
Abstract
Iron overload has been recognized as a risk factor for liver disease; however, little is known about its pathological role in the modification of liver injury. The purpose of this study is to investigate the influence of iron overload on liver injury induced by two hepatotoxicants with different pathogenesis in rats. Rats were fed a control (Cont), 0.8% high-iron (0.8% Fe), or 1% high-iron diet (1% Fe) for 4 weeks and were then administered with saline, thioacetamide (TAA), or carbon tetrachloride (CCl4). Hepatic and systemic iron overload were seen in the 0.8% and 1% Fe groups. Twenty-four hours after administration, hepatocellular necrosis induced by TAA and hepatocellular necrosis, degeneration, and vacuolation induced by CCl4, as well as serum transaminase values, were exacerbated in the 0.8% and 1% Fe groups compared to the Cont group. On the other hand, microvesicular vacuolation induced by CCl4 was decreased in 0.8% and 1% Fe groups. Hepatocellular DNA damage was increased by iron overload in both models, whereas a synergistic effect of oxidative stress by excess iron and hepatotoxicant was only present in the CCl4 model. The data showed that dietary iron overload exacerbates TAA- and CCl4-induced acute liver injury with different mechanisms.
Keywords
Introduction
Iron is one of the most abundant metals in the body and an essential element involved in important biochemical processes, working as a protein cofactor for hemoglobin and non-heme iron-containing proteins.45,63 In mammals, iron metabolism needs to be tightly regulated in the body to prevent from either iron deficiency or overload.12,48 Excess iron leads to transferrin saturation and non–transferrin-bound iron (NTBI) accumulation in the circulation and certain cell types including hepatocytes.17,21,39 NTBI is highly reactive and can induce oxidative stress, which is a pivotal trigger of several organ dysfunction including heart failure, liver failure, renal disease, and neurodegenerative disease. 37 The key step for the excessive production of oxidative stress is the Fenton reaction, resulting in the production of hydroxyl radicals and phospholipid peroxidation, oxidation of amino acid side chains, DNA strand breaks, and protein fragmentation.37,39
The liver plays a major role in iron homeostasis, particularly through the production of hepcidin, a negative regulator of systemic iron homeostasis. 39 Chronic liver disease (CLD) is caused by various types of liver injury such as viral infection, alcohol abuse, and metabolic dysfunction. CLD may decrease hepatic hepcidin synthesis, resulting in blood and hepatic iron overload. 39 Iron overload is observed in 10% to 30% of patients with CLDs including chronic hepatitis C (HCV), alcoholic liver disease (ALD), and nonalcoholic fatty liver disease (NAFLD),25,34,39 and it has been recognized as a risk factor for the progression of CLDs. In fact, iron depletion by phlebotomy improves biochemical and/or histological outcomes in patients with chronic HCV infection,31,53 ALD, 47 and NAFLD.28,32,47 However, another study suggests that phlebotomy does not improve pathology of NAFLD. 3 The role of iron overload in CLD progression is still debated.
Chemically induced liver injury models using rodents are useful for studying the role of iron in liver disease progression. Hepatic iron overload has been reported to alter susceptibility to hepatotoxicity in rodent models. Some studies have shown that iron overload exacerbates liver injury induced by administration of thioacetamide (TAA), 1 allyl alcohol, 42 and carbon tetrachloride (CCl4), 6 while other studies have shown that iron overload suppresses or does not alter liver injury induced by TAA 8 and acetaminophen. 2 The pathological role of iron overload in the modification of hepatotoxicity needs to be investigated in more detail. TAA and CCl4 are broadly utilized hepatotoxicants. Both toxicants can induce centrilobular (zone 3) liver injury by producing active metabolites in rodents,14,15,23,29 but the induced histopathological changes are different: Fatty change (microvesicular vacuolation) is observed in the CCl4 but not in the TAA model. 40 A comparative analysis of these two hepatotoxicants with a different mechanism of injury would be helpful to understand in more detail how iron overload modifies chemically induced hepatotoxicity. Therefore, the main purpose of this study is to investigate the influence of hepatic iron overload on liver injury induced by TAA and CCl4 in rats.
In addition, we previously reported an unexpected bleeding tendency with decreased activity of vitamin K-dependent coagulation factors in iron-overloaded rats fed with citrate iron-supplemented diets. 26 The coagulation abnormalities are considered to be caused by a combination of iron overload and latent vitamin K insufficiency. The vitamin K content in these diets (18.2 µg/100 g diet) is comparable to that in AIN-76 (17 µg/100 g diet) 5 but is lower than that in AIN-93M (83.4 µg/100 g diet) formula. 50 Because bleeding reduces serum iron levels, it may alter iron homeostasis and thereby susceptibility to hepatotoxicity as a confounding factor. Therefore, before starting the hepatotoxicity study, we investigated bleeding frequency and coagulation factor activity in rats fed diets with higher and lower vitamin K content, to establish a better and more stable model of diet-induced iron overload.
Materials and Methods
The experiments conducted in this study are reported in accordance with the Animal Research: Reporting of In Vivo Experiments (ARRIVE) guidelines (https://arriveguidelines.org). All interpretations for clinical and histopathological endpoints were conducted by board-certified toxicologic pathologists (diplomates of the Japanese Society of Toxicologic Pathology; Y.I. and T.I.).
Coagulation Study
Six-week-old male F344/DuCrlCrlj rats (Charles River Laboratories Japan, Yokohama, Japan) were divided into Cont+VK (n = 6), 0.8% Fe+VK (n = 6), and 1% Fe+VK (n = 5) groups. Cont+VK, 0.8% Fe+VK, and 1% Fe+VK groups were fed a diet containing 0.02%, 0.8%, and 1% Fe (supplemented from ferric citrate) with higher vitamin K supplementation (Oriental Yeast Co. Ltd., Tokyo, Japan) for 4 weeks, respectively. As shown in Supplemental Table S1, the diets with higher vitamin K supplementation contain 88.2 µg of vitamin K per 100 g of diet, which is comparable to that in the AIN-93M formula 51 and considered to be nutritionally sufficient. Grouping was conducted to ensure homogeneity of mean body weights at the beginning of the experiment. Two authors (S.F. and T.I.) were aware of the group allocation at the different stages of the experiments. A sample size per group was determined in terms of reduction (minimizing the number of animals) and experimental accuracy (minimizing the influence of individual variations). Food and water were provided ad libitum. All rats were included in the following analyses without any exclusion. After 4-week feeding, whole blood was collected from the abdominal aorta under deep isoflurane anesthesia, and serum was separated by centrifugation (1500 × g, 10 minutes). In addition, plasma was separated from the blood in a tube with sodium citrate (Terumo, Tokyo, Japan) by centrifugation (1500 × g, 10 minutes). The serum and plasma samples were subjected to biochemical (serum iron, total iron-binding capacity [TIBC, to calculate transferrin saturation], and albumin) and coagulation analyses (activity of coagulation factors II, VII, and VIII), respectively. The biochemical parameters and activity of coagulation factors were analyzed by SRL Inc. (Tokyo, Japan). Confounders were not controlled in this study; however, the influence of the order of treatments and measurements is considered minimal. The experiment was approved by the Institutional Animal Care and Use Committee (code no. 21-33) and was performed according to the Guidelines for Animal Experimentation of Osaka Metropolitan University.
Frequency of hemorrhage and hemorrhage-related death was evaluated in a series of our previous studies using iron- and vitamin K-modified diets (30 studies from 2012 to 2023), including data published previously. 26 Composition of the diets used in this analysis is shown in Supplemental Table S1. All animals included in this analysis are 6-week-old male F344/DuCrlCrlj rats at the beginning of the experiments. They were fed each diet for at least 4 weeks, and during the first 4 weeks of feeding, the number of animals with any of the following criteria was recorded and counted. The number of animals with (1) gross hemorrhage with no relation to experimental injection and/or (2) with hematological and biochemical findings suggestive of hemorrhage (decreased hematocrit value [HCT], albumin, and increased reticulocytes) was counted as “hemorrhage”. The number of dead animals without evidence of other identifiable cause than hemorrhage was counted as “hemorrhage-related death”.
