
Abstract
NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ (NSG) mice, lacking many components of a mature immune system, are at increased risk of disease. General understanding of potential pathogens of these mice is limited. We describe a high mortality disease outbreak caused by an opportunistic bacterial infection in NSG mice. Affected animals exhibited perianal fecal staining, dehydration, and wasting. Histopathologic lesions included a primary necrotizing enterocolitis, with inflammatory and necrotizing lesions also occurring in the liver, kidneys, heart, and brain of some mice. All affected individuals tested negative for known opportunistic pathogens of immunodeficient mice. We initially identified a member of Enterobacter cloacae complex (ECC) in association with the outbreak by traditional diagnostics. ECC was cultured from extraintestinal organs, both with and without histopathologic lesions, suggesting bacteremia. Infrared spectroscopy and MALDI-TOF mass spectrometry demonstrated that isolates from the outbreak shared molecular features and likely a common origin. We subsequently hypothesized that advanced sequencing methods would identify a single species of ECC associated with clinical disease. Using a novel targeted amplicon-based next-generation sequencing assay, we identified Enterobacter hormaechei in association with this outbreak. Knowledge of this organism as a potential opportunistic pathogen in NSG mice is critical for preclinical studies to prevent loss of animals and confounding of research.
Keywords
Introduction
Immunodeficient mice are an increasingly popular research model due to their utility in studying human immune responses and cancer, and their use has increased significantly in preclinical research for multiple applications.5,11,21,24,35,39,45,52 In preclinical studies in areas such as oncology and immunology, immunodeficient mice have been important to the understanding of mechanisms of disease, target identification and validation, and toxicologic effects of compounds. In addition, immunodeficient strains may recapitulate some of the immune dysfunction observed following immunosuppressive conditions in humans, such as chemotherapy, radiation therapy, surgery, infectious disease, or paraneoplastic syndromes,17,48 and therefore may serve as a better predictor of toxicokinetics and safety in these instances. 40
Possessing spontaneous or engineered genotypes that lead to underlying immune defects, immunodeficient mice pose a particular challenge for biosecurity in research facilities. These mice are susceptible to a wider range of agents that typically do not infect or cause clinical disease in immunocompetent mice 13 and therefore represent a significant source of confounding in preclinical studies that may lead to misinterpretation of clinical and pathology data.47,53 Newer immunodeficient mice, such as NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ (NSG) mice, have multiple mutations of the immune system which produce profound immune alterations and consequently need additional protections and surveillance to prevent contamination of the environment, exposure of animals to potential pathogens, and loss of animals and research data critical for preclinical studies in biomedical research.
NSG mice were first introduced in 2005 as a new class of immunodeficient mice to facilitate engraftment of human hematopoietic stem cells. 42 NSG mice are now often used for patient-derived xenografts (PDXs) and increasingly serve as platforms for the development of mice with additional immune system mutations to further facilitate PDX engraftment. 33 In preclinical research, NSG mice have been used to test vaccine safety in the context of immunodeficient patients and the toxicity of human cell-based therapies.24,45 Based on these research applications, NSG mice and other polygenic variants will likely continue to increase in popularity and value. However, immunodeficient mice created with multiple genetic mutations, including NSG mice, are very expensive to procure and often engrafted with tumors directly harvested from human patients and therefore irreplaceable. Owing to the value of these mice, there is a great need to protect them from disease and scientific perturbation.
The list of microbial organisms to which NSG and other polygenic immunodeficient mice are susceptible remains incomplete. NSG mice at Jackson Laboratories reportedly developed spontaneous illness with pathologic evidence of nephritis and bacteremia due to ascending infections. 12 Kidney culture revealed multiple individuals that were positive for opportunistic bacterial agents including Enterococcus and Klebsiella. Another outbreak of acute mortality in NSG mice was attributed to a human-related virulent strain of Clostridioides difficile and associated with antibiotic treatment, 28 while another study reported that aging female NSG-SGM3 mice developed pleuropneumonia and bronchopneumonia due to opportunistic infection with Rodentibacter pneumotropicus. 37 More recently, Klebsiella pneumoniae was implicated as causing typhlocolitis, septicemia, and systemic disease including pneumonia and meningoencephalitis in a breeding colony of NSG mice. 44
A critical component of laboratory animal medicine is the protection of animal health and integrity of research by ensuring animals are free of infectious diseases. 32 Laboratory animal facilities have relied on diagnostic and surveillance methods including serology and polymerase chain reaction (PCR) to exclude harmful pathogens.2,7 While these methods have largely shaped traditional biosecurity programs, these assays define animal microbial status based on current understanding of infectious diseases of rodents, but fail to detect unrecognized agents or microbes not previously of concern. Modern techniques such as metagenomics and next-generation sequencing (NGS) assays reveal a wide array of host-associated organisms that can influence the outcome of disease as well as potential microbial effects on the studied phenotype that are currently less understood.7,27,38 As the host microbiome can lead to biotransformation, modified absorption, and altered toxicity of compounds, incorporation of these tools in preclinical studies may be critical to the characterization of the microbial populations within study animals. 40
We experienced large-scale morbidity and mortality within a group of NSG mice that was unassociated with any known murine pathogens. An organism belonging to Enterobacter cloacae complex (ECC) was isolated by culture from animals with observed disease, and we hypothesized that this isolate was associated with the clinical disease. We utilized traditional diagnostic methods coupled with modern techniques to investigate the microbial landscape associated with infection. We hypothesized that clinical disease was associated with an overall expansion of a single species belonging to ECC which could be determined by a targeted amplicon-based NGS assay.
Materials and Methods
Husbandry
All activities involving animals were approved by the IACUC at University of Michigan, which is AAALACi accredited. NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ (NSG, Strain #:005557) mice were purchased from Jackson Laboratories (Bar Harbor, ME) and bred onsite in a breeding colony that received immunodeficient mice only from approved vendors. Colonies were maintained for 3 to 4 generations and refreshed with new vendor animals approximately every 2 years. Mice were strictly used for breeding and no experiments were performed in this barrier. Postweaning progeny were transferred to separate barrier facilities within the campus for researcher use. Mice were housed on individually ventilated cage (IVC) racks (Allentown, Allentown, NJ) in cages with corn cob bedding (Bed-o’Cobs, The Andersons, Maumee, OH). Animals were fed ad libitum rodent chow (5L0D, LabDiet, St Louis, MO) and water. Water from a municipal source was either triple filtered (experimental animals) or reverse osmosis-treated or deionized (breeding colony) before it was provided to animals. Cages, bedding, feed, and water were autoclaved before being given to NSG mice. Cages were changed biweekly and water bottles were changed weekly. Mice were exposed to a 12 hr:12 hr light:dark cycle and humidity and temperature levels within the recommended ranges of the Guide for the Care and Use of Laboratory Animals. 32 Using monthly and quarterly surveillance testing of dirty bedding sentinels and environmental samples, mice were consistently free of the following agents: lymphocytic choriomeningitis virus, mouse adenovirus, Mycoplasma pulmonis, Theiler’s murine encephalomyelitis virus (TMEV), pneumonia virus of mice (PVM), reovirus, Sendai virus, mouse hepatitis virus (MHV), minute virus of mice (MVM), mouse parvovirus, mouse rotavirus, ectromelia virus, mouse polyomavirus, pinworms (Aspiculuris tetraptera, Syphacia obvelata), fur mites (Mycoptes musculinus, Myobia musculi, Radfordia affinis), and Corynebacterium bovis. The breeding colony and the barrier containing experimental animals operated under the same pathogen exclusion status. Mice with xenografts were housed under ABSL-2 conditions.
