis/Single Cell Necrosis in Cold Saline–Perfused Brain
Apoptosis/single cell necrosis in the brains of cold saline–perfused NHPs occurs reproducibly as rare (ie, “minimal”), randomly distributed cells in selected forebrain domains. The finding is most common in the superficial cerebral cortex (characterized by dispersed round interneurons interlaced with glial cells and fine capillaries) and less often hippocampus (in or near the cornu ammonis [CA] fields). These brain regions are regularly sampled in NHPs using current “best practice” recommendations 8 (sections 3A and 3B for the cerebral cortex and hippocampus, respectively), so the finding will be detectable—if it occurs—with routine brain sampling. Importantly, these rare apoptotic cells are difficult to discern using low-power objectives (2X, 4X, and 5X), so microscopic evaluation to verify their presence will typically need to be performed at 10X and often 20X.
In conventional paraffin-embedded, hematoxylin and eosin (H&E)–stained sections, the affected cells mainly exhibited classic end-stage features of death by apoptosis (Figure 1) although some characteristics overlapped with those of single cell necrosis. 14 Principal morphologic traits of apoptosis included cytoplasmic hypereosinophilia, nuclear condensation (pyknosis) and/or fragmentation (karyorrhexis), irregular plasma membrane contours, and shrunken profiles; classical apoptotic bodies (Figure 1C) are exceedingly rare. Typically, the affected cells were either located centrally within a clear, colorless space or attached to one edge of the space. Occasional cells displayed some attributes of single cell necrosis, including enlarged cell and nuclear sizes, cytoplasmic pallor, and nuclear fragmentation or dissolution (karyolysis). Accordingly, the combined diagnosis “apoptosis/single cell necrosis” (A/SCN) was used as there was no need to differentiate between the two processes in generating the pathology data sets. 14 The nearby neural parenchyma was characterized by unaffected (“normal”) neurons with large, round nuclei and scant, pale basophilic cytoplasm surrounded by a continuous field of pale eosinophilic neuropil. Despite their presence in neuronal layers, the identities of affected elements were uncertain. Pyknotic nuclei often exhibited a fusiform shape, a profile consistent with a glial lineage (for cells located in the parenchymal fields, occasionally next to neurons) or possibly pericytes (when found near