
Abstract
Biotherapeutic modalities such as cell therapies, gene therapies, nucleic acids, and proteins are increasingly investigated as disease-modifying treatments for severe and life-threatening neurodegenerative disorders. Such diverse bio-derived test articles are fraught with unique and often unpredictable biological consequences, while guidance regarding nonclinical experimental design, neuropathology evaluation, and interpretation is often limited. This paper summarizes key messages offered during a half-day continuing education course on toxicologic neuropathology of neuro-targeted biotherapeutics. Topics included fundamental neurobiology concepts, pharmacology, frequent toxicological findings, and their interpretation including adversity decisions. Covered biotherapeutic classes included cell therapies, gene editing and gene therapy vectors, nucleic acids, and proteins. If agents are administered directly into the central nervous system, initial screening using hematoxylin and eosin (H&E)-stained sections of currently recommended neural organs (brain [7 levels], spinal cord [3 levels], and sciatic nerve) may need to expand to include other components (e.g., more brain levels, ganglia, and/or additional nerves) and/or special neurohistological procedures to characterize possible neural effects (e.g., cell type-specific markers for reactive glial cells). Scientists who evaluate the safety of novel biologics will find this paper to be a practical reference for preclinical safety testing and risk assessment.
Keywords
Introduction
Hundreds of novel therapeutics are currently under development globally to treat severe neural diseases, most of which are products of biological origin (i.e., “bio-derived”) designed to target a specific molecular target or signaling pathway involved in the disease pathogenesis. The principal bio-derived modalities of current interest are cell therapies (e.g., chimeric antigen receptor-expressing [CAR] T-cells, stem cells secreting growth factors, or neurotransmitters); gene therapies (e.g., adeno-associated virus [AAV] and retroviral vectors and clustered regularly interspaced short palindromic repeats [CRISPR] gene editing); nucleic acids (e.g., antisense oligonucleotides [ASOs], messenger RNAs [mRNAs], micro RNAs [miRNAs]; and small interfering RNAs [siRNAs]); and proteins (e.g., antibody-based fusion proteins, antibody fragments, and carrier proteins to facilitate blood-brain barrier [BBB] penetration). The unique attributes of novel biotherapeutics generally dictate a case-by-case approach to designing nonclinical safety assessment programs. In this regard, key aspects to consider include the differing impacts of these modalities on cell function, effects related to comparatively high test article doses (e.g., substantiated for some gene therapy vectors due to variable numbers of empty or partial viral particles 40 ), routes of administration that introduce test articles directly into the central nervous system (CNS), the need to determine biodistribution (“drug distribution”) rather than pharmacokinetics, and the direct and indirect actions of biomolecules on gene expression (“pharmacologic properties”). The variable toxicologic profiles require adjustments to nonclinical safety studies to ensure that unexpected neural effects (whether “on target” or “off target”) are detected.
The challenges related to evaluating such questions led to the organization of a half-day continuing education (CE) course discussing the “Toxicologic Neuropathology of Novel Biotherapeutics” by the Special Interest Group for Neuropathology (SIGN) of the Society of Toxicologic Pathology (STP). The course was presented at the 42nd annual STP symposium meeting for a mixed audience of toxicologic pathologists, toxicologists, and scientists engaged in the preclinical evaluation of test articles destined to treat human neurological diseases. The course objective was to provide an overview of the chemical and pharmacological properties as well as suitable preclinical study designs (emphasizing pathology analysis and interpretation) for four classes of innovative biotherapeutics: (1) cell and gene therapies (main talk delivered by Dr. Sarah Cramer with a subsequent overview of major considerations for routes of biotherapeutic administration given by Dr. Elizabeth Galbreath), (2) ASOs (Dr. Jessica Grieves), (3) engineered Fc (“fragment, crystallizable”) transport vehicles (Dr. René Meisner), and (4) siRNAs (Dr. Arlin Rogers). A final conceptual talk on gliosis in preclinical toxicity studies (Dr. Brad Bolon) discussed the interpretation of this frequent observation when investigating the safety of bio-derived materials introduced to the CNS by direct delivery. This paper summarizes key messages outlined in these lectures as an aid to toxicologic pathologists and toxicologists who contribute to preclinical studies for bio-derived test articles.