Pathology
Diagnostic necropsies were performed on clinically affected and unaffected mice after humane euthanasia with carbon dioxide asphyxiation, and on mice that were recently found dead (Table 1). Of the clinically unaffected mice, necropsies were performed on cage mates of clinically affected mice (n = 2) and on mice from cages without signs of clinical illness (n = 6). Tissues (brain, heart, lungs, kidneys, adrenal glands, liver, spleen, stomach, small and large intestines, mesenteric lymph nodes, salivary glands, uterus, cervix, ovaries, oviducts, bulbourethral glands, epididymis, prostate, seminal vesicles, testes, hindlimb [femur, tibia, stifle joint, skeletal muscle, and peripheral nerves], sternum) were collected and were either frozen at −20°C for microbiologic testing or fixed in 10% neutral buffered formalin (NBF). Select fixed tissues were routinely trimmed and embedded in paraffin. Paraffin-embedded sections (5 μm) were mounted on glass slides and stained with hematoxylin and eosin for microscopic evaluation by a boarded veterinary pathologist (MH).
Summary of lesions and E. cloacae complex cultures in outbreak colony.
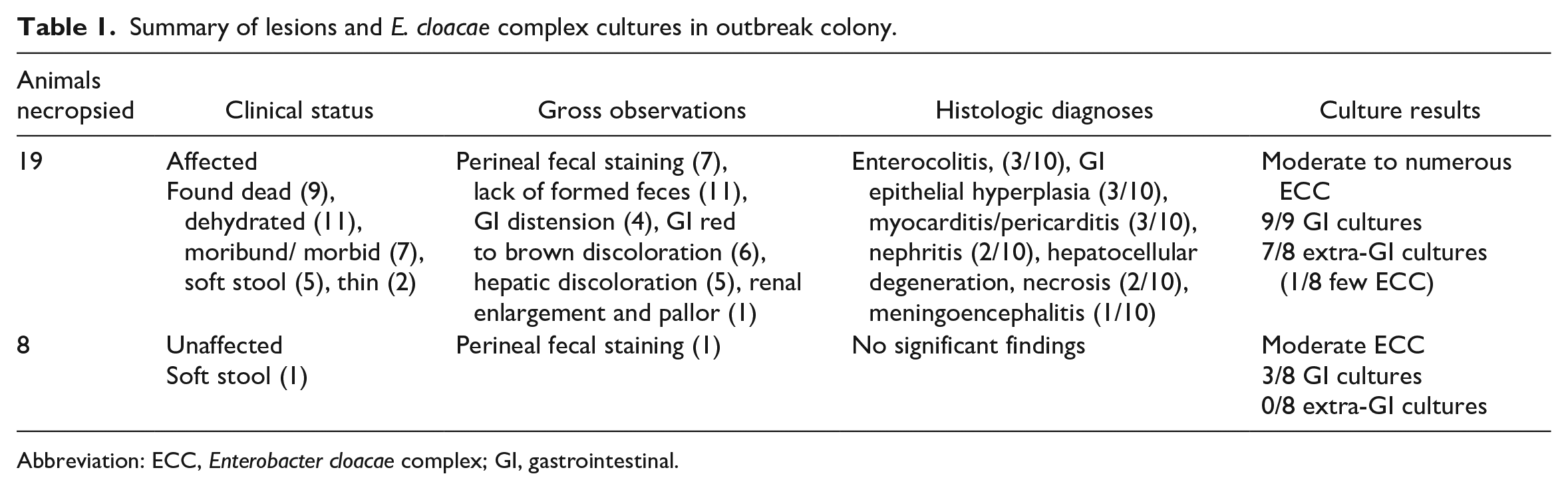
Abbreviation: ECC, Enterobacter cloacae complex; GI, gastrointestinal.
Molecular Diagnostics
Samples (feces, pelt and oral swabs, gastrointestinal contents, and lung tissue) were collected from a subset of clinically normal mice housed in the same room as the clinically affected mice (n = 45 cages) and from clinically affected NSG mice (n = 5) and submitted to Charles River Laboratories (CRL) (Wilmington, MA). Samples were tested by PCR for viruses including mouse parvovirus, murine norovirus, mouse coronavirus, murine rotavirus, mouse theilovirus, rodent chaphamaparvovirus, adenovirus types 1 and 2, reovirus types 1, 2, 3, and 4, pneumonia virus of mice, Sendai virus, ectromelia virus, lymphocytic choriomeningitis virus, bacterial agents including Helicobacter, Citrobacter rodentium, Mycoplasma pulmonis, Streptobacillus moniliformis, Rodentibacter heylii, Rodentibacter pneumotropicus, Clostridium piliforme, Pseudomonas aeruginosa, Salmonella, Campylobacter, Bordetella bronchiseptica, Bordetella pseudohinzii, Corynebacterium kutscheri, Corynebacterium bovis, Staphylococcus aureus, Streptococcus pneumoniae, Klebsiella pneumoniae, Klebsiella oxytoca, beta hemolytic Streptococcus groups A, B, C, G, Proteus mirabilis, and parasites including fur mites (Myobia, Mycoptes, Radfordia), pinworms (Aspiculuris, Syphacia), Giardia, Spironucleus muris, Cryptosporidium, Entamoeba, Pneumocystis, Demodex, Tritrichomonas. Additional clinically affected animals were tested for Clostridioides difficile, Pneumocystis, and C. bovis by PCR at CRL and for clostridial toxins by ELISA at Michigan State University Veterinary Diagnostic Laboratory (MSU VDL).
Bacteriology
Tissue samples (lung, kidney, liver, spleen, small and large intestine), cecal contents, and feces were submitted to MSU VDL for aerobic bacterial culture.
Infrared Spectroscopy
Bacterial isolates used for infrared (IR) spectroscopy analysis were grown on TSAB media (Remel, Lenexa, KS) at 37°C for 24 hours. Sample processing and spectra acquisition was performed as per manufacturer (Bruker Daltonics, Billerica, MA) recommended protocol. In brief, a loopful of bacteria was suspended in freshly prepared 70% ethanol. Molecular grade water was added to the suspension to make a total volume of 100 µL. The suspension was homogenized, and 15 µL was placed in triplicates on a silicon plate (Bruker Daltonics) and dried at room temperature for 30 minutes. Infrared spectra were captured and visualized using the Opus software v8.2 (Bruker Daltonics). The spectra in the area of polysaccharides (1300-800 cm−1) were vector normalized, and the second derivative was used to amplify differences between isolates.
Matrix-Assisted Laser Desorption/Ionization-Time of Flight (MALDI-TOF) Analysis
Bacterial protein spectra were captured using MALDI-TOF MS (Microflex LT, Bruker Daltonics) following manufacturer recommended protocol. A dendrogram was constructed using Compass Explorer v4.1 (Bruker Daltonics) with default parameters (Biotyper MS dendrogram creation v1.4, Bruker Daltonics).
rpoB PCR and Sequencing
Partial rpoB gene for the β subunit of bacterial RNA polymerase was amplified using the primers CM7 and CM31b. 30 The PCR amplicon was cleaned up with ExoSAP-IT Express (Thermo Fisher Scientific Inc., Waltham, MA) and the DNA was sequenced (Eurofins Genomics, Louisville, KY). Consensus sequences from the forward and reverse primer were assembled using Geneious Prime Software (“Geneious Prime 2020, version 2020.2” https://www.geneious.com) and the sequences analyzed for bacterial species identification using Nucleotide BLAST. 3
E. cloacae Complex and E. hormaechei PCR
Semiquantitative TaqMan PCR (qPCR) was performed on samples collected from clinically affected (n = 12) and unaffected animals (n = 13) to determine the presence and abundance of ECC. Enterobacter cloacae complex includes the following species: Enterobacter asburiae, E. cancerogenus, E. cloacae, E. hormaechei, E. ludwigii, E. kobei, E. nimipressuralis, and E. mori. They are considered genetically close, retaining roughly 60% DNA:DNA homology. 10 The broadly reactive ECC assay targeted the PTS sorbitol transporter subunit IIC srlA gene conserved across all species listed above and including the following additional Enterobacter species: E. bugandensis, E. chengduensis, E. sichuanensis, E. oligotrophica, and E. roggenkampii. All are newly described species of Enterobacter, and their relevance to immunodeficient animal disease is currently unknown. However, the assay was inclusive of these agents because of the increased susceptibility of immunodeficient animals to disease. Although the sorbitol transport machinery is found in Escherichia coli, Klebsiella, Raoutella, Citrobacter, and other bacteria, the assay was specific to a conserved region found in Enterobacteriaceae.