Hepatotoxicity Study
Six-week-old male F344/DuCrlCrlj rats (Charles River Laboratories) were divided into control (Cont), 0.8% high-iron (0.8% Fe), and 1% high-iron (1% Fe) groups. Grouping was conducted to ensure homogeneity of mean body weights at the beginning of the experiment. Three authors (Y.I., T.K., and T.I.) were aware of the group allocation at the different stages of the experiments. Rats in the Cont group were fed a control diet containing 0.02% Fe (Oriental Yeast Co. Ltd.), while rats in the 0.8% and 1% Fe groups were fed high-iron diets containing 0.8% and 1% Fe (supplemented from ferric citrate, Oriental Yeast Co. Ltd.) for 4 weeks, respectively. All diets contain 88.2 µg of vitamin K per 100 g of diet, comparable to the AIN-93M formula, 50 as shown in Supplemental Table S1. A sample size per group was determined in terms of reduction (minimizing the number of animals) and experimental accuracy (minimizing the influence of individual variations). Food and water were provided ad libitum. After 4-week feeding, saline (p.o.), TAA (100 mg/kg, i.p., FUJIFILM Wako, Osaka, Japan), or CCl4 (0.75 mL/kg, p.o., FUJIFILM Wako) were administered to rats of each group: Saline-Cont (n = 4), Saline-0.8% Fe (n = 4), Saline-1% Fe (n = 4), TAA-Cont (n = 4), TAA-0.8% Fe (n = 4), TAA-1% Fe (n = 4), CCl4-Cont (n = 6), CCl4-0.8% Fe (n = 5), CCl4-1% Fe (n = 6). Rats were euthanized under deep isoflurane anesthesia; the blood and liver were collected 24 hours after administration. The dose and time point of sampling after dosing were determined on the basis of the results of the author’s previous studies showing that hepatocellular injury peaks at 24 hours after TAA or CCl4 administration.27,42 Absolute and relative liver weights were examined. Rats were maintained in specific pathogen-free conditions with controlled temperature (21 ± 3°C) and 12-hour light-dark cycle. Confounders were not controlled in this study; however, the influence of the order of treatments and measurements is considered minimal as far as observing the data obtained. The experiment was approved by the Institutional Animal Care and Use Committee (code nos. 21-33 and 22-38) and was performed according to the Guidelines for Animal Experimentation of Osaka Metropolitan University.
Blood Biochemistry
The serum was separated from the blood collected from the abdominal aorta by centrifugation (1500 × g, 10 minutes). The serum samples (Saline-Cont, n = 4; Saline-0.8% Fe, n = 4; Saline-1% Fe, n = 4; TAA-Cont, n = 4; TAA-0.8% Fe, n = 4; TAA-1% Fe, n = 4; CCl4-Cont, n = 6; CCl4-0.8% Fe, n = 5; CCl4-1% Fe, n = 6) were subjected to biochemical analyses (serum iron, TIBC, aspartate aminotransferase [AST], alanine aminotransferase [ALT], and albumin), conducted by SRL Inc.
Histopathology and Immunohistochemistry
The left lateral lobe of the liver was fixed in 10% neutral-buffered formalin for less than 24 hours, routinely processed, embedded in paraffin, cut at 5 µm, and stained with hematoxylin and eosin (H&E) for histopathological examination. Sections were also stained with Perls iron histochemistry with or without 3,3′-diaminobenzidine (DAB) enhancement as described previously.7,42 For immunohistochemistry, the following primary antibodies were used: rabbit monoclonal anti-NAD(P)H quinone dehydrogenase 1 (NQO1; clone 1D12, 1:1000; Merck, Darmstadt, Germany) and rabbit monoclonal anti-phospho (Ser139)-histone H2AX (γH2AX; clone 20E3, 1:500; Cell Signaling Technology, Danvers, MA, USA). After autoclave pretreatment at 121°C for 10 minutes, sections were incubated with the primary antibody at room temperature for 1 hour, followed by incubation with peroxidase-conjugated secondary antibody (Simplestain MAX-PO; Nichirei, Tokyo, Japan) for 1 hour. Signals were visualized with a DAB substrate kit (Nichirei).
Artificial Intelligence–Assisted Image Analysis
Whole slide images were obtained using digital slide scanners (Aperio GT450; Leica Biosystems, Wetzlar, Germany, or VS-120; Evident Corporation, Tokyo, Japan), and the following analyses were conducted using HALO (v3.3.2541; Indica Labs, New Mexico, USA): The H&E images of the TAA model were classified into necrosis, hepatocyte (without necrosis), and other (including portal and glass area) areas as shown in Figure 3A-B and Supplemental Figure S2. The percentage of necrosis area in the parenchymal area (a sum of necrosis and hepatocyte areas) was calculated in each animal.
The H&E images of the CCl4 model were classified into necrosis (coagulation necrosis of hepatocytes with pyknosis, karyorrhexis, and/or karyolysis), degeneration (eosinophilic changes of hepatocytes with normal-appearing, hypochromatic, and/or swollen nuclei, often accompanied with single-cell necrosis), vacuolation (vacuolated hepatocytes with swollen cytoplasm, often referred to as “fatty degeneration” in the rodent CCl4 model), microvesicular vacuolation (of hepatocytes), infiltration (aggregation of inflammatory cells in the hepatic lobule), hepatocyte (without aforementioned morphological change), and other (including portal area and glass) areas as shown in Figure 3C-F and Supplemental Figure S3. The percentage of necrosis, degeneration, vacuolation, microvesicular vacuolation, and infiltration area in the parenchymal area (a sum of necrosis, degeneration, vacuolation, microvesicular vacuolation, infiltration, and hepatocyte areas) was calculated, as well as the percentage of combined necrosis, degeneration, and vacuolation area in the parenchymal area.
The images of γH2AX and NQO1 immunohistochemistry were classified into parenchymal area, including both normal and degenerative/necrotizing hepatocytes, and non-parenchymal areas as shown in Figure 5. The percentage of γH2AX-positive nuclei in the total nuclei of the parenchymal area was calculated. The percentage of NQO1-positive area in the parenchymal area was also calculated.
Classifier (v3.3.2541, Densenet V2) was used for the classification of the H&E and immunohistochemistry images. Multiplex IHC (v3.1.4) was used for nuclear segmentation and counting γH2AX-positive hepatocyte nuclei. Area Quantification (v2.2.1) was used for quantification of NQO1 immunohistochemistry. All image analyses were conducted using whole slide images of the left lateral lobe. Validation for each classifier and quantification analyses was conducted by confirming the appropriateness of the analyzed images by board-certified pathologists (Y.I. and T.I.).
Malondialdehyde Assay
Hepatic malondialdehyde (MDA) content was measured by the thiobarbituric acid–reactive substances (TBARS) method with an MDA Assay Kit (Northwest Life Science Specialities, Vancouver, Canada) according to the manufacturer’s instructions. The left lateral lobe, snap frozen in liquid nitrogen, was used for this assay.
Oxidized Glutathione/Reduced Glutathione Assay
Hepatic reduced glutathione (GSH) and oxidized glutathione (GSSG) were measured using a GSSG/GSH Quantification kit (DOJINDO Laboratories, Tokyo, Japan) according to the manufacturer’s instructions. The left lateral lobe, snap frozen in liquid nitrogen, was used for this assay. The ratio of GSSG/GSH was calculated.
Statistical Analysis
Data are presented as mean ± standard deviation (SD) with individual data points. Statistical analyses were performed using one-way or two-way analysis of variance (ANOVA) followed by Tukey’s multiple comparison by Prism software (ver. 9.5.1.; GraphPad, San Diego, CA, USA). A value of P < .05 was considered statistically significant.
Results
Bleeding Frequency by High-Iron Diets With Lower Vitamin K Supplementation Is Suppressed by Higher Vitamin K Supplementation
The authors previously showed that dietary iron overload with lower vitamin K supplementation induces bleeding tendency with decreased activity of vitamin K-dependent coagulation factors in rats. 26 To suppress the bleeding tendency in iron-overloaded rats, the authors produced an improved rat model of dietary iron overload with higher vitamin K supplementation. After 4 weeks of feeding, increases in serum iron, transferrin saturation, and albumin were observed in 0.8% and 1% Fe+VK groups (Figure 1A-C). The activity of coagulation factors II and VII decreased with increasing dietary iron contents (Figure 1D-E), but factor VIII did not change among groups (Figure 1F). The data showed that even with nutritionally sufficient vitamin K content, the activity of vitamin K-dependent coagulation factor was decreased in iron-overloaded rats.