capillaries). The absence of cells exhibiting cytoarchitectural features indicative of early degeneration (eg, altered cytosolic and/or nucleoplasm colors, small cytoplasmic vacuoles, or subtle changes to chromatin structure) is not unexpected in these animals due to the delay in cell preservation associated with immersion fixation and the rarity of affected cells in any brain domain.
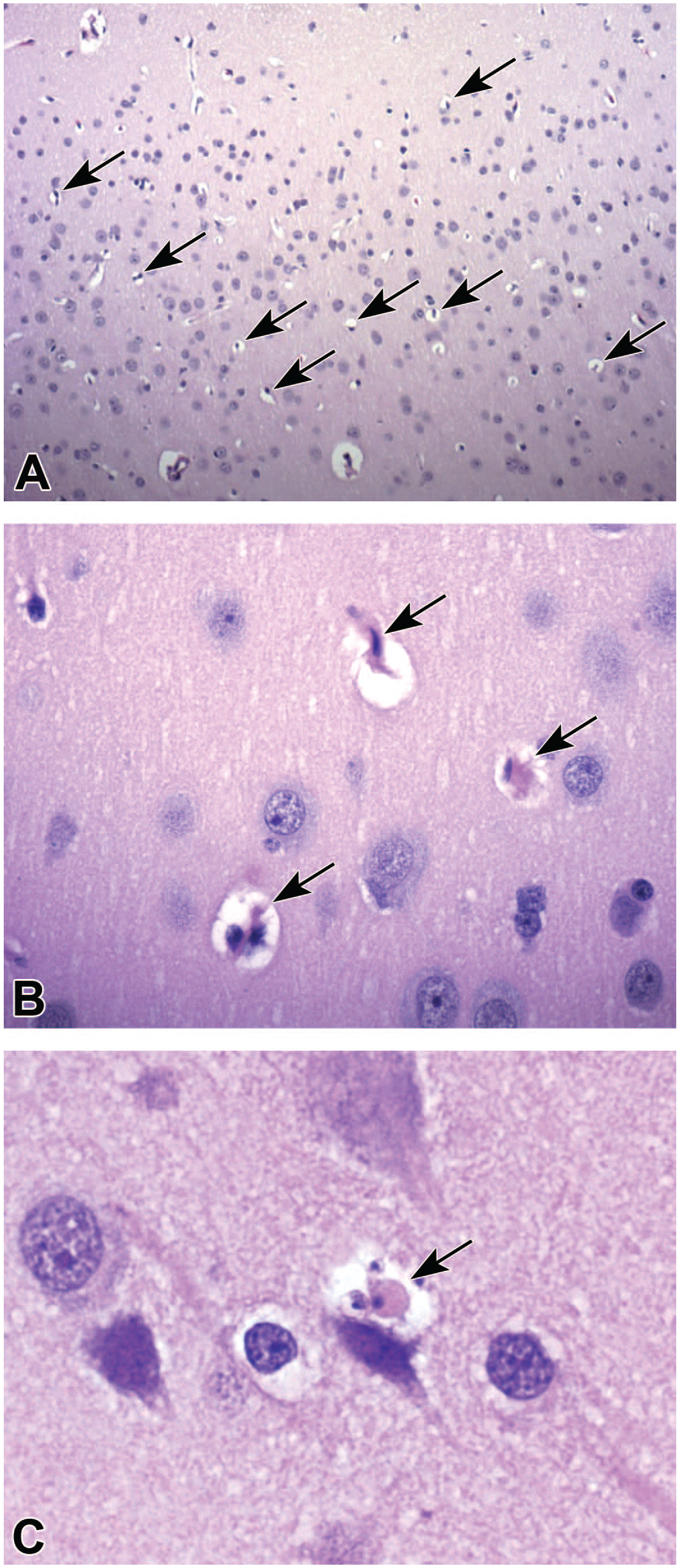
Representative appearance of apoptosis/single cell necrosis (arrows) in the superficial cerebral cortex of vehicle-treated cynomolgus monkeys perfused intravenously at necropsy with ice-cold (2°C-8°C) 0.9% saline followed by tissue trimming in a chilled brain matrix prior to delayed immersion fixation in neutral buffered 10% formalin. Panel A: Affected cells are scattered randomly in low numbers throughout the affected cortex. Panel B: Affected cells most often exhibit features of apoptosis, including hypereosinophilic cytoplasm, condensation of nuclear chromatin, and colorless pericellular spaces. Panel C: Classical apoptotic bodies with fragmented nuclei and cytoplasmic blebs are quite rare. Stain: H&E. Original objective magnifications: Panel A = 10X; Panels B and C = 40X.
Neural cell A/SCN was not accompanied by other tissue reactions. Immunohistochemical (IHC) methods to detect glial fibrillary acidic protein (GFAP, for reactive astrocytes) or ionized calcium-binding adaptor molecule 1 (IBA1, for reactive microglia and macrophages) did not demonstrate co-localization of either biomarker in the affected brain domains (unless the numbers of intraparenchymal macrophages are increased, such as in animals treated with an antisense oligonucleotide [ASO] TA, where scattered ASO-laden macrophages may be distributed randomly throughout brain parenchyma). Mononuclear cell infiltrates (ie, a common finding in NHPs treated with biologic TAs, 1 where an “infiltrate” is a leukocyte accumulation that does not damage the organ parenchyma 23 ) or inflammation (ie, where “inflammation” denotes leukocyte accumulation that is injuring the parenchyma 23 ) is not seen in brain regions with A/SCN. We have not seen more widespread neuronal degeneration in brain regions with A/SCN while reviewing H&E-stained sections by fluorescent microscopy using a fluorescein isothiocyanate (FITC) filter.22,34 To date, we have not been presented with serial brain sections stained to specifically highlight cell death (eg, Fluoro-Jade B or C 8 ).