A second TaqMan PCR assay was developed to identify E. hormaechei species by targeting the pyrroloquinoline quinone biosynthesis protein gene pqqB.
Both TaqMan assays were qualified for use through a series of primer and probe optimization, specificity, sensitivity, and finally, screening with Sanger sequencing target confirmation steps.
Fecal samples processed for PCR analysis were extracted using an automated, magnetic-based total nucleic acid extraction system. After extraction, samples were plated for TaqMan PCR. Any positive samples were re-extracted to confirm PCR status by a second, independent run. System suitability controls were included in each PCR run to verify performance and provide confidence in PCR assay results. In addition to verifying that PCR reagents and equipment are functioning properly, these controls generate the reference fluorescence values used to interpret sample results. In addition, sample-suitability controls were included, notably, a Spike Control. A negative result for the spike, in which a known amount of exogenous template is added to the sample and tested by a specific assay, indicates the presence of PCR inhibitors, requiring dilution or re-extraction of the sample prior to retesting. A spike assay was performed for each sample that was screened.
Surveillance for ECC
To survey for the presence of ECC within our facility’s rodent colonies, fecal samples were collected from mice located in different animal housing rooms in different facilities across our campus, including the breeding colony. Samples were collected from the following strains C.B-Igh-1b/IcrTac-Prkdcscid or C.B-17 scid (n = 17), Nu/J (n = 32), B6.129S7-Rag1tm1Mom/J or Rag1 KO (n = 3), NOD.Cg-Prkdcscid/J or NOD scid (n = 11), NOD.Cg-Prkdcscid Il2rgtm1Sug/JicTac or NOG (n = 2), and NSG mice (n = 27). Fecal samples were collected and pooled from 3 animals in each cage, with the unit of analysis being the cage. Copy numbers of ECC in the fecal samples were quantified by qPCR as described previously.
The prevalence of ECC was also assessed by Charles River Laboratories. Fecal pellets, oral swabs, and pelt swabs were collected from immunocompetent and immunodeficient strains within the vendor’s facilities. Similar samples were also collected from external users. Copy numbers of ECC in the samples were quantified by qPCR as described previously.
Microbiome Analysis
Fecal samples were collected from clinically normal (n = 13) and affected mice (n = 5), and DNA was extracted using the DNeasy PowerLyzer PowerSoil kit (Qiagen, Hilden, Germany) as optimized by the NGS laboratory. The V3-V4 region of the 16S ribosomal RNA gene was amplified for characterization of the bacterial microbiome. 18 Briefly, after V3-V4 amplicon generation, samples were cleaned using a magnetic bead-based clean up strategy (AMPure, Beckman Coulter, Brea, CA) followed by indexing and amplification (Illumina, San Diego, CA), a second round of clean-up, and finally, bioanalyzer analysis (Agilent Technologies, Santa Clara, CA) and SYBR Green-based qPCR quantification (Kapa Biosystems, Wilmington, MA). Libraries were equimolar pooled and loaded onto an Illumina MiSeq v3 2x300 flow cell.
Next-Generation Sequencing (NGS) Assay
Owing to the extensive number of species represented within ECC and their overall genetic similarities, it was deemed impractical to run individual Sanger sequencing reactions and thus, a targeted amplicon-based NGS assay was designed to detect and differentiate complex members. Representative genomes from each species were aligned using LASTZ in Geneious (“Geneious Prime 2022, current version 2023.1” https://www.geneious.com). The alignment was manually evaluated for alternating regions of high homology and high diversity. The tsx gene was identified to fit such criteria. Further BLAST analysis of both the primers and the entirety of the region revealed that the regions upstream and through tsx would make a good fit for an assay.3,54 Primers were designed to include the Illumina adapter region (Supplemental Table S1).
A synthesized positive template control encompassing the targeted gene region was used as a control sample. Amplicon generation, library preparation and sequencing were performed as described for the 16S V3-V4 sequencing.
Samples were analyzed using the NGS amplicon analysis workflow in Geneious Prime 2023.0.1 (https://www.geneious.com). Briefly, paired-end reads were paired, trimmed for adapters and quality using BBDuk v38.84, and merged using BBMerge v38.84. The trimmed and merged reads were then size filtered to remove any short reads. The remaining reads were then BLAST against the ECC database and the NCBI nucleotide database (accessed May-June 2022). 31 The results were used to create a reference library for classification. The reads were then classified against the reference library to determine identity using the Geneious Sequence Classifier, with a minimum percentage identity of 90% and query identity of 90%.
Statistical Methods
Statistical and visual analyses were performed using R version 4.2.2 (Comprehensive R Archive Network [CRAN], https://cran.r-project.org) in RStudio version 2022.12.0. Statistical significance for analyses was defined as a P-value less than 0.05. Two-sided t or two-sample Welch t-tests were used for two-way analysis. Spearman correlation was used for two-way comparisons. For 16S sequencing, Benjamini-Hochberg correction was used to adjust P-values for multiple comparison testing between taxons.
16S sequences were processed using mothur v1.47.1 and as previously described. 25 Briefly, contigs were assembled from paired end reads and were subsequently screened for size, ambiguous base calls, homopolymers (>8 bp), and chimeras. Contigs were aligned to the Silva 16S rRNA reference database. Contigs of up to 3% dissimilarity were grouped using a de novo nearest neighbor method into operational taxonomic units and taxonomic assignments were made based on the Ribosomal Database Project (RDP) database. 16S data were analyzed using the R packages phyloseq v1.42.0 and vegan v2.6-4.
Spectroscopy data were analyzed using the R package ChemoSpec v6.1.4.
Results
An outbreak occurred over a 6-month period and 74 mice died in a group of NSG mice with an average monthly census of 35 cages housed in a single room operating under ABSL-2 conditions (Figure 1). This group of mice belonged to a research investigator using human-derived xenograft tumor models for developing novel cancer treatment approaches. Tumors used for xenotransplantation were passaged in rodents. Overall incidence of disease appeared consistent across the group of experimental animals and was independent of sex or xenotransplantation of the mice (P = .3994, .5992). Clinical signs were characterized by an acute onset of morbidity and mortality within a 24-hour period, and most affected animals were found dead despite appearing normal on preceding routine health checks. Deceased animals had evidence of nonspecific disease including loose stool, dehydration, and wasting on postmortem examination.