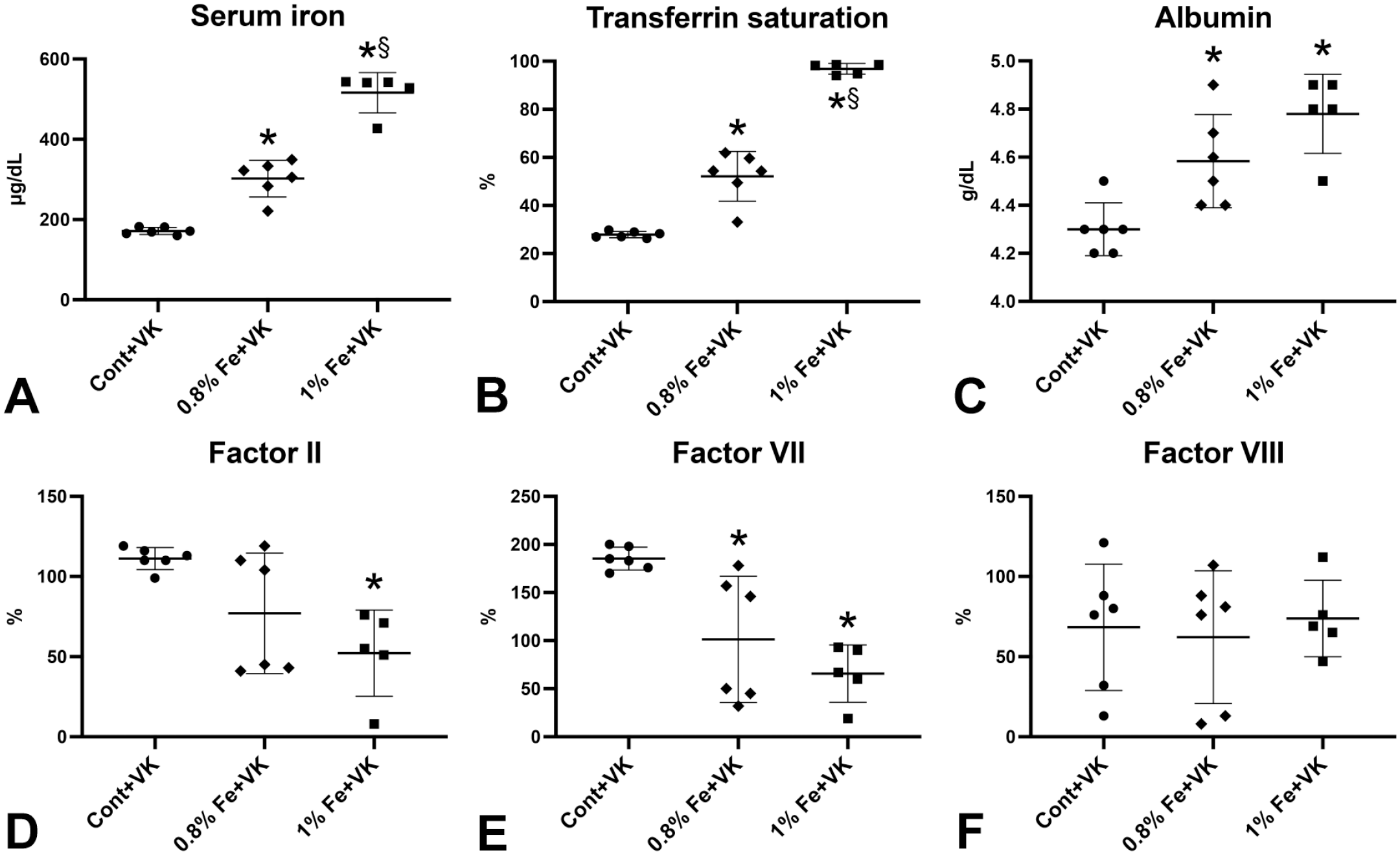
Blood biochemistry and activity of coagulation factors in rats fed high-iron diets with higher vitamin K supplementation. (A-C) Serum iron, transferrin saturation, and albumin increase with dietary iron content. (D-F) Coagulation factors II and VII (vitamin K-dependent factors), but not coagulation factor VIII (vitamin K-independent factor), decrease in 1% Fe+VK groups compared with Cont+VK group.
To investigate the effect of higher vitamin K supplementation on the clinical phonotypes, the incidence of hemorrhage and/or death within the first 4 weeks of experiment was evaluated in a series of the authors’ previous studies using iron- and vitamin K-modified diets (Supplemental Table S2). In rats fed iron-modified diets with lower vitamin K supplementation, hemorrhage and/or hemorrhage-related death were recorded in 8.6% and 16.9% of the 0.8% and 1% iron groups, respectively (Supplemental Table S2). Hemorrhage was frequently seen in thymus, lungs, and nasal cavity as described in the authors’ previous study. 26 In contrast, there was no incidence of hemorrhage and hemorrhage-related death in any groups of the iron-modified diet with higher vitamin K supplementation (Supplemental Table S2). The data showed that rats with dietary iron overload and higher vitamin K supplementation would be a more stable model for investigation of pathological role of iron overload in liver diseases.
Dietary Iron Overload Exacerbates Both TAA- and CCl4-Induced Acute Liver Injury With Different Histopathological Changes
In the hepatotoxicity study, the diets with higher vitamin K supplementation were used to eliminate bleeding as a confounding factor. After 4-week feeding of the iron-modified diets, decreased body weight and increased relative liver weight were observed in 0.8% and/or 1% Fe groups (Supplemental Table S3), which is consistent with the authors’ previous study using rats with dietary iron overload. 22 In the Saline group, food intake and calorie intake were higher in 0.8% and 1% Fe groups than those in the Cont group between weeks 3 and 4 (data not shown). In the Saline groups, serum iron, transferrin saturation, and hepatic iron accumulation, demonstrated by DAB-enhanced Perls iron stain, were increased with dietary iron content (Figure 2A-C and Supplemental Figure S1A-C). In the Saline-1% Fe group, iron accumulation is more intense in periportal hepatocytes than in centrilobular hepatocytes (Figure 2C and Supplemental Figure S1C), as described in the authors’ previous study.8,42 Iron accumulation is also intense in sinusoidal cells, morphologically suggestive of Kupffer cells, in Saline-0.8% and Saline-1% Fe groups (Supplemental Figure S1B, C, F). No significant change in serum albumin was seen between the Saline groups (Figure 2F). Serum AST or ALT was not elevated in the Saline groups with iron overload (Figure 2E-F). After TAA or CCl4 administration, increased serum ALT and AST with decreased serum iron, transferrin saturation, and/or albumin were observed compared with the Saline group (Figure 2A-B, D, E-F). Iron accumulation was more diffuse and more intense in non-injured hepatocytes after TAA or CCl4 administration in the iron-overloaded rats (TAA- or CCl4-0.8% Fe group) than that in the control rats (Saline-0.8% Fe group) (Supplemental Figure S1B, D, E). Interestingly, serum ALT and AST were higher in CCl4-0.8% Fe, TAA-0.8%, and TAA-1% Fe groups than in the control (0.02% Fe) group, respectively (Figure 2E-F). The data showed that dietary iron overload exacerbates TAA- and CCl4-induced acute hepatotoxicity.