Common Study Design Parameters Correlated With Apoptosis/Single Cell Necrosis in Nonhuman Primate Brain
The presence of A/SCN in the brains of cold saline–perfused NHPs was associated with certain nonclinical study design parameters used consistently in combined biodistribution/toxicity studies employed for adeno-associated virus (AAV)-based gene therapy vectors and ASO gene modulation therapies. Factors listed here have been compiled by reviewing design details in the study protocols for 6 combined studies (4 for AAV and 2 for ASO agents) that were prepared by 4 test facilities.
Affected NHPs were all young adult cynomolgus monkeys (Macaca fascicularis). Animals of both Cambodian and Mauritian origin, both male and female, were represented. This pattern indicates that cold saline–associated A/SCN does not reflect enhanced sensitivity based on genetic or hormonal states.
Affected animals were sedated and then euthanized using conventional procedures (eg, intramuscular [IM] injection with ketamine followed by intravenous [IV] injection with pentobarbital sodium, sometimes together with other injectable sedative or anesthetic agents). Immediately thereafter, transcardial perfusion was performed with an ice-cold (2°C-8°C) fluid, typically 0.9% (“physiological”) saline but sometimes ribonuclease-free phosphate-buffered saline (PBS), with both solutions constituted at pH 7.4. The osmolarity of the saline variants was not stated in the protocols, but these solutions are formulated to be isotonic (ie, having an osmolarity matching that of human plasma). Three protocols did not specify the perfusion time or volume, instead stating that perfusion was continued until the returning fluid was clear. In 2 instances, the protocol noted a relatively narrow range for both the perfusion time (7.5-10 minutes) and perfusion volume (1.5-2 L). In 1 case, the protocol noted that the perfusion time was 5 or more minutes but did not state the perfusion volume.
When saline perfusion was completed, necropsies included an undefined but extended interval between brain removal and initial tissue fixation. In most protocols, no details were given regarding the length of this delay in fixation. However, in 4 protocols, a sequence of complex tasks were performed in approximately the order listed here. Brains and spinal cords were removed and sampled first to facilitate the collection and processing of specimens for biodistribution analysis of the TAs in selected CNS structures (the number and location of which were not mentioned in the study protocols). Brains were weighed and photographed next to a ruled scale. Next, brains were either placed in a prechilled brain matrix or first submerged in ice-cold PBS for up to 30 minutes, although most typically for 10 to 15 minutes, before being transferred to a prechilled brain matrix. Brains then were cut in the coronal plane into approximately 4-mm-thick slices that were then arranged on an ice-cold cutting board (infrequently positioned over an ice-filled tray to sustain cutting board chilling), labeled to permit ready identification of each slice, and then photographed. Brain slices were then divided along the midline and the halves from one hemisphere (usually the left) were processed for freezing (to support analysis of TA biodistribution and/or other molecular endpoints); in our experience, the slices destined for molecular analyses are processed first (to minimize TA degradation), whereas the slices slated for microscopic evaluation remain on the cutting board until the prosectors can devote time and attention to preparing them. Finally, halves from the other brain hemisphere were fixed by immersion in neutral buffered 10% formalin (NBF, presumably a conventional premixed commercial formulation containing approximately 1% methanol as a stabilizing agent) at room temperature for 36 to 48 hours before being transferred to PBS (pH and temperature at initial tissue contact was not stated) for storage at 2°C to 8°C until shipment to a remote histology facility. The length of this storage period was not indicated in any protocol.
The authors now work solely as bench microscopists and thus cannot comment on the precise length of time between the initiation of transcardial saline perfusion and the complete fixation of brain tissue at the various test facilities. An estimate based on the protocol details noted previously, and the authors’ past experiences with NHP necropsies, would suggest a range from 20 to 75 minutes between the start of transcardial perfusion and the placement of brain slices into NBF. Thereafter, complete fixation of lipid-rich brain tissue requires 24 hours or more. 24 Based on these presumptions, a reasonable means for mitigating A/SCN would be to provide sufficient prosectors at necropsies so that brain slices slated for microscopic evaluation may be placed in a fixative immediately after their collection.