General Considerations for Preclinical Evaluation of Novel Biotherapeutics
Treating CNS diseases requires that drugs enter the brain in sufficient doses to achieve therapeutic effects. The major obstacle to successful drug entry into the CNS is the selective permeability of the BBB. This physiological hurdle prevents hydrophilic substances, charged molecules, proteins, and peptides from entering the brain. The BBB is present throughout the brain and spinal cord except for the circumventricular organs, where the absence of a BBB permits the passage of polypeptide hormones from the blood into selected brain nuclei.63,157 While damage to the BBB is associated with aging and pathological states such as Alzheimer’s disease and infection,7,124 the healthy BBB is composed of a single layer of non-fenestrated endothelial cells that are connected by tight junctions and supported by a basement membrane, pericytes, and a dense mesh of astrocyte endfeet. 1 Pericytes function to decrease transcytosis and induce polarization of the astrocyte endfeet, while the endfeet control intercellular tightening and polarized expression of capillary endothelial cell transporters.3,164 This BBB effectively excludes macromolecules to protect the CNS, and therefore effectually precludes entry of systemically administered bio-derived therapeutics at therapeutically relevant concentrations; it is estimated that only 0.1% of circulating antibodies cross the intact BBB. 128 A similar blood-nerve barrier protects nerves, while most somatic and autonomic ganglia lack a tight blood-ganglion barrier.20,132,161 Accordingly, delivery of bio-derived therapeutics to most of the CNS and peripheral nervous system (PNS) requires specialized test articles or physical procedures to cross various neurovascular barriers. These include transient chemical or osmotic disruption of the BBB, non-viral (e.g., lipid nanoparticles) or viral-based (e.g., AAV vectors) carriers, and/or direct delivery into the neural (usually brain) parenchyma or cerebrospinal fluid (CSF). 5 This requirement complicates the design of preclinical studies needed to support the development of cell and gene therapies, nucleic acids, and proteins.
Basic Principles
Product development considerations for novel bio-derived test articles intended to treat neural diseases differ in some important respects from those relevant to traditional small molecule and biologic (e.g., nucleic acids and proteins) drugs. Cell and gene therapies often can be given only once (due to immune reactions against the foreign antigens), and expression of the gene product or gene editing generally cannot be reversed once administered. An additional concern for cell and gene therapies is that engrafted cells or integrating gene therapy (GTx) vectors may be oncogenic (i.e., induce neoplasms) or have unintended disruption of gene function. Nucleic acids and engineered proteins may be delivered more than once (in both preclinical testing and clinical usage), are frequently immunogenic, and may require direct delivery of high concentrations to the CNS sometimes with relatively narrow therapeutic margins. The nature of the preclinical program for such novel biotherapeutics depends on many factors including the nature of the test article, intended indication, known class effects for similar products, and the ability to monitor potential clinically translatable neural effects that can be monitored with minimally invasive methods such as electrophysiology, non-invasive imaging, or serum or CSF biomarker levels.
Several key questions must be addressed to examine the suitability of novel cell and gene therapies as biotherapeutic candidates. Initial in vitro and/or ex vivo studies may be used to characterize the properties of the test article (e.g., cell lineage and proliferative capacity, transgene sequence, cellular biopotency assays). Exploratory in vivo studies may be conducted to select the optimal test system for preclinical testing (e.g., animal model of disease for combined evaluation of efficacy and toxicity and/or in healthy animals solely for safety assessment). Ideal in vivo study designs will incorporate biodistribution (i.e., location and expression of implanted cells or administered GTx vectors), persistence, efficacy, and safety (i.e., local and systemic toxicity, including tumorigenicity).