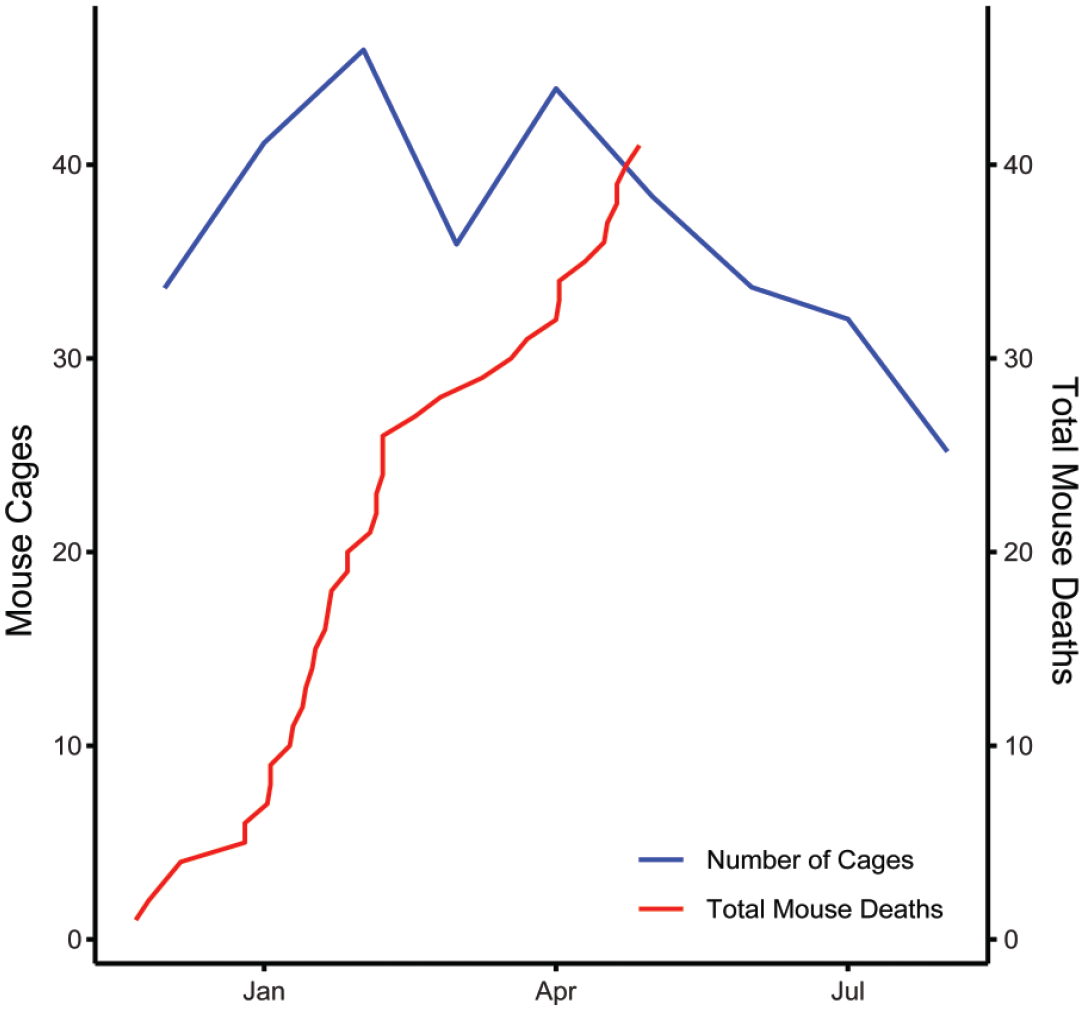
A progressive outbreak of acute fatal disease occurred over several months in a group of NSG mice housed in a single room. Room census as defined by the number of cages is also shown. NSG indicates NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ.
Diagnostic necropsies were performed on clinically affected (n = 19) and unaffected animals (n = 8). Table 1 summarizes gross pathology including perineal fecal staining, gastrointestinal (GI) distension and discoloration, and hepatic pallor and discoloration. Microscopic lesions in clinically affected animals included necrotizing enterocolitis, characterized by loss of villus architecture in the small intestine accompanied by mixed inflammatory infiltrates within the lamina propria and glandular degeneration in the colon (Figure 2A, B). Gram staining of gastrointestinal tissues demonstrated numerous Gram-negative bacterial rods colonizing the mucosal surface (Figure 2C). In other tissues including the liver, kidney, and brain, there were variable inflammatory cell lesions, including an increased frequency of random, mixed cell inflammatory infiltrates and associated single hepatocyte necrosis in the liver (Figure 2D), minimal to mild neutrophilic infiltrates in the renal medullary tubules (Figure 2E), and suppurative meningitis in the brain (Figure 2F), consistent with a systemic inflammatory response and/or sepsis in affected animals. Histologic lesions were not observed in clinically unaffected animals.
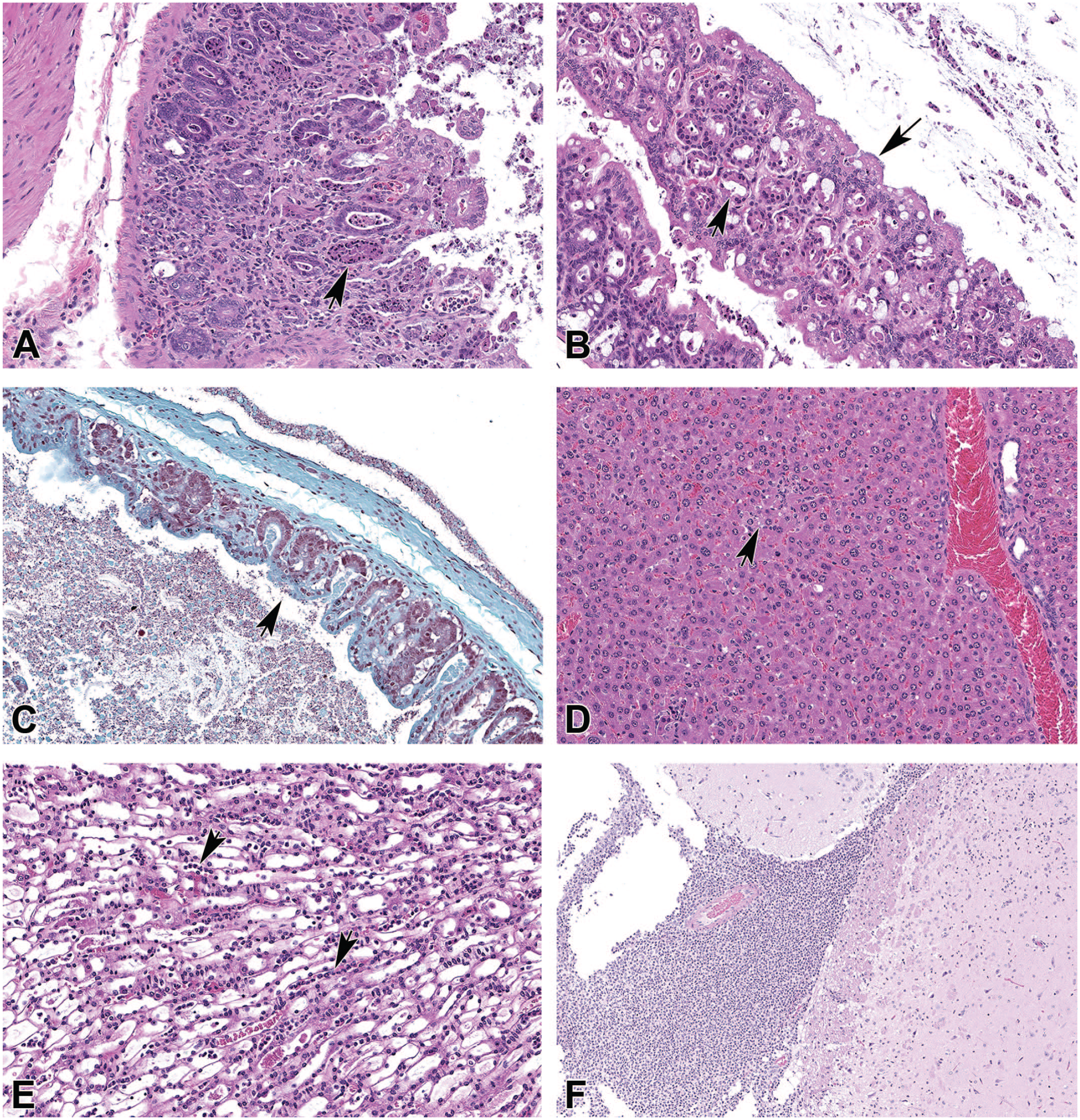
Histopathologic features of clinically affected mice involved in the outbreak. Hematoxylin and eosin (A) Necrotizing enteritis characterized by loss of villus architecture, presence of necrotic cellular debris within crypts (arrowhead), and a mixed inflammatory infiltrate within the lamina propria. (B) Necrotizing colitis characterized by degeneration of glandular epithelium (arrowhead), scalloping of the mucosal surface with adherent bacteria (arrow) and presence of necrotic cellular debris. (C) Gram stain, colon: numerous Gram-negative bacilli are present colonizing the mucosal surface (arrowhead). (D) Liver. Diffuse Kupffer cell hyperplasia and multifocal foci of single to multiple degenerate hepatocytes with presence of small neutrophilic infiltrates (arrowhead). (E) Kidney. Numerous neutrophils present within medullary tubules and capillaries (arrowheads). (F) Brain. Marked suppurative meningoencephalitis characterized by the presence of marked neutrophilic inflammatory infiltrates diffusely expanding the meninges and multifocally infiltrating the underlying neuropil.