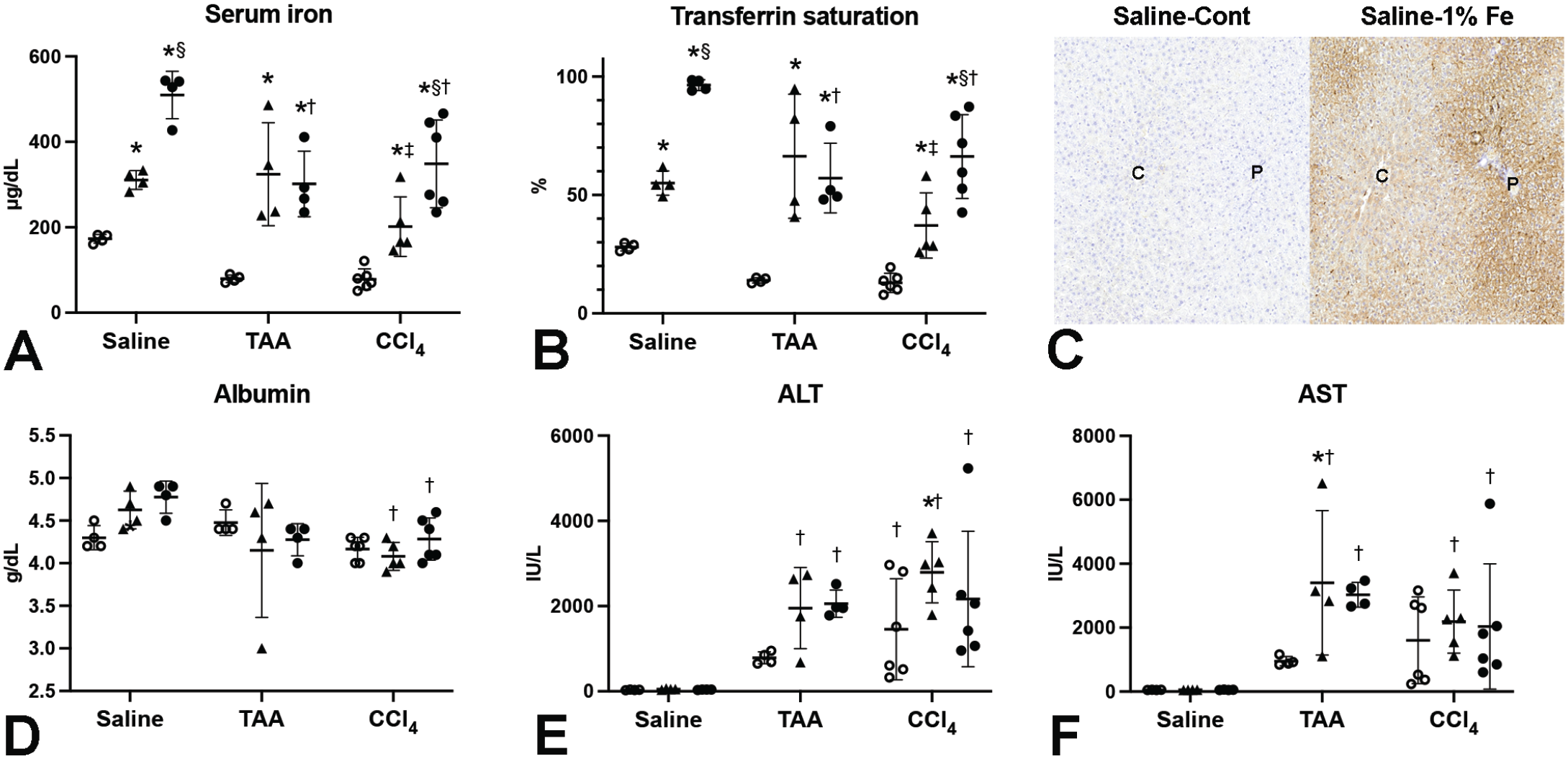
Blood biochemical parameters of rats fed high-iron diets with higher vitamin K supplementation at 24 hours after single dosing of saline, TAA, or CCl4. (A, B) Serum iron and transferrin saturation increase in 0.8% Fe and 1% Fe groups regardless of the TAA or CCl4 dosing. (C) Representative image of DAB-enhanced Perls iron stain. Marked iron accumulation is observed in the liver of 1% Fe group; left panel, Saline-Cont; right panel, Saline-1% Fe. (D) Serum albumin shows an increasing trend in 0.8% and 1% Fe groups compared with Cont group in Saline-treated rats, and it decreases after CCl4 administration in 0.8% Fe and 1% Fe groups. (E, F) Serum ALT and AST increase after TAA or CCl4 dosing, with the levels higher in 0.8% Fe and 1% Fe groups than in Cont group. C, central vein; P, portal area. ○, Cont; ▲, 0.8% Fe; •, 1% Fe. *P < .05 vs Cont. §P < .05 vs 0.8% Fe. †P < .05 vs Saline. ‡P < .05 vs. TAA, by Tukey’s multiple comparison (Saline-Cont, n = 4; Saline-0.8% Fe, n = 4; Saline-1% Fe, n = 4; TAA-Cont, n = 4; TAA-0.8% Fe, n = 4; TAA-1% Fe, n = 4; CCl4-Cont, n = 6; CCl4-0.8% Fe, n = 5; CCl4-1% Fe, n = 6).
To characterize the morphological changes associated with the exacerbation of TAA- and CCl4-induced hepatotoxicity by the iron overload, histopathological findings were classified and quantified with an artificial intelligence (AI)-based image analysis (Figure 3A-F and Supplemental Figures S2 and S3). The TAA model is characterized by coagulation necrosis of centrilobular hepatocytes (Figure 3A-B and Supplemental Figure S2G-L). Necrosis area was increased after TAA dosing with a strong positive correlation with serum ALT values (r = 0.94, P < .0001), suggesting that necrosis is the primary change responsible for the deviation of liver enzymes in the TAA model (Figure 4A-B). There was an increasing trend in necrosis area with increasing the dietary iron concentrations in rats with TAA dosing (Figure 4A).
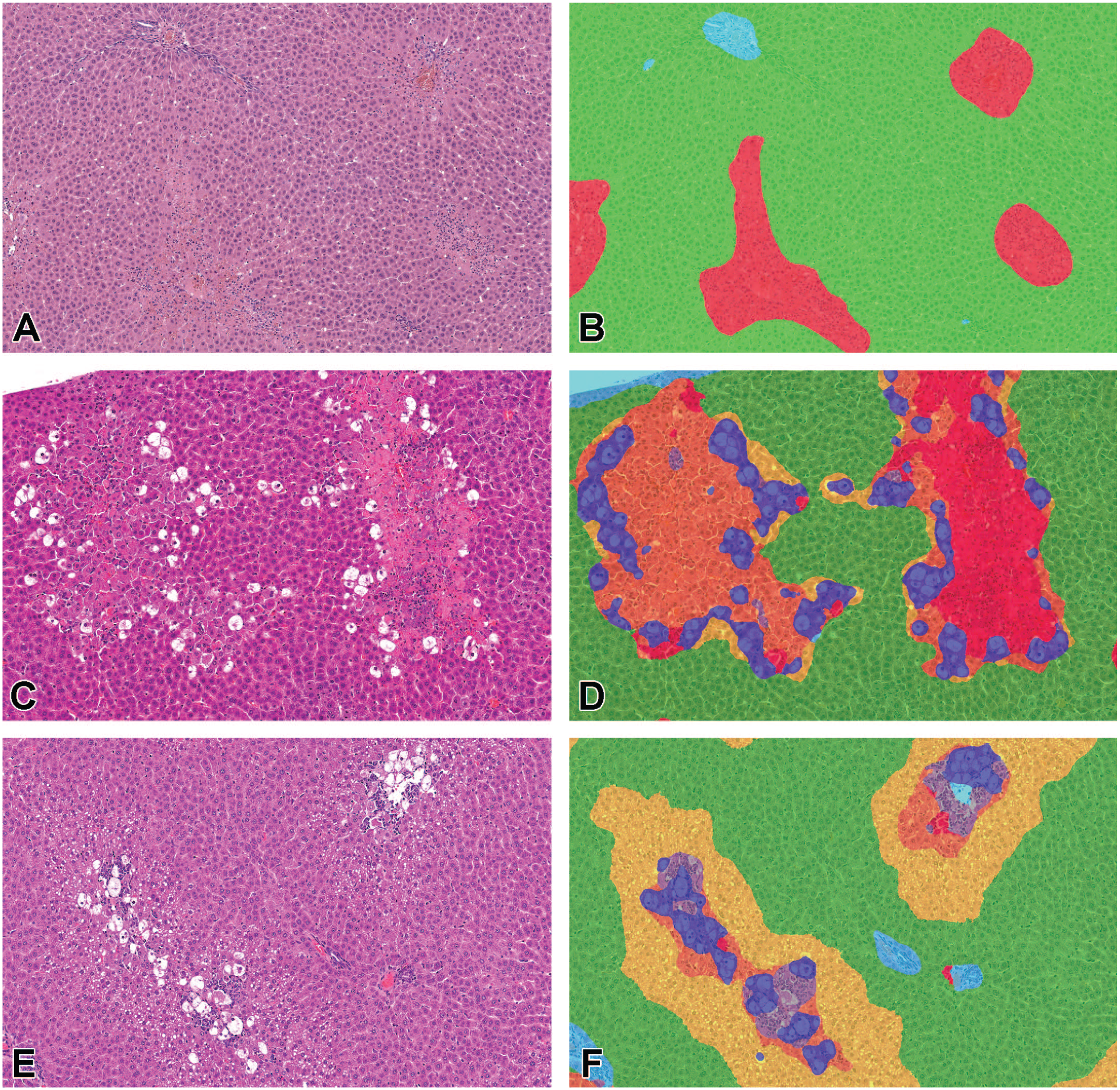
Histopathological findings (A, C, E) and images after AI-based image analysis (B, D, F) in rats fed high-iron diets with higher vitamin K supplementation at 24 hours after single dosing of TAA or CCl4. (A, B) Liver, TAA-0.8% Fe group. Coagulation necrosis is observed in the centrilobular hepatocytes after TAA administration. The HE image is classified into necrosis (red), hepatocyte without necrosis (green), and other area (light blue). (C-F) Liver: CCl4-1% Fe group (C, D) and CCl4-Cont group (E, F). Coagulation necrosis, degeneration, vacuolation, and microvesicular vacuolation are observed in the centrilobular hepatocytes by CCl4 administration. The H&E images are classified into necrosis (red), degeneration (orange), vacuolation (blue), microvesicular vacuolation (yellow), infiltration (gray), hepatocyte without any findings (green), and other areas (light blue).