Potential Mechanisms for Apoptosis/Single Cell Necrosis in Brain for Combined Biodistribution/Toxicity Studies
Several plausible mechanisms might explain the presence of rare A/SCN in brains of NHPs from combined biodistribution/toxicity studies. Some are intrinsic in the common experimental design for such studies, whereas others represent possible biological attributes of living tissue. As yet, no evidence exists to distinguish among these causes, so further investigation will be needed to discriminate whether one or several of them or some other process is responsible for the finding.
Common Study Design Parameters
One similarity linking all 6 studies was cold saline perfusion followed by substantial handling over many minutes prior to delayed fixation by immersion. In this scenario, cold shock—perhaps in combination with burgeoning tissue acidity and hypoxia related to cessation of blood flow—might act as a terminal stressor responsible for driving rare TA-stressed neural cells to initiate a self-directed death program leading to apoptosis. The importance of thermal stress for studies with this experimental design is suggested by the observation that many cells exhibiting A/SCN were localized near or at a short distance from capillaries, and thus were likely to have experienced rapid cooling. A possible confounding factor is that cold solutions may produce localized vasoconstriction, leading to multifocal restricted perfusion of various brain regions including the cerebral cortex. 16 Such vasoconstriction might exacerbate progressive microenvironmental shifts (depletion of glucose, oxygen, and other essential nutrients, and/or a rise in tissue acidity) that can injure terminally stressed neural cells.
A second study design feature reported for 4 of the 6 studies was terminal sedation with ketamine alone or ketamine with another agent (in both cases given by IM injection). The ketamine dose was not recorded for 2 studies that used the agent “to effect,” whereas in the other 2 studies ketamine was administered at 10 to 15 mg/kg when used as a standalone agent or 7.5 to 12 mg/kg when used in combination with another sedative (with either option being permitted as a prelude to administration of the euthanasia solution [pentobarbital sodium, given alone or with phenytoin sodium]). The sedation protocol was not listed for the other 2 studies, but in our experience with numerous other NHP toxicity studies, it seems likely that ketamine was used. Ketamine acts as a noncompetitive N-methyl-D-aspartate (NMDA) receptor antagonist and, depending on the situation and dose, is reported to have neuroprotective or neurotoxic effects. 12 Ketamine administration (5-10 mg/kg IM) lowers the serum glucose concentration in rhesus macaques (Macaca mulatta), 2 which might be detrimental to brain cell survival as many neural cell populations require a consistent supply of glucose. More importantly, ketamine is known to induce neuronal apoptosis in developing (7-day-old) rats when given repeatedly at 20 mg/kg (7 subcutaneous [SC] doses at 90-minute intervals) but not in rats treated repeatedly at 10 mg/kg (7 SC doses at 90-minute intervals) or once SC at 20 mg/kg. 36 Ketamine also incites neuronal degeneration in the cerebral cortex of neonatal (≤7-day-old) rhesus macaques administered a preoperative dose of 2 mg/kg as an IV bolus followed by a 0.5 mg/kg/hr infusion during an invasive surgical procedure. 41 Older (35-day-old) rhesus macaques exhibit reduced neural cell death compared with neonatal (5-day-old) animals. 39 Long-term ketamine exposure (6 months at 1 mg/kg IV) does cause enhanced cell death in the cerebral cortex of adolescent cynomolgus monkeys, but this effect does not develop after a shorter (1-month) exposure. 40 For this reason, ketamine neurotoxicity as a direct (neural cell injury) and/or indirect (glucose depletion) effect seems unlikely as a mechanism by which terminal A/SCN might arise in these studies.