Diagnostic testing of clinically affected and normal animals within the outbreak room was negative for known pathogens of immunodeficient mice including rodent chaphamaparvovirus (murine chapparvovirus), Pneumocystis carinii, Rodentibacter heylii, R. pneumotropicus, Clostridioides difficile, and clostridial toxins. Broad spectrum culture was performed on feces, cecal contents, small intestine, large intestine, kidney, liver, and/or spleen from normal and clinically affected animals. Enterobacter cloacae complex was isolated by culture from feces of both populations, although at higher frequency in clinically affected animals (Figure 3A). Interestingly, only animals with clinical disease had positive organ cultures from nongastrointestinal sites. Positive cultures often correlated with the presence of inflammatory lesions in organs suggesting septicemia, or in some cases in histologically normal organs, which was suggestive of bacteremia. Antibiotic resistance profiles were nearly identical across all culture isolates (Figure 3B). Isolates were generally resistant to β-lactam antibiotics including amoxicillin and clavulanic acid, ampicillin, cefazolin, and cephalexin with one isolate exhibiting intermediate resistance to cefpodoxime. Isolates were generally susceptible to cefovecin, tetracyclines, fluoroquinolones, and trimethoprim sulfamethoxazole.
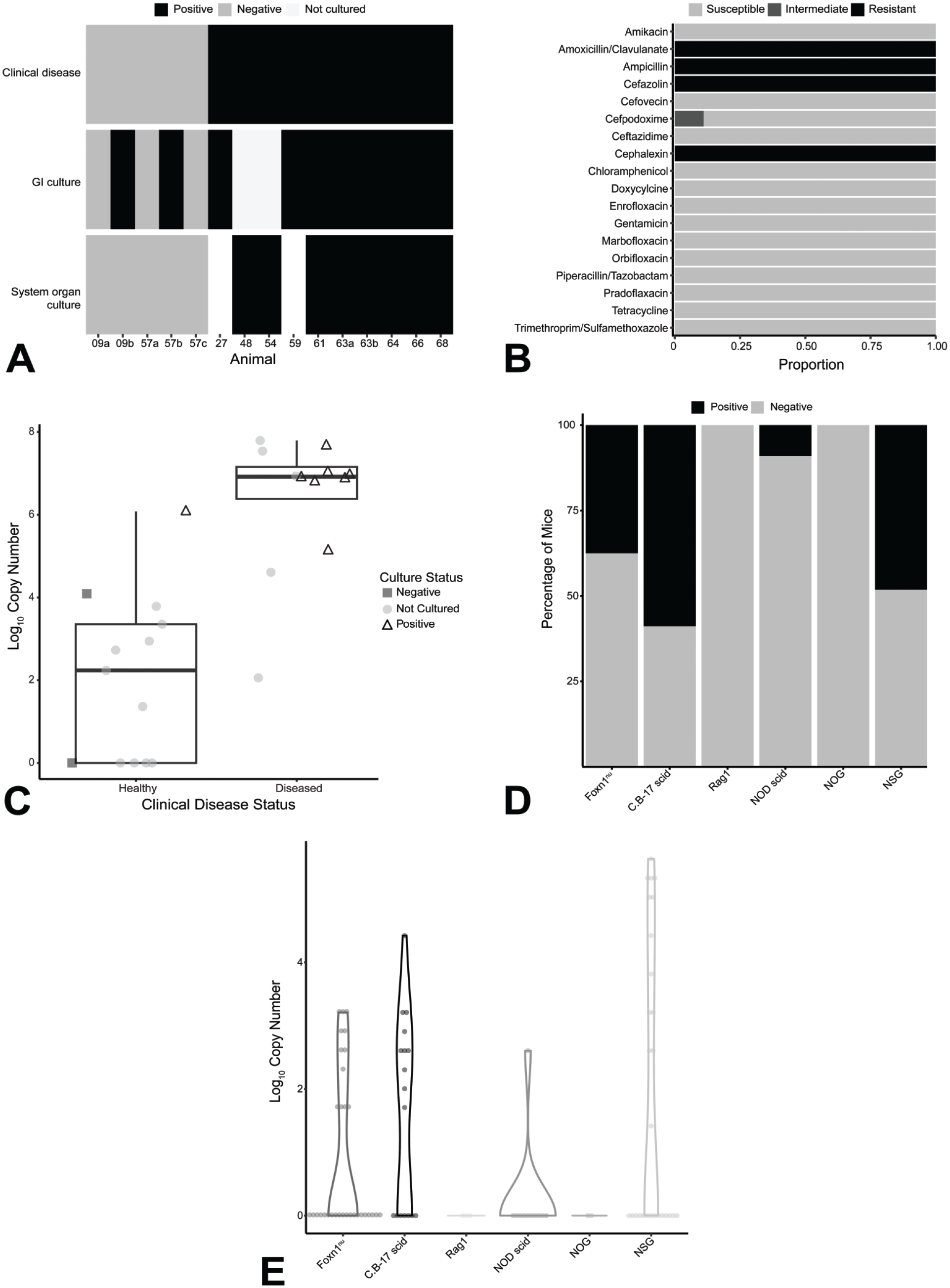
Bacterial culture and molecular quantification of ECC. (A) The heat map shows that diseased animals were more likely to have positive cultures from gastrointestinal and extra-gastrointestinal organs. (B) Antibiotic sensitivity demonstrated by ECC cultures isolated from the outbreak. (C) By PCR, diseased and culture positive animals hosted significantly greater copy numbers of ECC; P = .0005. (D) Prevalence of ECC in different immunocompromised strains within the facility. (E) NSG mice generally hosted greater copy numbers of ECC than other strains of mice. ECC indicates Enterobacter cloacae complex; NSG, NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ; PCR, polymerase chain reaction.
To determine the prevalence of ECC in the affected population, we tested feces and gastrointestinal organ samples by PCR. Mice with clinical disease had significantly higher ECC copy numbers compared to unaffected mice from the same room (Figure 3C, P = .0005). Fecal samples were collected from several immunodeficient strains of healthy mice within different facilities on our campus, including the breeding colony, to evaluate the overall prevalence of ECC within our facilities. ECC complex was detected by PCR in all immunodeficient strains we examined. C.B-17 scid mice had the highest proportion of animals carrying ECC (Figure 3D), whereas NSG mice carried the highest copy numbers of ECC (Figure 3E). ECC copy numbers of healthy NSG mice included in this survey were on average lower than those of clinically affected NSG mice in the outbreak. ECC was detected by PCR in C.B-17 scid and NSG mice in our breeding colony, with ECC being more prevalent in C.B-17 scid mice (Supplemental Figure S1). Finally, samples collected from healthy mice at Charles River Laboratories and samples submitted from external users to Charles River Laboratories were tested for ECC. Overall, ECC was prevalent in immunodeficient mice with similar prevalence observed between vendor animals and samples submitted from external users (Supplemental Table S2). Median ECC copy numbers were generally on the same order as those of clinically unaffected animals that tested positive for ECC within this outbreak (Supplemental Table S3).