Existing industry best practices,27,28 regulatory guidance, 60 and internationally harmonized recommendations 85 that provide for standard neuropathology evaluation of test articles with no or unknown impact on neural tissues (i.e., conventional “general toxicity screening”) may not be sufficient for novel biotherapeutics intended to have neuroactive properties and/or that must be administered directly into the nervous system. Damage to neural tissue caused by the delivery procedure (e.g., needle tracks due to unavoidable physical damage [microtrauma] related to intracerebroventricular [ICV], intraparenchymal [IPa], or intrathecal [IT] injections) or known class effects of a particular test article (e.g., neuronal necrosis in dorsal root ganglia [DRG] associated with AAV-based GTx vectors) typically will necessitate “expanded neurohistopathology” (as mentioned in various guidance documents61,86 and scientific papers26,53 and outlined in current industry “best practice” recommendations27,28) to fully characterize the nervous system effects produced by biotherapeutic test articles. Expanded evaluation may consist of additional sampling (usually implemented as more brain levels, further collection of DRG, and multiple nerves; special neurohistology methods (usually to detect cell death and/or reactive glia and in some cases in situ localization of on- and off-target gene expression; or both additional sampling and special methods. Study pathologists and sponsors retain considerable flexibility in deciding how the neuropathology evaluation should be designed, and whether or not expanded neuropathology should be implemented from the beginning of a study or only added if an indication of potential neurotoxicity appears in the clinical observations and/or microscopic findings seen in hematoxylin and eosin (H&E)-stained sections. From a practical perspective, study protocols for preclinical studies of novel biotherapeutics should generally include expanded sampling at necropsy (so tissues that might be needed are retained, thereby avoiding the need for a full-scale follow-up study) and the option to incorporate special neurohistology methods if deemed necessary by the study pathologist. Risks that may be associated with the last-minute implementation of such special methods primarily include delays in study timelines required to arrange access to resources and personnel to perform such procedures, the indeterminate time needed to develop novel assays, and pathologist availability. Nonetheless, the overarching concern in such scenarios is the demonstrate test article safety rather than to sidestep the risk of delayed timelines necessitated by the additional work.
Frequent Findings Related to Novel Biotherapeutics