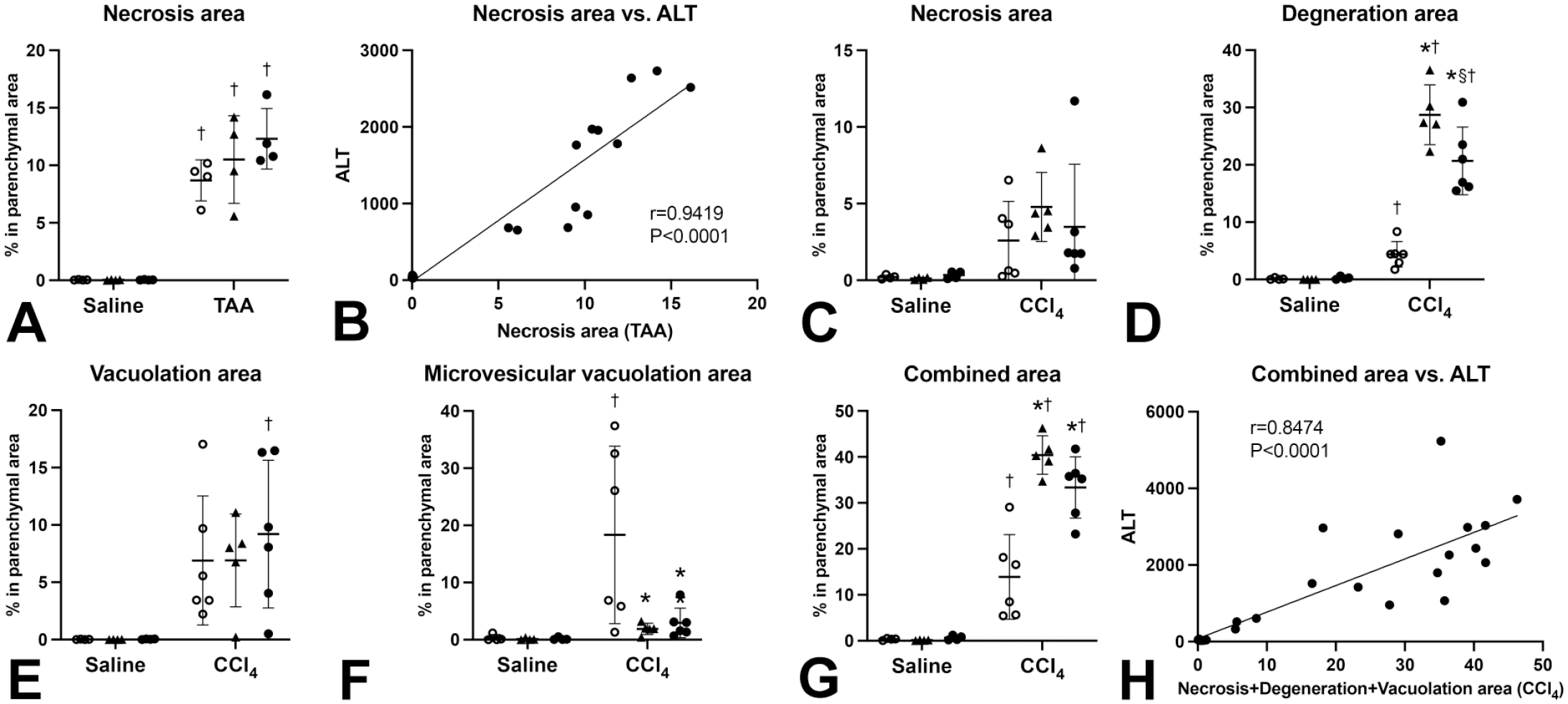
Quantification of histopathological findings in rats fed high-iron diets with higher vitamin K supplementation at 24 hours after single dosing of TAA or CCl4. (A, B) Necrosis area increased after TAA dosing with a strong positive correlation with serum ALT. The necrosis area shows an increasing trend with dietary iron content in TAA-treated rats. (C-H) Necrosis, degeneration, vacuolation, and microvesicular vacuolation area increase after CCl4 dosing. Degeneration area further increases, and microvesicular vacuolation area inversely decreases with iron overload. Combined necrosis, degeneration, and vacuolation area increases after CCl4 dosing and further increases with iron overload. The combined area is positively correlated with serum ALT. ○, Cont; ▲, 0.8% Fe; •, 1% Fe.
The CCl4 model was characterized by coagulation necrosis, degeneration, vacuolation, and microvesicular vacuolation of hepatocytes with infiltration of inflammatory cells including mononuclear cells and neutrophils (Figure 3C-F and Supplemental Figure S3G-L). Necrosis area (r = 0.86, P < .0001), degeneration area (r = 0.71, P < .0001), and vacuolation area (r = 0.67, P < .0001) were positively correlated with serum ALT values, while microvesicular vacuolation area (r = −0.069, P = .72) or infiltration area (r = 0.34, P = .073) was not correlated with ALT. Degeneration, vacuolation, and microvesicular vacuolation area increased after CCl4 dosing (Figure 4D-F). Dietary iron overload further increased degeneration area and inversely decreased microvesicular vacuolation area in rats with CCl4 dosing (Figure 4D, F). Interestingly, degeneration area was greater in 0.8% iron than in 1% iron group. Combined necrosis, degeneration, and vacuolation area increased after CCl4 dosing with a positive correlation with serum ALT (r = 0.85, P < .0001) and further increased with iron overload (Figure 4G-H). The data showed that dietary iron overload modified TAA- and CCl4-induced histopathological findings.
Dietary Iron Overload Enhances TAA- and CCl4-Induced DNA Damage and Induces NQO1 Expression After TAA Injury
In order to further characterize hepatocellular injury in the study model, immunohistochemistry for γH2AX, a marker for DNA damage, 44 was performed. γH2AX immunolabeling was observed mainly in the nucleus of hepatocytes within the necrosis area of TAA model and within the necrosis and degeneration areas of CCl4 model (Figure 5A-F and Supplemental Figure S4). The percentage of γH2AX-positive hepatocytes was increased in 0.8% and 1% Fe groups after TAA and CCl4 dosing (Figure 6A), with a positive correlation with necrosis area in the TAA model (r = 0.88, P < .0001), with combined necrosis and degeneration area in the CCl4 model (r = 0.82, P < .0001), and with serum ALT in both models (TAA: r = 0.95, P < .0001; CCl4: r = 0.89, P < .0001). In addition, γH2AX-positive hepatocytes increased in the CCl4-0.8% group compared with the CCl4-Cont group, similar to serum ALT and degeneration area in the CCl4 model. The data suggest that DNA damage, indicated by γH2AX immunohistochemistry, is one of the main causes for hepatocellular death and is enhanced by iron overload in both models.