High hydrostatic pressure (HHP) has been shown to induce cell death in skin, with the death process depending on the extent of the pressure elevation. Application of HHP at 200 megapascals (MPa) for 10 minutes leads to cell necrosis (with retention of extracellular matrix structure), whereas HHP of 50 MPa for 36 hours induces widespread apoptosis instead. 26 These pressures, which are used deliberately in biomedical engineering to produce cell-depleted tissue grafts, far exceed the hydrostatic pressures employed during routine intravascular perfusion at necropsy; the preferred pressure range when performing whole-body perfusion fixation for nonclinical studies is 120 to 150 mm Hg (ie, approximating systolic blood pressure during life), 7 which equates to about 0.016 to 0.020 MPa. Elevated intravascular perfusion pressure substantially above the upper limit of the in-life physiological range may disrupt blood-brain barrier (BBB) integrity and induce both vasoconstriction and vasodilation. 37 The consistent lack of capillaries that were blood-filled (ie, local perfusion failure), collapsed (ie, vasoconstriction), or widely dilated (ie, vasodilation) suggests that perfusion pressures were not excessive in affected cerebrocortical and hippocampal domains of the NHPs reviewed for these 6 studies. Although the protocols for the 6 NHP studies in this data set did not list perfusion pressure(s) used in introducing the cold saline, experienced test facilities generally incorporate pressure ranges in the vicinity of 0.015 to 0.025 MPa applied for about 7.5 to 15 minutes in standard operating procedures. Taken together, these details suggest that HHP is not a principal factor in driving A/SCN in nonclinical studies that include cold saline perfusion as a design feature.
Confounding Biological Processes
A potential explanation for A/SCN is that the finding represents a spontaneous change. Neuronal death is reported at low levels in brains of healthy but aged rats 42 and elderly humans.25,35 To our knowledge, a baseline rate of neural cell death has not been established for brains of healthy NHPs, such as the young adult cynomolgus monkeys from the current 6 studies, but the anticipated number of affected cells should be quite low; one apoptotic cell or fewer per square millimeter is the reported number for aged humans. 25 Conceptually, A/SCN might be a regular feature in routine formalin-fixed, paraffin-embedded NHP brain sections as pathology evaluation in conventional nonclinical studies typically involves screening tissue in a less systematic manner, often with little to no use of high-magnification objectives (20X and 40X) needed to reliably detect sporadic neural cell death of the sort observed in the current 6 studies. The present authors were sensitized to the need for such high-magnification analysis because biologic and gene therapy TAs assessed in combined biodistribution/toxicity studies sometimes induce very subtle cellular changes. 1 That said, the authors do not recall seeing A/SCN in NHP toxicity studies that employ study designs in which cold saline perfusion was omitted (as samples for biodistribution analysis do not need to collected) but which in other respects are substantially equivalent in design to those of the 6 studies described here.
Future Mechanistic Investigations
Further work is necessary to assess whether one or several of the mechanisms described previously actually contribute to A/SCN in the NHP brain. For instance, cold shock might be investigated by evaluating the incidence of A/SCN across a variety of perfusion conditions and/or necropsy modifications that facilitate more rapid brain fixation of slices slated for microscopic evaluation. Contributions by ketamine might be examined by selecting other sedative regimens but retaining cold saline perfusion. Perfusion pressure might be evaluated by varying the rate and/or volume of solution (held at a constant temperature) instilled during a given span of time. Background incidences of A/SCN for distinct brain regions can be assessed across test species, different ages, and both sexes to ascertain whether this finding occurs normally in healthy, purpose-bred animals used in nonclinical studies.
Furthermore, supplemental work is necessary to ascertain whether the most appropriate diagnosis for this finding is apoptosis, single cell necrosis, or A/SCN. At present, A/SCN conforms to current diagnostic nomenclature, given the morphologic changes seen in the affected cells in H&E-stained sections. 14 The rarity of A/SCN will likely make such follow-up investigations quite challenging with conventional morphologic endpoints (eg, assessing additional molecular markers of apoptosis in serial tissue sections). “Omics” data (eg, single-cell transcriptomics) might prove to be more successful in providing a definitive answer.
Implications of Apoptosis/Single Cell Necrosis in Cold Saline–Perfused Brain
Several additional considerations must be contemplated when interpreting the significance of A/SCN in cold saline–perfused NHP brain. To date, the authors have not been presented with brain sections from similarly designed nonclinical studies performed in other laboratory animal species (eg, rat, dog, or minipig), so whether or not rare A/SCN develops in other test species remains an open question.