Primary injury to neural organs associated with the administration of bio-derived test articles during preclinical studies exhibits a number of fairly consistent patterns. The spectrum of findings reflects both test article-related changes (potential or definitive) superimposed over procedure-induced effects, especially for agents administered by direct CNS delivery. For example, ICV or IPa injection into the brain produces a linear injection track associated with the insertion of the flexible cannula or rigid needle (Figure 1). A comparable injection track may be created if intracisternal (intra-cisterna magna [ICM]) injection or IT injection into the lumbar cistern induces physical trauma in the underlying brainstem (ICM) or spinal cord (IT), respectively. Such tracks are characterized by local, sometimes locally extensive tissue disruption that may be accompanied by hemorrhage (with intact erythrocytes for acute lesions or hemosiderin accumulation in macrophages for previous events) and/or cavity formation (i.e., neural tissue loss) in the parenchyma, sometimes with accumulation of “gitter” cells (lipid-laden phagocytes) within the defect (Figure 1). The extent of parenchymal involvement at the administration site (i.e., where test article depot is delivered into tissue, not the injection track penetrating a narrow column of tissue) depends on multiple factors including the nature of the test article, the dose, the rate and volume of carrier infusion, and the size of the injection device. With respect to parenchymal involvement, the interpretation of microscopic findings may be aided if ancillary information (e.g., biodistribution data, in-life neurological signs, and surgical records) is available to the pathologist during their evaluation. For IPa injection sites, tissue disruption greater than that seen in control animals has been observed with some test articles. Explanations for this exacerbation include potential idiosyncratic differences in the on- or off-target effects, chemical attributes, immunogenicity, or dose-related responses of test articles as well as the rate and/or volume (i.e., pressure) of administration and larger cannula sizes. 52 Long-term cannula placement in the vertebral canal may yield an annular ring of fibrosis (to wall off the foreign body) or less often localized inflammation at the cannula tip, sometimes with compression of the underlying spinal cord. 34 Microscopic effects on the adjacent compressed spinal cord or spinal nerve roots (e.g., nerve fiber degeneration and/or gliosis) are usually modest and occur most often in rats, 34 which have much smaller IT volumes compared with non-rodents and humans. 158
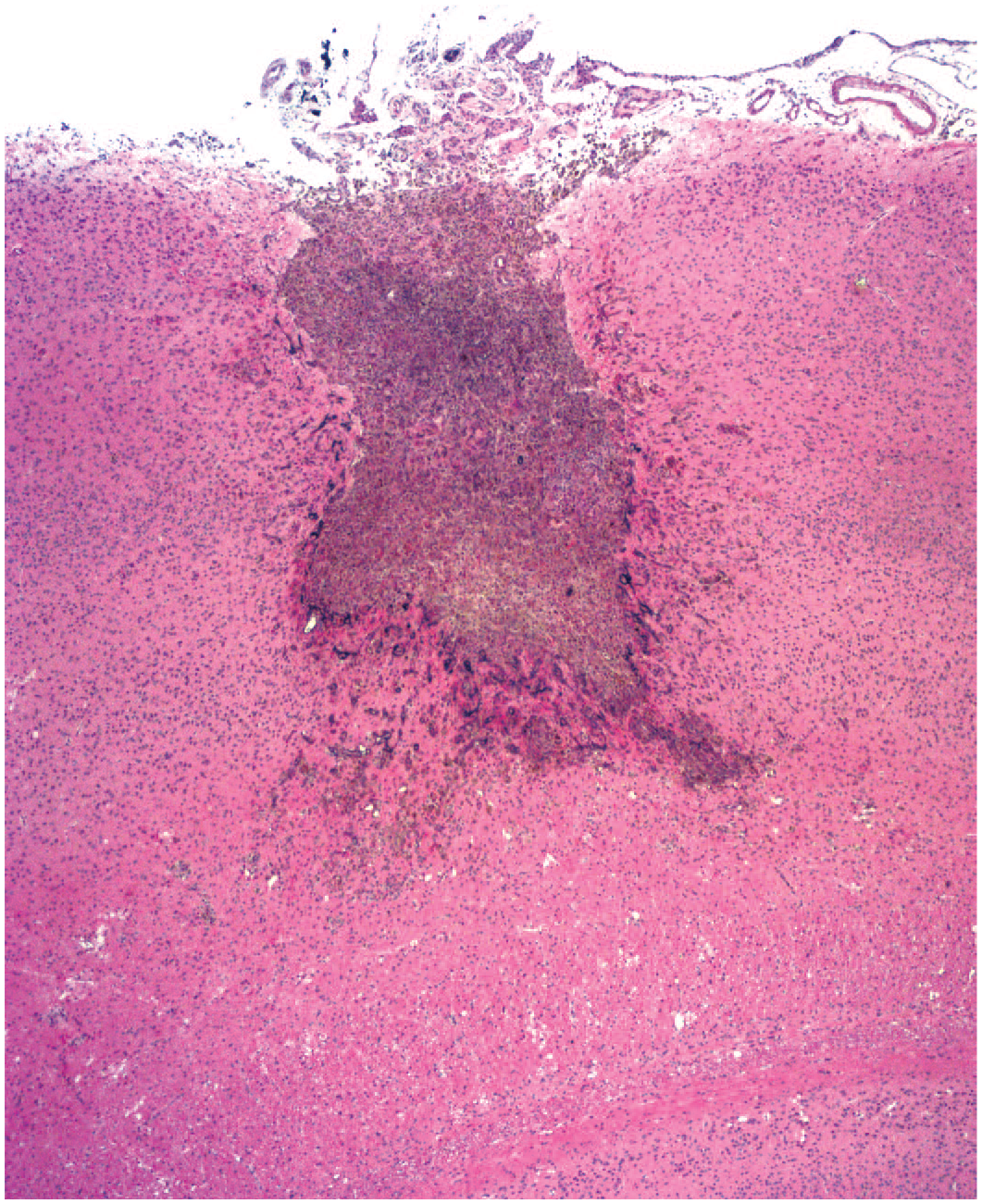
Injection site in the superficial cerebral cortex associated with intraparenchymal injection of a gene therapy test article. The rectangular linear defect (INHAND diagnosis = “necrosis”) extends to the middle of the gray matter and is filled with pigment-laden phagocytes (“gitter cells”); additional pigment-laden cells are scattered in the parenchyma lateral and deep to the defect as well as in the meninges. Test system: juvenile cynomolgus macaque. Stain: hematoxylin and eosin. Original objective magnification: 2×. INHAND indicates International Harmonization of Nomenclature and Diagnostic Criteria for Lesions.