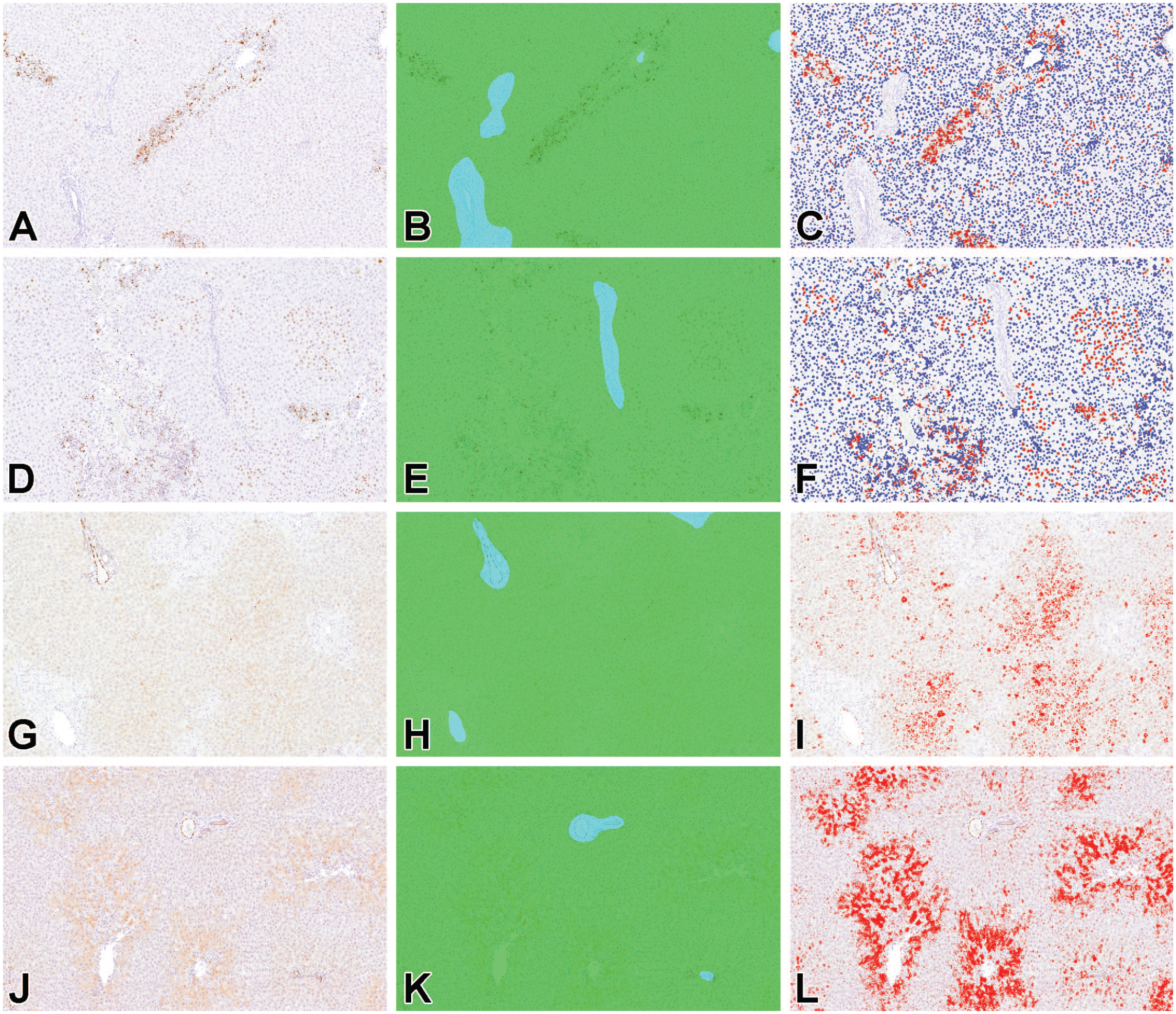
Representative images of immunohistochemistry for γH2AX (A-F) and NQO1 (G-L) and images after AI-based image analysis. After TAA (A-C) or CCl4 (D-F) administration, γH2AX immunoreactivity is observed mainly in the nucleus of necrotized or degenerated hepatocytes of iron-overloaded rats, whereas NQO1 immunoreactivity is observed mainly in non-injured hepatocytes. Left panels (A, D, G, J), original images of immunohistochemistry; middle panels (B, E, H, K), classified images with parenchymal (green) or non-parenchymal area (light blue); right panels (C, F, I, L), analyzed images with positive (red) and negative (blue) signals. Liver: TAA-1% Fe (A-C), CCl4-1% Fe (D-F), TAA-0.8% Fe (G-I), and CCl4-0.8% Fe (J-L).
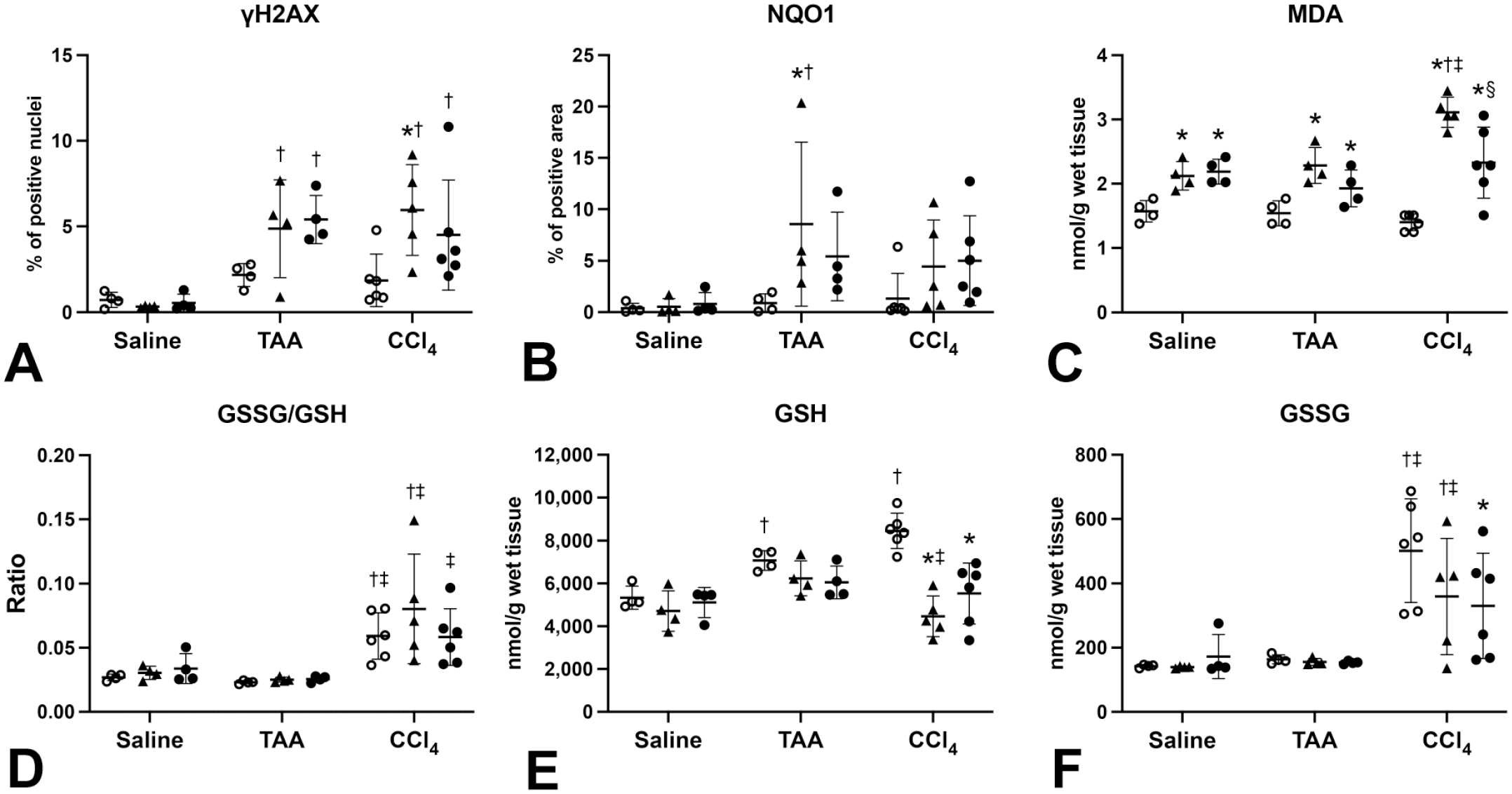
Quantification for γH2AX and NQO1 immunohistochemistry with oxidative stress–related parameters of rats fed high-iron diets with higher vitamin K supplementation at 24 hours after single dosing of saline, TAA, or CCl4. (A) γH2AX-positive hepatocyte nuclei (%) increase after TAA or CCl4 dosing, with higher percent observed in the CCl4-0.8% Fe group than in the CCl4-Cont group. TAA-0.8% Fe, TAA-1% Fe, and CCl4-1% groups also show higher percent increase than their corresponding Cont group. (B) NQO1-positive area (%) increases in the TAA-0.8% Fe group compared with both TAA-Cont and Saline-0.8% Fe groups. (C) Dietary iron overload increases hepatic MDA content, which is further increased by a combination of 0.8% iron diet and CCl4 treatment. (D-F) Both hepatic GSH content and GSSG/GSH ratio increase after CCl4 dosing, and iron overload decreases hepatic GSH in the CCl4 model. ○, Cont; ▲, 0.8% Fe; •1% Fe. *P < .05 vs Cont. §P < .05 vs 0.8% Fe. †P < .05 vs Saline, ‡P < .05 vs TAA, by Tukey’s multiple comparison (Saline-Cont, n = 4; Saline-0.8% Fe, n = 4; Saline-1% Fe, n = 4; TAA-Cont, n = 4; TAA-0.8% Fe, n = 4; TAA-1% Fe, n = 4; CCl4-Cont, n = 6; CCl4-0.8% Fe, n = 5; CCl4-1% Fe, n = 6).