We used additional molecular techniques to further characterize the relationship between the isolates obtained from clinically affected individuals. Infrared spectroscopy, which characterizes organisms based on their component carbohydrates, lipids, and proteins, was performed on culture isolates obtained from clinical (n = 13) and subclinical mice (n = 3), as well as several ECC control isolates originating from other sources. There was a high degree of similarity among the clinical and subclinical isolates, with these isolates clustering together relative to the control isolates of varying species (Figure 4A, B). Matrix-assisted laser desorption/ionization-time of flight (MALDI-TOF) mass spectrometry likewise demonstrated a high degree of similarity between clinical isolates compared to the control isolates (Supplemental Figure S2). Polymerase chain reaction amplification and Sanger sequencing of the rpoB gene locus of select isolates showed the clinical isolates to be more closely related to each other than to the control isolates (Figure 4C). The close relationship between the clinical isolates, as confirmed by these three assays, suggested that a single etiologic agent was responsible for the outbreak. Subsequent alignment of the rpoB sequences identified the clinical isolates as Enterobacter hormaechei, a member of ECC.
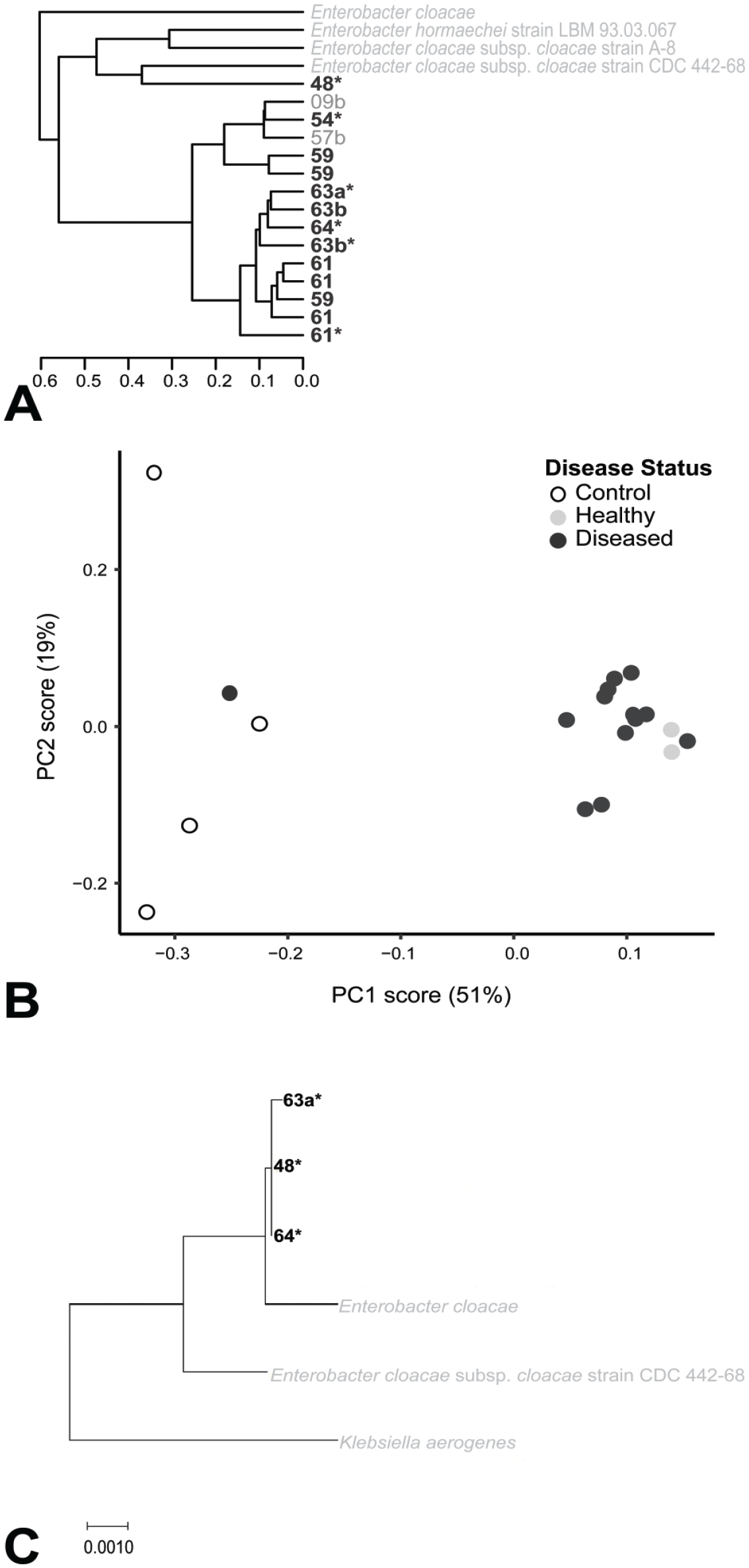
Microbiological analysis of cultured isolates from the outbreak. Isolates from the outbreak are identified by number. Control isolates are identified by name. Isolates from healthy animals are written in lighter gray. Isolates from diseased animals are written in dark gray and in bold. Isolates were derived from gastrointestinal tissue or nongastrointestinal organ tissue (asterisk). (A) Clustering of isolates based on IR spectroscopy. Isolates from the outbreak generally clustered together and separate from the control isolates. (B) Principal component analysis demonstrated similar clustering of isolates from the outbreak compared to control isolates. (C) rpoB sequencing demonstrated similar clustering of isolates from the outbreak compared to control isolates.
We sought to identify whether there were differences in microbial populations of clinically normal and abnormal animals that could be associated with clinical outcome. 16S rRNA sequencing was performed on fecal samples collected from clinically normal and affected animals in the outbreak room and from clinically normal animals in the breeding colony. The relative abundances of the top 10 bacterial genera or other taxa and of the bacterial genera or other taxa with greater than 1% mean relative abundance are shown (Figure 5A, B). Bacteria of the Enterobacter genus and an unclassified Enterobacteriaceae were significantly more abundant in the microbiomes of clinically affected animals than in those of the clinically normal animals (P = .0057, .0055). True abundance of Enterobacter spp. was also higher in clinically affected animals than in clinically normal animals (Supplemental Figure S3). Muribaculaceae, Ruminococcaceae, Duncaniella, Bacteroides, Paramuribaculum, Lachnospiraceae, and Lactobacillus were significantly more abundant in the microbiomes of clinically normal animals than in those of the clinically affected animals (Figure 5B). Interestingly, clinically normal vendor animals housed in the outbreak room (Ja-Jd) hosted proportionately greater abundances in bacterial family Muribaculaceae and genus Alistipes and lacked genus Duncaniella compared to clinically normal experimental animals also housed in the outbreak room (57a-57c, 7408-7545) (Figure 5A). Clinically affected animals likewise hosted bacterial populations that clustered together and hence were more similar to each other than to bacterial populations hosted by clinically normal animals (Figure 5C).