Neuron degeneration and necrosis is a common primary change induced by bio-derived test articles. Affected sites depend on the test article. For example, AAV agents may induce necrosis of ganglionic neurons (e.g., DRG, trigeminal ganglia, and sympathetic ganglia) regardless of the administration route. This change typically is most pronounced following direct delivery into the CSF via the ICV and IT routes but does occur after systemic (IV) injection; the finding can be minimal or absent following IPa administration because test article entry into the CSF is variable and possibly limited so that ganglionic exposure may be reduced or eliminated. In contrast, parenchymal neurons are impacted more often following IPa delivery. Special techniques (e.g., Fluoro-Jade staining) may be used to highlight necrotic CNS neurons, but this procedure rarely is helpful in ganglia. 28 Degenerating neurons are often highlighted in conventional H&E-stained tissue sections by using fluorescent light in combination with the fluorescein isothiocyanate (FITC) filter,90,131 although this effect may yield inconsistent success (R. H. Garman, personal communication, October 2023).
Nerve fiber degeneration as a secondary effect of primary neuronal damage is another common finding for some classes of biotherapeutic test articles. This finding may occur in various regions (as described below) depending on the population(s) of affected neurons. For example, this change may occur in the brain (in the white matter of the cerebral crura and pyramids) due to minimal injury to the ventral forebrain following IPa injection of any biologic (e.g., cells or AAV-based GTx vectors) that impacts trigeminal neurons. In contrast, biotherapeutic-related nerve fiber degeneration in the spinal cord occurs most often in the white matter of the dorsal and lateral funiculi (Figure 2) or spinal nerve roots as a result of gangliotoxic (e.g., AAV-induced) secondary disintegration of ascending axons that originate from the DRG sensory neurons (for the dorsal funiculi) or possibly spinal cord interneurons innervated by DRG sensory neurons (for the lateral funiculi).20,25,83 Nerve fiber degeneration in nerves typically represents a secondary effect of damage to dendrites of ganglionic neurons or axons of spinal cord motor neurons. This nerve finding reflects the inability of the affected neurons to provide metabolic support to the distal portions of long cell processes. If tissue (brain, spinal cord, or nerves) from both sides of the body is evaluated, nerve fiber degeneration related to test article administration is often but not always bilateral, though it frequently is asymmetric.20,25,83 Degeneration that is unilateral in nature is most often associated with an ipsilateral injection site in the brain or long-term cannula placement in the brain or over the caudal spinal cord and adjacent spinal nerve roots (Figure 2), unilateral effects may be observed with any route of administration for AAV-based GTx vectors as DRG effects typically are not detected in all DRG and for a given DRG pair are not necessarily bilateral. 20 Primary damage to CNS and PNS neurons, CNS neurites, and CNS myelin is not subject to effective repair and therefore is inherently adverse. 117 In contrast, nerve fiber degeneration in nerves may be partially or completely restored in time. Adverse findings of limited degree (as communicated in a pathology report) may be acceptable in the context of a regulatory risk assessment for a novel biotherapeutic being developed to treat a severe or life-threatening neural or neuromuscular disease. Nonetheless, clinical awareness of the potential for effects from repeated lifetime administration is warranted.