NQO1 is an antioxidant enzyme and a target molecule of NF-E2-related factor 2 (Nrf2), a transcription factor that regulates gene expression of antioxidant and xenobiotic metabolizing enzymes in response to various environmental stresses.18,54 NQO1 immunolabeling was observed mainly in hepatocytes around necrosis area of the TAA model and hepatocytes in and around degeneration area of the CCl4 model (Figure 5G-L and Supplemental Figure S5). The NQO1-positive area was increased in TAA-0.8% Fe group compared with the Saline-0.8% Fe and TAA-Cont groups (Figure 6B and Supplemental Figure S5B, D, E). There were also increasing trends in TAA-1% Fe, CCl4-0.8%, and CCl4-1% Fe groups compared with their corresponding Saline and Cont groups (Figure 6B and Supplemental Figure S5B, D, E). The NQO1-positive area had a weak positive correlation with serum ALT (r = 0.36, P = .022). There was no statistically significant change in the NQO1-positive area in the CCl4 model. The data suggest that expression of the antioxidant enzyme NQO1 is increased within the hepatic lesion after TAA or CCl4 dosing in rats with dietary iron overload.
Dietary Iron Overload Enhances Oxidative Stress After CCl4 but Not TAA Dosing
Excess iron in the blood and organs causes extensive oxidative stress via generation of hydroxyl radicals by the Fenton reaction.37,39 Oxidative stress is the major cause of DNA damage and also a strong inducer of Nrf2 activation.16,58 MDA is an end-product of lipid peroxidation and is therefore regarded as an oxidative stress marker. 9 GSH is the major antioxidant and plays crucial roles in detoxifying xenobiotic and endogenous compounds. GSSG is formed from GSH under oxidative stress, and the ratio of GSSG to GSH reflects redox status in cells and tissues.43,49,65 In order to investigate the status of oxidative stress in the TAA and CCl4 models by dietary iron overload, hepatic MDA, GSH, and GSSG content and the GSSG-GSH ratio were analyzed. In the Saline and TAA groups, hepatic MDA content increased with dietary iron overload (Figure 6C). However, hepatic MDA did not increase significantly by TAA dosing (Figure 6C). Hepatic GSH content increased after TAA dosing, but hepatic GSSG or GSSG-GSH ratio did not change in the TAA model (Figure 6D-F), suggesting that TAA administration increases hepatic GSH store but does not affect the redox status.
In the CCl4 group, hepatic MDA increased in the 0.8% Fe group, with its level being higher than that in the Saline and TAA groups (Figure 6C). Interestingly, the MDA level was higher in the CCl4-0.8% Fe group than in CCl4-Cont and CCl4-1% Fe groups. The hepatic GSSG-GSH ratio increased after CCl4 dosing (Figure 6D). Both hepatic GSH and GSSG increased after CCl4 dosing and then decreased with iron overload (Figure 6E-F). These data suggest that CCl4 administration with iron overload alters redox status to oxidative condition and induces oxidative stress with increasing lipid peroxidation.
TAA and CCl4 are activated by CYP2E1, and their active metabolites are ultimate hepatotoxicants.14,41 To investigate whether metabolic enzyme activity is altered by iron overload, hepatic CYP2E1 activity was measured using samples of Saline groups as these samples reflect hepatic condition before dosing of TAA or CCl4. No significant changes were observed in the hepatic CYP2E1 activity between Cont, 0.8% Fe, and 1% Fe groups, but there was a decreasing trend in the 1% Fe group compared with the Saline-Cont and Saline-0.8% Fe groups (Supplemental Figure S6).
Discussion
The pathological role of iron overload in the modification of hepatotoxicity remains largely unknown. Our data show that dietary iron overload exacerbates TAA- and CCl4-induced acute liver toxicity with different mechanisms.
The data obtained from Saline groups represent baseline conditions of the liver in rats with different iron status. Compared to the Saline-Cont group, increased serum iron, transferrin saturation, albumin, and relative liver weights and accumulation of iron in the liver were observed in the Saline-0.8% and 1% Fe groups, which is similar to author’s previous study in rats fed a high-iron diet with lower vitamin K supplementation. 26 Increased relative liver weights are considered to be related with an accumulation of iron in the liver. Feeding of 0.8% and 1% iron diets with higher vitamin K supplementation still decreased the activity of vitamin K-dependent coagulation factors, but did not induce hemorrhage in this study, suggesting that influence of coagulation abnormality as a confounding factor on author’s models may be minimal. The previous studies on rabbits 62 and humans 59 have demonstrated an early coagulopathy without severe hepatic dysfunction in acute iron poisoning. Such coagulopathy is considered to be caused by high susceptibility of serine protease activity (factor II, VII, IX, X, XI, and XII) to NTBI.13,52 Dietary iron overload increased hepatic MDA content but did not induce hepatic injury in saline-treated rats. These data suggest that dietary iron overload in this study induces mild (non-hepatotoxic) oxidative stress. On the other hand, the dietary iron overload exacerbates TAA- and CCl4-induced acute liver injury.
TAA is activated by CYP2E1 to its S-oxide (TASO) and then to its chemically reactive S, S-dioxide (TASO2).14,23 TASO and TASO2 are ultimate hepatotoxicants and cause centrilobular necrosis of hepatocytes by covalent binding to proteins and lipids19,23,33,60 and/or induction of oxidative stress such as lipid peroxidation.30,55,56,57 Oxidative stress such as generation of hydroxyl radicals and other electrophilic molecules can cause DNA damage including double-strand breaks.16,38 In the TAA model, dietary iron overload enhances TAA-induced hepatocellular injury with increased DNA damage and hepatic MDA content. One of the possible mechanisms for the exacerbation of TAA-induced liver injury by iron overload is enhanced oxidative stress by excess iron. However, the values of hepatic MDA and GSSG-GSH ratio were comparable to those of the Saline group with iron overload, suggesting an absence of the synergistic effect of TAA dosing and dietary iron overload on oxidative stress. Ackerman et al 1 also reported that hepatic iron overload induced exacerbation of TAA-induced acute liver injury. Their data are considered to support the results since a decrease in hepatic GSH content after TAA administration, an oxidative stress indicator, was rather milder in iron-overloaded rats than in control rats, 1 suggesting that there is no or minimal synergistic effect on oxidative stress.
Another possible mechanism is increased production of active metabolites. NQO1 is an antioxidant enzyme and is also a target molecule of Nrf2, a master transcriptional regulator of cellular response to electrophilic stress as well as oxidative stress.54,58 Iron overload increased NQO1 expression in hepatocytes with iron accumulation after TAA administration, suggesting the possibility of Nrf2 activation. As mentioned earlier, TAA-induced liver injury is mainly caused by its active metabolite TASO2, an electrophile that can easily react with biomolecules. 60 Although direct evidence for Nrf2 activation by TASO2 has not been reported, electrophilic stress is well known to induce Nrf2 activation by inhibiting the binding of Nrf2 to its suppressor Keap1. 58 Sensing mechanism of Keap1 for hydroxyl radical is distinct from that for electrophilic Nrf2 inducers. 58 It is possible that the dietary iron overload may induce greater activation of Nrf2 in response to TAA-induced electrophilic and/or oxidative stress than in control rats. However, hepatic CYP2E1 activity did not increase in iron-overloaded rats. There may be other xenobiotic metabolizing enzymes (eg, flavin-containing monooxygenases) related to metabolic activation of TAA. 36 Further toxicokinetic study on active metabolites or related enzyme activities would clarify whether such electrophilic stress increases with TAA-induced liver injury in iron-overloaded rats. Taken together, the data suggest that oxidative stress is less involved in the exacerbation of TAA-induced liver injury by iron overload.