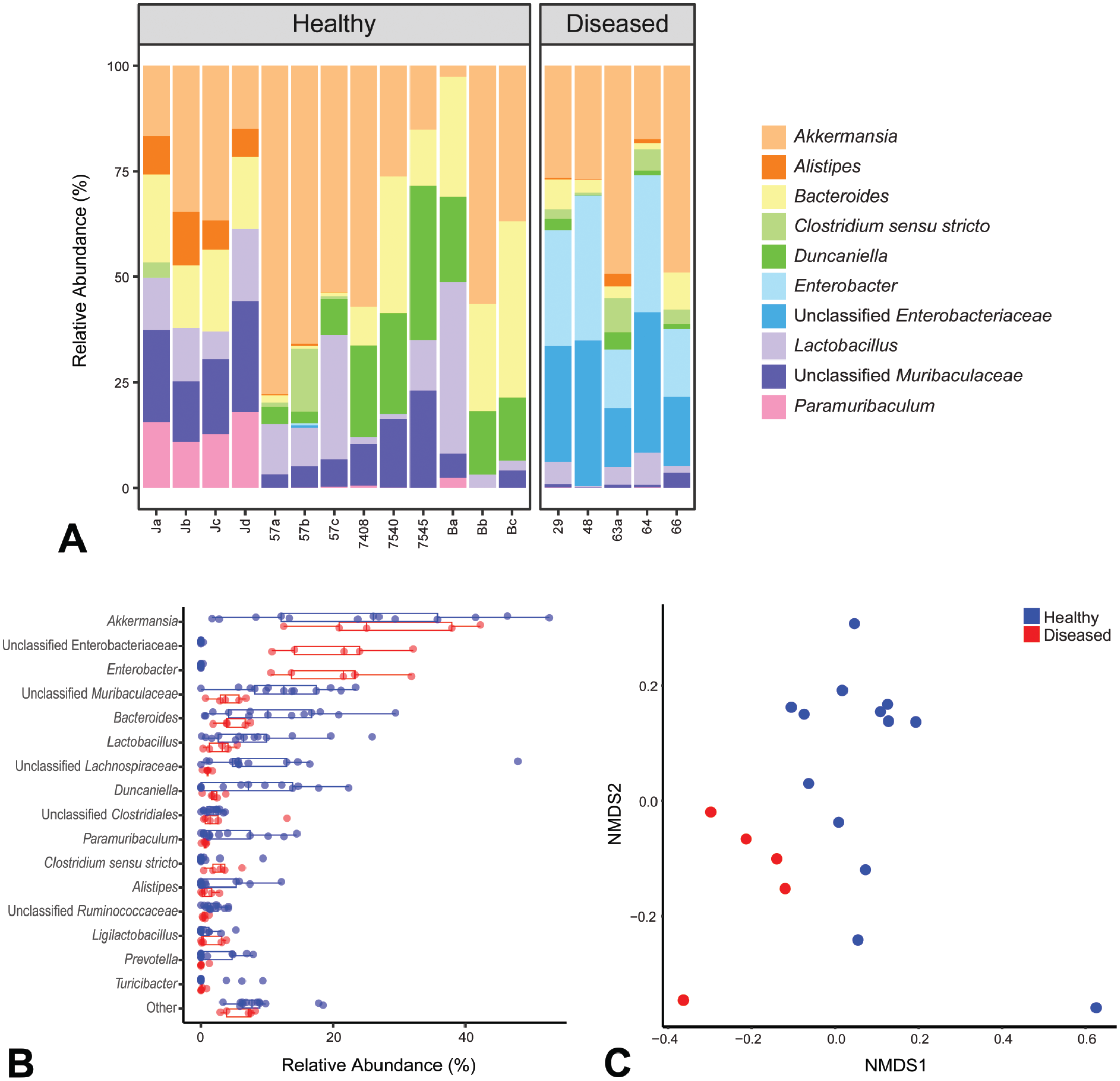
16S sequencing (A) The top 10 bacterial genera or other taxa are shown. Diseased animals had greater relative abundance of Enterobacter spp. and unclassified Enterobacteriaceae spp. than healthy animals. Healthy animals from the vendor (Ja-Jd) had qualitatively different microbiome composition than healthy animals derived from the breeding colony. (B) Bacterial genera or other taxa are compared in healthy and diseased animals. Genera or other taxa of less than 1% mean relative abundance were pooled as “Other.” Diseased animals had significantly greater relative abundance of Enterobacter and unclassified Enterobacteriaceae spp. than healthy animals. (C) Diseased animals had more similar microbiomes that were distinct from those of healthy animals.
As ECC consists of a complex of 22 species in the Enterobacter genus, 51 we next developed a targeted amplicon-based NGS assay to differentiate and quantify the Enterobacter spp. present in a sample. Fecal samples were collected from clinically affected and normal animals from the outbreak room as well as clinically normal animals sourced from a vendor and housed within the same room. Enterobacter hormaechei reads were overrepresented in clinically affected animals but not in clinically normal animals (Figure 6A). Unbiased clustering on overall read prevalence for the various ECC species differentiated clinically affected animals from clinically unaffected animals. Interestingly, clinically normal animals hosted elevated numbers of unclassified species; these unidentified populations were significantly reduced in clinically affected animals. Polymerase chain reaction amplification of E. hormaechei in the same samples showed clinically affected animals having significantly higher copy numbers compared to those of most clinically normal animals (Figure 6B, P = .0453). Furthermore, copy numbers of E. hormaechei derived from NGS were highly correlated with copy numbers derived from the E. hormaechei PCR assay (Figure 6C, P = 5.293e−05, rho = .8064).

NGS sequencing (A) Number of sequences of Enterobacter species in healthy and diseased animals represented by a heat map. Diseased animals had greater abundance of E. hormaechei than healthy animals. Healthy animals had proportionately more sequences for unclassified organisms. (B) Diseased and culture positive animals likewise had increased PCR copy numbers of E. hormaechei compared to healthy and culture-negative animals; P = .0453. (C) Numbers of E. hormaechei sequences determined by NGS and PCR were correlated; P = 5.293e−05, rho = .8064. EH indicates E. hormaechei; NGS, next-generation sequencing; PCR, polymerase chain reaction.
Discussion
Our facility experienced a high mortality disease outbreak in which an opportunistic bacterial infection led to significant morbidity and mortality due to bacteremia and likely septicemia, in a group of NSG mice. Using a novel targeted NGS assay, we identified Enterobacter hormaechei in association with this outbreak. E. hormaechei, a Gram-negative, rod-shaped bacteria, has not been documented previously as a causative agent of disease in NSG or immunodeficient mice. Most strains of E. hormaechei have been isolated from samples derived from humans. 10 Enterobacter hormaechei has been reported to cause septicemia in humans, predominantly in immunocompromised and neonatal patients,26,56 and has been associated with extraintestinal disease in several domestic species.15,41,46 In addition, E. hormaechei subsp. steigerwaltii was implicated in the exacerbation of graft-versus-host disease in irradiated mice receiving hematopoietic stem cell transplantation due to disruption in the gut microbiome. 6 In humans, E. hormaechei is an emerging cause of nosocomial infections in the hospital setting with many isolates having multidrug resistance phenotypes.10,55 In contrast, isolates from this outbreak, resistant to penicillins and a subset of cephalosporins, demonstrated a resistance profile intrinsic to ECC and lacked a multidrug resistance phenotype.4,29 Lack of multidrug resistance may suggest a nonnosocomial or nonxenograft origin. Whole genome sequencing and analysis would confirm types of antibiotic resistance genes and bacterial strain.4,55 Overall, recognition of E. hormaechei as a potential opportunistic pathogen affecting immunodeficient mice will help improve biosecurity and screening measures and reduce outbreaks of disease, as well as prevent the loss or misinterpretation of preclinical in vivo data when using immunodeficient mouse models.
Identifying unknown pathogens in NSG mice remains a significant challenge. Herein, we demonstrate an approach to identifying novel agents of clinical significance, in this case E. hormaechei, using traditional and more modern diagnostic tools. Historically, traditional microbiological assays including MALDI-TOF and even more advanced methods such as 16S rRNA sequencing had difficulty distinguishing member species within ECC.16,34 We used these methods to establish our ECC isolates to be highly similar, suggesting these isolates had a common origin. These methods also found an association between clinical disease and a general expansion of ECC. We hypothesized a targeted amplicon-based NGS assay would identify a single species of ECC associated with the outbreak. The NGS assay showed, independent of culture-based diagnostics, clinically affected animals had increased reads for the single species E. hormaechei, supporting our hypothesis, while unaffected animals had increased reads for unclassified organisms. The NGS assay therefore allowed us to identify a novel organism with respect to disease in NSG mice.
Application of next-generation sequencing assays for the diagnosis of infectious disease is growing. NGS technology allows for sequencing and analysis of many samples in parallel.20,36 NGS technology provides a unique advantage over cultural isolation since detection methods do not rely on oxygen, thermal, or biochemical requirements, which are often unknown for fastidious organisms.8,9 Targeted NGS uses a defined set of primers or probes to amplify and examine specific parameters of interest. 49 Assays can be multiplexed to screen for many infectious agents in one run.1,23 In addition to its value for detection and identification of pathogenic organisms, NGS can be used to examine genes for antibiotic resistance and to type organisms for understanding transmission and epidemiology of an outbreak.8,20 Here we employed targeted NGS to speciate the member of ECC we initially isolated by culture. Targeted NGS has been used to diagnose other human and veterinary diseases,23,14,19,22,50 and serves to increase sensitivity and specificity of diagnostics and reduce cost and time when applying NGS technology.