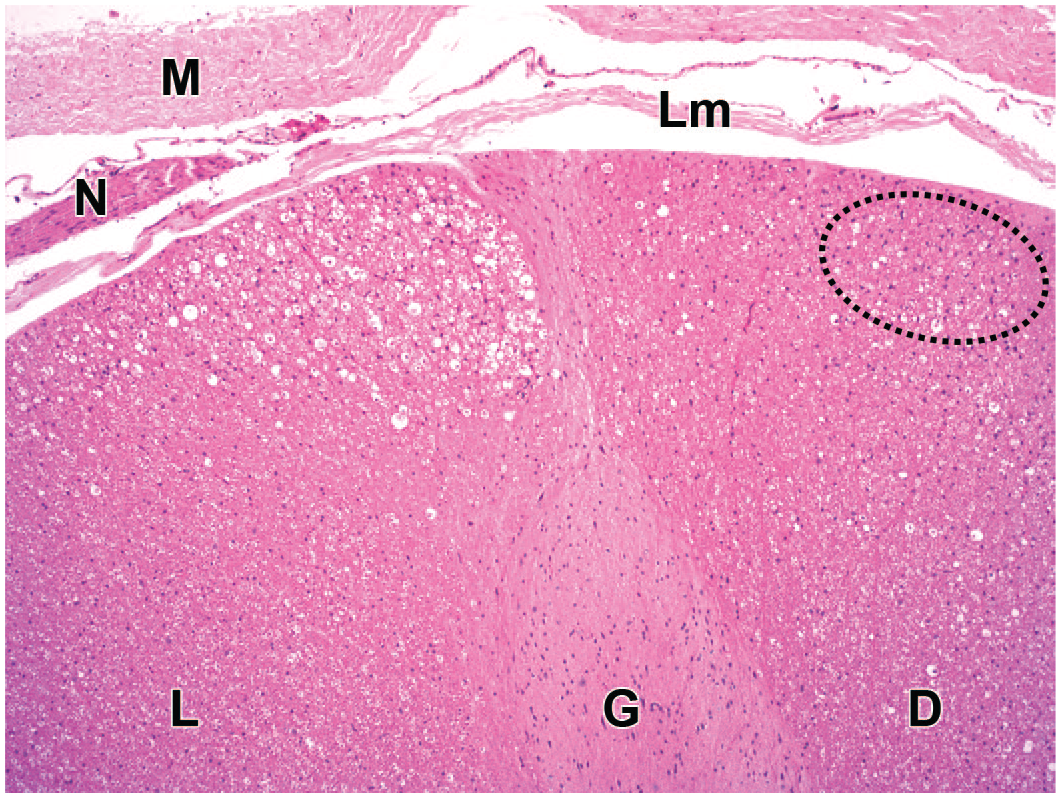
Nerve fiber degeneration (INHAND diagnosis = “degeneration, nerve fiber”) in the dorsal aspect of the lumbar spinal cord characterized by multiple vacuoles, sometimes containing cell debris or phagocytes (“gitter cells”). The triangular region with numerous large vacuoles in the lateral funiculus (L) was associated with the placement of a flexible polyurethane catheter. The scattered small vacuoles in the dorsal funiculus (D) reflect axonal loss following administration of an adenovirus-associated (AAV) gene therapy vector; the increased number of nuclei in the affected region of the dorsal funiculus (oval) represents a modest increase in the number of glial cells (INHAND diagnosis: “gliosis, not otherwise specified”) as a secondary reaction to the nerve fiber degeneration. Test system: juvenile Beagle dog. Stain: hematoxylin and eosin. Original objective magnification: 4×. G indicates gray matter of the dorsal horn; INHAND, International Harmonization of Nomenclature and Diagnostic Criteria for Lesions; Lm, leptomeninges; M, dura mater; N, dorsal spinal nerve root.