First and foremost, cells exhibiting A/SCN in NHP brain occur commonly in vehicle-treated control animals, and the severity in controls is uniformly minimal. Therefore, the effect is interpreted to be a procedural artifact and not evidence of TA-related cytotoxicity.
Second, the extent of A/SCN may be slightly increased in severity (from minimal to mild) by some neuro-targeted TA (eg, ASOs) that exhibit pharmacologic activity for the NHP molecular pathway. This effect is evident especially for higher and/or more frequently administered doses of TA. The authors posit that the pathogenesis for this modest exacerbation of A/SCN in the face of high TA tissue levels results from exuberant intracellular accumulation of the TA leading to neural cell stress, after which cold saline inflicts a final blow that impels a small proportion of the stressed cells to initiate the cascade of molecular events that ultimately culminate in self-programmed cell death.
A third consideration is the preferred diagnostic terminology when recording this finding. Based on the morphological appearance, the only options are “apoptosis” and “single cell necrosis,” which are differentiated in H&E-stained tissue sections by their cytoarchitectural features, or the combined diagnosis “apoptosis/single cell necrosis” in situations when recording cell death does not require that its mechanism be known. 14 “Apoptosis” is assigned when cells exhibiting the following features are visible: cell shrinkage; hypereosinophilic cytoplasm; condensed, often fragmenting nuclei; and clear, colorless, pericellular spaces. In contrast, “single cell necrosis” is diagnosed when the affected elements exhibit cell and nuclear enlargement, pale eosinophilic cytoplasm, and variable nuclear fragmentation or complete nuclear dissolution (karyolysis); inflammation (leukocyte accumulation resulting from leakage of intracellular contents) may or may not be seen with “single cell necrosis” but does not occur with “apoptosis.” Based on these features, the morphology of the bulk of affected neural cells in cold saline–perfused brain (Figure 1) appears to warrant a diagnosis of “apoptosis.” This choice makes mechanistic sense as apoptosis represents a self-directed suicide program that is often selected by cells undergoing substantial stress. That said, the cytoarchitectural features of a few affected cells had characteristics that more closely resembled “single cell necrosis.” For this reason, the authors use the diagnosis “apoptosis/single cell necrosis” in documenting this finding, doing so because the cause of the death was immaterial for risk assessment as its occurrence in control animals shows that it is not a direct consequence of TA exposure.
A final consideration is appropriate interpretation and communication of rare A/SCN when evaluating brain sections from nonclinical studies that incorporate ice-cold saline perfusion and variably delayed immersion fixation. The presence of this finding in control animals and treated animals will often obviate the need to include “apoptosis/single cell necrosis” as a diagnosis in the final pathology data set with a corresponding discussion as an artifact in the report narrative. Exacerbation of cold saline–associated A/SCN in treated animals compared with the extent of the finding in controls may need to be incorporated in the pathology data set and described in the narrative in which instance carefully formulated explanatory text will be necessary to support the interpretation of this finding as enhanced occurrence of a likely procedural artifact and not a TA-related effect.
In the authors’ view, A/SCN of minimal severity in the NHP brain, seemingly associated with cold saline perfusion, should not impact development of biologics, gene therapies, and similar products as the finding appears to be a procedural artifact. That said, pathologists who perform primary evaluations and pathology peer reviews need to be aware and willing to assess brain sections using high-power objectives to confirm that A/SCN occurs across all treatment groups including controls, and to see whether the finding is increased in treated animals at higher doses. In our opinion, occurrence of minimal A/SCN in all groups including controls is not adverse by definition as it is not a TA-related effect. Modest exacerbation (relative to controls) of A/SCN in animals given a TA that accumulates in a neural cell population should be noted as potential evidence of neural cell stress, but the finding as a standalone effect should not be interpreted as TA-related neurotoxicity.
Footnotes
Acknowledgements
The authors gratefully thank Ms Beth Mahler for her assistance in optimizing the layout and resolution of the figure.
Author Contributions
The authors are solely responsible for the contents and crafting of this article.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.