In this study, we found a significant association between an abundance of E. hormaechei and disease in animals. Although Koch’s postulates were not approached in this investigation, the use of NGS provided additional support that other bacteria previously reported to contribute to disease were not detected and could not have contributed to the disease state. Furthermore, the outbreak was successfully contained with administration of fluoroquinolones (enrofloxacin) based on the identification and antibiotic sensitivity testing of ECC. Surveillance within our barrier facilities identified ECC in healthy immunodeficient mice, including those within our breeding colony. Healthy mice hosted lower copy numbers than did clinically affected animals from the outbreak. Therefore, we suspect E. hormaechei to be an opportunist of severely immunodeficient animals. Our data could not establish whether a different organism incited or facilitated disease prior to expansion of E. hormaechei. We were unable to determine the underlying factors that predisposed this group of animals to clinical disease associated with E. hormaechei. Future studies to fulfill Koch’s postulates would be beneficial to characterize the pathogenic potential and infective dose of E. hormaechei, the natural history of disease and lesion development, as well as disease susceptibility in relation to specific host immune deficiencies. More research is also needed into methods of exclusion of ECC and E. hormaechei and prevention of disease by these agents within vulnerable populations.
We identified and characterized an unprecedented outbreak of E. hormaechei associated with high mortality in NSG mice. Modern genetic techniques used in the characterization of this outbreak will continue to progress our knowledge of the causes and epidemiology of disease outbreaks, particularly in animal models that have increased susceptibility to nontraditional pathogens. This knowledge serves to inform surveillance practices for immunodeficient rodent colonies, diagnostic and treatment approaches when faced with a similarly presenting outbreak, and interpretation of research results.
Supplemental Material
sj-docx-1-tpx-10.1177_01926233241231286 – Supplemental material for Next-Generation Sequencing-Based Identification of Enterobacter hormaechei as Causative Agent of High Mortality Disease in NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ (NSG) Mice
Supplemental material, sj-docx-1-tpx-10.1177_01926233241231286 for Next-Generation Sequencing-Based Identification of Enterobacter hormaechei as Causative Agent of High Mortality Disease in NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ (NSG) Mice by Catherine Si, Kourtney Nickerson, Taylor Simmons, Parker Denton, M. Russell Nichols, Robert C. Dysko, Mark Hoenerhoff, Rinosh Mani, Cheryl Woods, Kenneth S. Henderson and Zachary T. Freeman in Toxicologic Pathology
Supplemental Material
sj-docx-2-tpx-10.1177_01926233241231286 – Supplemental material for Next-Generation Sequencing-Based Identification of Enterobacter hormaechei as Causative Agent of High Mortality Disease in NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ (NSG) Mice
Supplemental material, sj-docx-2-tpx-10.1177_01926233241231286 for Next-Generation Sequencing-Based Identification of Enterobacter hormaechei as Causative Agent of High Mortality Disease in NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ (NSG) Mice by Catherine Si, Kourtney Nickerson, Taylor Simmons, Parker Denton, M. Russell Nichols, Robert C. Dysko, Mark Hoenerhoff, Rinosh Mani, Cheryl Woods, Kenneth S. Henderson and Zachary T. Freeman in Toxicologic Pathology
Supplemental Material
sj-docx-3-tpx-10.1177_01926233241231286 – Supplemental material for Next-Generation Sequencing-Based Identification of Enterobacter hormaechei as Causative Agent of High Mortality Disease in NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ (NSG) Mice
Supplemental material, sj-docx-3-tpx-10.1177_01926233241231286 for Next-Generation Sequencing-Based Identification of Enterobacter hormaechei as Causative Agent of High Mortality Disease in NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ (NSG) Mice by Catherine Si, Kourtney Nickerson, Taylor Simmons, Parker Denton, M. Russell Nichols, Robert C. Dysko, Mark Hoenerhoff, Rinosh Mani, Cheryl Woods, Kenneth S. Henderson and Zachary T. Freeman in Toxicologic Pathology
Supplemental Material
sj-docx-4-tpx-10.1177_01926233241231286 – Supplemental material for Next-Generation Sequencing-Based Identification of Enterobacter hormaechei as Causative Agent of High Mortality Disease in NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ (NSG) Mice
Supplemental material, sj-docx-4-tpx-10.1177_01926233241231286 for Next-Generation Sequencing-Based Identification of Enterobacter hormaechei as Causative Agent of High Mortality Disease in NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ (NSG) Mice by Catherine Si, Kourtney Nickerson, Taylor Simmons, Parker Denton, M. Russell Nichols, Robert C. Dysko, Mark Hoenerhoff, Rinosh Mani, Cheryl Woods, Kenneth S. Henderson and Zachary T. Freeman in Toxicologic Pathology
Supplemental Material
sj-tif-5-tpx-10.1177_01926233241231286 – Supplemental material for Next-Generation Sequencing-Based Identification of Enterobacter hormaechei as Causative Agent of High Mortality Disease in NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ (NSG) Mice
Supplemental material, sj-tif-5-tpx-10.1177_01926233241231286 for Next-Generation Sequencing-Based Identification of Enterobacter hormaechei as Causative Agent of High Mortality Disease in NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ (NSG) Mice by Catherine Si, Kourtney Nickerson, Taylor Simmons, Parker Denton, M. Russell Nichols, Robert C. Dysko, Mark Hoenerhoff, Rinosh Mani, Cheryl Woods, Kenneth S. Henderson and Zachary T. Freeman in Toxicologic Pathology
Supplemental Material
sj-tif-6-tpx-10.1177_01926233241231286 – Supplemental material for Next-Generation Sequencing-Based Identification of Enterobacter hormaechei as Causative Agent of High Mortality Disease in NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ (NSG) Mice
Supplemental material, sj-tif-6-tpx-10.1177_01926233241231286 for Next-Generation Sequencing-Based Identification of Enterobacter hormaechei as Causative Agent of High Mortality Disease in NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ (NSG) Mice by Catherine Si, Kourtney Nickerson, Taylor Simmons, Parker Denton, M. Russell Nichols, Robert C. Dysko, Mark Hoenerhoff, Rinosh Mani, Cheryl Woods, Kenneth S. Henderson and Zachary T. Freeman in Toxicologic Pathology
Supplemental Material
sj-tif-7-tpx-10.1177_01926233241231286 – Supplemental material for Next-Generation Sequencing-Based Identification of Enterobacter hormaechei as Causative Agent of High Mortality Disease in NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ (NSG) Mice
Supplemental material, sj-tif-7-tpx-10.1177_01926233241231286 for Next-Generation Sequencing-Based Identification of Enterobacter hormaechei as Causative Agent of High Mortality Disease in NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ (NSG) Mice by Catherine Si, Kourtney Nickerson, Taylor Simmons, Parker Denton, M. Russell Nichols, Robert C. Dysko, Mark Hoenerhoff, Rinosh Mani, Cheryl Woods, Kenneth S. Henderson and Zachary T. Freeman in Toxicologic Pathology
Footnotes
Acknowledgements
The authors would like to acknowledge Charles River Laboratories, Michigan State University’s Veterinary Diagnostic Laboratory, the ULAM Pathology Core for their diagnostic support as well as Drs. Ingrid Bergin and Kate Eaton for their assistance with interpretation of histopathology and diagnostic advice. The authors would like to thank the Merajver lab for their collaboration in handling this outbreak.
Authors’ Contributions
CS, KN, TS, MRN, CW, KH, RM, and ZTF planned and conducted data collection; RCD contributed to clinical and diagnostic planning; MH performed histopathological interpretation; CS, PD, and ZTF performed graphical and statistical analysis; the manuscript was written by CS and ZTF with all authors reviewing prior to submission.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: KN, CW, and KSH are employees of Charles River Laboratories.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the ULAM Cohen Comparative Medicine Research Award and Charles River Laboratories.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.