Secondary gliosis may result from procedure-induced or test article-related injury to one or more elements of the brain or spinal cord parenchyma (e.g., neurons, neurites [axons and dendrites], and myelin) but more frequently occurs as a reactive change to altered microenvironmental conditions in the absence of any light microscopic evidence of parenchymal damage. 25 In such instances, reactive glia often are invisible in H&E-stained sections but are clearly evident in serial sections immunolabeled to demonstrate glial fibrillary acidic protein (GFAP, an astrocyte marker) or ionized calcium-binding adaptor molecule 1 (Iba1, a microglia/macrophage marker) (Figure 3). Glial reactions represent a challenge in making adversity decisions regarding the neural effects of novel biotherapeutics. Homeostatic (“resting”) astrocytes and microglia have small cell bodies with many processes supporting numerous fine branches, while reactive (“activated”) astrocytes and microglia have larger cell bodies and thicker branches (“hypertrophy”) and may congregate in locally greater numbers (“hyperplasia”) (Figure 3).119,146 If only one glial cell type is responding, microglia are involved more often because they are the most dynamic cells in the CNS.119,150 Microglial reactions arise more rapidly and regress more quickly compared with astrocytic responses. 25 In the PNS, gliosis in ganglia is diagnosed as increased satellite glial cellularity, while gliosis in somatic nerve trunks is designated as increased Schwann cellularity. (Axons in autonomic nerves have limited myelin, so the apparent increase in Schwann cell numbers in these small-diameter nerves represents their normal appearance.) Gliosis in the CNS but especially in the ganglia of the PNS is often accompanied by the accumulation of mononuclear leukocytes (e.g., T-lymphocytes and/or macrophages with fewer B-lymphocytes).21,75,78,166 Injury to the neural parenchyma is the main consideration in making adversity decisions for preclinical studies, while glial reactions in the absence of overt parenchymal damage are typically limited in severity and thus can be considered to represent non-adverse (i.e., adaptive) responses related to the expected physiological function of glial cells faced with altered microenvironmental conditions.38,104,119,146
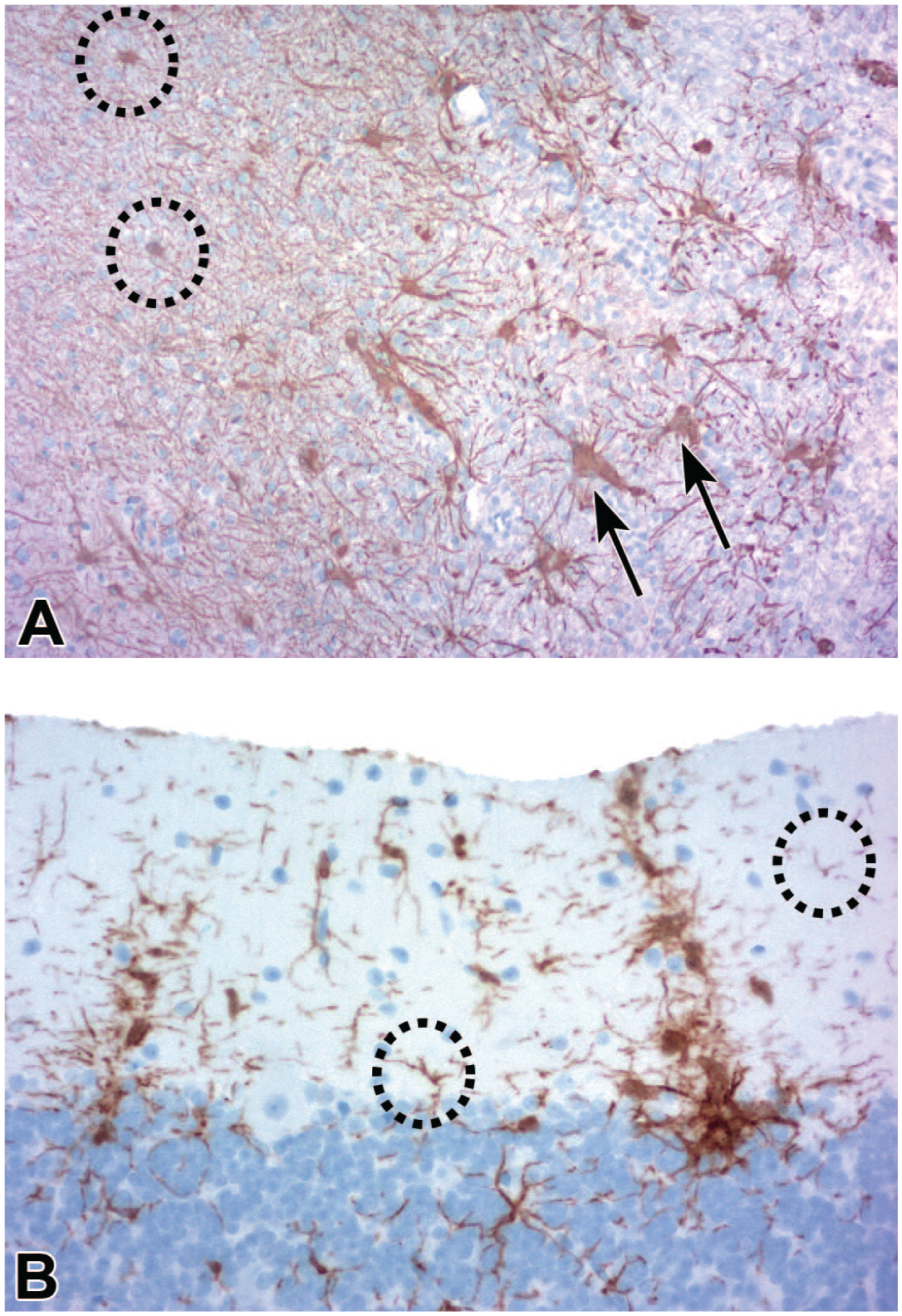