CCl4-induced hepatotoxicity is initiated by the generation of its reactive metabolites trichloromethyl radical (•CCl3) and trichloromethyl peroxyradicals (•O-O-CCl3). The former radical binds covalently to cellular macromolecules and thereby inhibits lipoprotein secretion leading to steatosis induction, while both radicals can cause lipid peroxidation.11,41,61 In the CCl4 model, dietary iron overload enhances hepatocellular injury with increased DNA damage and hepatic MDA content. Compared to the TAA model, induction of oxidative stress was more prominent in iron-overloaded rats after CCl4 administration, suggesting a synergistic effect on oxidative stress. Many studies have shown that antioxidants suppress CCl4 hepatotoxicity.4,24,35,64 These findings suggest that increased lipid peroxidation is the major cause of exacerbation of CCl4 hepatotoxicity by iron overload.
Interestingly, the AI-based analysis revealed that dietary iron overload increases degeneration area and inversely decreases microvesicular vacuolation in the CCl4 model. CCl4 induces hepatocellular necrosis and steatosis simultaneously, and steatosis is considered to be a result of covalent binding by •CCl3 that affects lipid metabolism. In contrast, •O-O-CCl3, formed from •CCl3 in high-oxygen-pressure conditions causes lipid peroxidation. 11 Our previous study using high-iron diets showed a slightly increased blood hemoglobin with increased mean corpuscular volume (MCV) and mean corpuscular hemoglobin (MCH) in dietary iron-overload rats, 26 as reported in human patients with iron overload.10,20 Since increased blood hemoglobin is a determinant of arterial oxygen content, 52 the iron-overloaded rats may have high arterial oxygen content compared to control rats. Given that the formation of •O-O-CCl3 occurs with high oxygen pressure, increased hemoglobin can facilitate production of •O-O-CCl3 from •CCl3. Our data suggest that iron overload might accelerate formation of •O-O-CCl3 from •CCl3, resulting in the decreased microvesicular vacuolation and increased lipid peroxidation.
Another interesting finding in the CCl4 model was the absence of iron concentration–dependent increase in liver injury. Hepatocellular injury including DNA injury, hepatic lipid peroxidation, and increased GSSG-GSH ratio were more prominent in the 0.8% Fe group than in the 1% Fe group. Transferrin saturation is an indicator of systemic iron homeostasis, and it was higher in the 1% Fe group (almost saturated) than in the 0.8% group, suggesting that NTBI may increase in the 1% Fe group. In general, increased NTBI induces oxidative stress via production of hydroxyl radicals by the Fenton reaction; 46 however, feeding of 1% high-iron diet alone did not induce hepatotoxicity in this study model. It is hypothesized that persistent non-hepatotoxic oxidative stress may cause the liver to acquire some stress tolerance. Nrf2 activation is a promising candidate for stress tolerance; however, there was no significant change in NQO1 immunoreactivity between CCl4-0.8% and CCl4-1% Fe groups. It is possible that other downstream molecules of Nrf2 may be involved in the decreased oxidative injury in the CCl4-1% Fe group. Further investigation is needed to elucidate the mechanism of stress tolerance in CCl4-induced liver injuries with iron overload.
The limitation of this study is that the molecular mechanism underlying the exacerbation of TAA- and CCl4-induced liver injury by iron overload is not fully investigated. Further studies on toxicokinetics of the active metabolites, cell death pathways (ferroptosis, necroptosis, and apoptosis), and stress response (Keap1-Nrf2 system) are required to overcome the limitation.
In summary, our data showed that dietary iron overload exacerbates TAA- and CCl4-induced acute liver injury with different mechanisms: Oxidative stress is mainly involved in the CCl4 model, but less involved in the TAA model. Further investigation of the molecular mechanism underlying the modification of chemically induced hepatotoxicity would help to understand the diverse roles of iron overload in liver diseases.
Supplemental Material
sj-docx-1-tpx-10.1177_01926233241235623 – Supplemental material for Difference in the Mechanism of Iron Overload–Enhanced Acute Hepatotoxicity Induced by Thioacetamide and Carbon Tetrachloride in Rats
Supplemental material, sj-docx-1-tpx-10.1177_01926233241235623 for Difference in the Mechanism of Iron Overload–Enhanced Acute Hepatotoxicity Induced by Thioacetamide and Carbon Tetrachloride in Rats by Yohei Inai, Takeshi Izawa, Tomomi Kamei, Sho Fujiwara, Miyuu Tanaka, Jyoji Yamate and Mitsuru Kuwamura in Toxicologic Pathology
Supplemental Material
sj-tif-2-tpx-10.1177_01926233241235623 – Supplemental material for Difference in the Mechanism of Iron Overload–Enhanced Acute Hepatotoxicity Induced by Thioacetamide and Carbon Tetrachloride in Rats
Supplemental material, sj-tif-2-tpx-10.1177_01926233241235623 for Difference in the Mechanism of Iron Overload–Enhanced Acute Hepatotoxicity Induced by Thioacetamide and Carbon Tetrachloride in Rats by Yohei Inai, Takeshi Izawa, Tomomi Kamei, Sho Fujiwara, Miyuu Tanaka, Jyoji Yamate and Mitsuru Kuwamura in Toxicologic Pathology
Supplemental Material
sj-tif-3-tpx-10.1177_01926233241235623 – Supplemental material for Difference in the Mechanism of Iron Overload–Enhanced Acute Hepatotoxicity Induced by Thioacetamide and Carbon Tetrachloride in Rats
Supplemental material, sj-tif-3-tpx-10.1177_01926233241235623 for Difference in the Mechanism of Iron Overload–Enhanced Acute Hepatotoxicity Induced by Thioacetamide and Carbon Tetrachloride in Rats by Yohei Inai, Takeshi Izawa, Tomomi Kamei, Sho Fujiwara, Miyuu Tanaka, Jyoji Yamate and Mitsuru Kuwamura in Toxicologic Pathology
Supplemental Material
sj-tif-4-tpx-10.1177_01926233241235623 – Supplemental material for Difference in the Mechanism of Iron Overload–Enhanced Acute Hepatotoxicity Induced by Thioacetamide and Carbon Tetrachloride in Rats
Supplemental material, sj-tif-4-tpx-10.1177_01926233241235623 for Difference in the Mechanism of Iron Overload–Enhanced Acute Hepatotoxicity Induced by Thioacetamide and Carbon Tetrachloride in Rats by Yohei Inai, Takeshi Izawa, Tomomi Kamei, Sho Fujiwara, Miyuu Tanaka, Jyoji Yamate and Mitsuru Kuwamura in Toxicologic Pathology
Supplemental Material
sj-tif-5-tpx-10.1177_01926233241235623 – Supplemental material for Difference in the Mechanism of Iron Overload–Enhanced Acute Hepatotoxicity Induced by Thioacetamide and Carbon Tetrachloride in Rats
Supplemental material, sj-tif-5-tpx-10.1177_01926233241235623 for Difference in the Mechanism of Iron Overload–Enhanced Acute Hepatotoxicity Induced by Thioacetamide and Carbon Tetrachloride in Rats by Yohei Inai, Takeshi Izawa, Tomomi Kamei, Sho Fujiwara, Miyuu Tanaka, Jyoji Yamate and Mitsuru Kuwamura in Toxicologic Pathology
Supplemental Material
sj-tif-6-tpx-10.1177_01926233241235623 – Supplemental material for Difference in the Mechanism of Iron Overload–Enhanced Acute Hepatotoxicity Induced by Thioacetamide and Carbon Tetrachloride in Rats
Supplemental material, sj-tif-6-tpx-10.1177_01926233241235623 for Difference in the Mechanism of Iron Overload–Enhanced Acute Hepatotoxicity Induced by Thioacetamide and Carbon Tetrachloride in Rats by Yohei Inai, Takeshi Izawa, Tomomi Kamei, Sho Fujiwara, Miyuu Tanaka, Jyoji Yamate and Mitsuru Kuwamura in Toxicologic Pathology
Supplemental Material
sj-tif-7-tpx-10.1177_01926233241235623 – Supplemental material for Difference in the Mechanism of Iron Overload–Enhanced Acute Hepatotoxicity Induced by Thioacetamide and Carbon Tetrachloride in Rats
Supplemental material, sj-tif-7-tpx-10.1177_01926233241235623 for Difference in the Mechanism of Iron Overload–Enhanced Acute Hepatotoxicity Induced by Thioacetamide and Carbon Tetrachloride in Rats by Yohei Inai, Takeshi Izawa, Tomomi Kamei, Sho Fujiwara, Miyuu Tanaka, Jyoji Yamate and Mitsuru Kuwamura in Toxicologic Pathology
Footnotes
Acknowledgements
We thank Tomoe Matsuo, Nana Hamachi, Youko Igakura, and Tomoko Sakamoto for their excellent technical support.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work is supported by JSPS KAKENHI (Grant Number 20K06415).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.