Glial reactions associated with direct administration of gene therapy test articles into the central nervous system. (A) Margin of an intraparenchymal injection site in the striatum showing normal tissue (brown neuropil on left) compared with a zone of pallor (pale basophilic cell field on right) representing the border of a cavity (INHAND diagnosis = “necrosis”) filled with microglia-derived phagocytes (“gitter cells”). Homeostatic (“resting”) astrocytes (dotted circles) have small cell bodies and thin processes, while reactive (“activated”) astrocytes (arrows) are enlarged and have fewer but thicker processes; the distance between the reactive cells indicates that hypertrophy is not accompanied by astrocytic proliferation. Method: GFAP with hematoxylin counterstain. Original objective magnification: 10×. (B) Increased numbers of reactive (“activated”) microglial cells (brown; INHAND diagnosis = “microgliosis”) in the molecular layer of the cerebellar cortex are characterized by large cell bodies with thick processes. The overlapping domains of the labeled cells in the linear columns are indicative of proliferation. Homeostatic (“resting”) microglia (dotted circles) have small bodies and thin processes. Method: Iba1 with hematoxylin counterstain. Original objective magnification: 20×. Test system: juvenile cynomolgus macaque. GFAP indicates glial fibrillary acidic protein (an astrocyte marker); Iba1, ionized calcium-binding adaptor molecule 1 (a microglia/macrophage marker); INHAND, International Harmonization of Nomenclature and Diagnostic Criteria for Lesions.
Biotherapeutics commonly incite leukocyte entry and aggregation following direct CNS delivery, especially after repeated dosing. Common areas of leukocyte accumulation include sites of immune surveillance in the choroid plexus, meninges, or perivascular (Virchow-Robin) spaces of the brain or spinal cord. The leukocyte accumulations usually are mononuclear cell infiltrates, where “infiltrates” is the INHAND (International Harmonization of Nomenclature and Diagnostic Criteria for Lesions) nomenclature for leukocyte accumulation without parenchymal damage and “mononuclear cell” describes foci of intermingled lymphocytes (mainly T-cells with fewer B-cells) and macrophages (Figure 4). Less often, leukocyte aggregates develop as mononuclear cell inflammation (where “inflammation” is the INHAND term for leukocyte aggregates causing tissue injury), possibly associated with increased local production of cytokines, or occasionally mixed cell infiltrates (Figure 5) or inflammation (where “mixed cell” refers to scattered granulocytes [eosinophils and/or neutrophils] in combination with mononuclear cells).
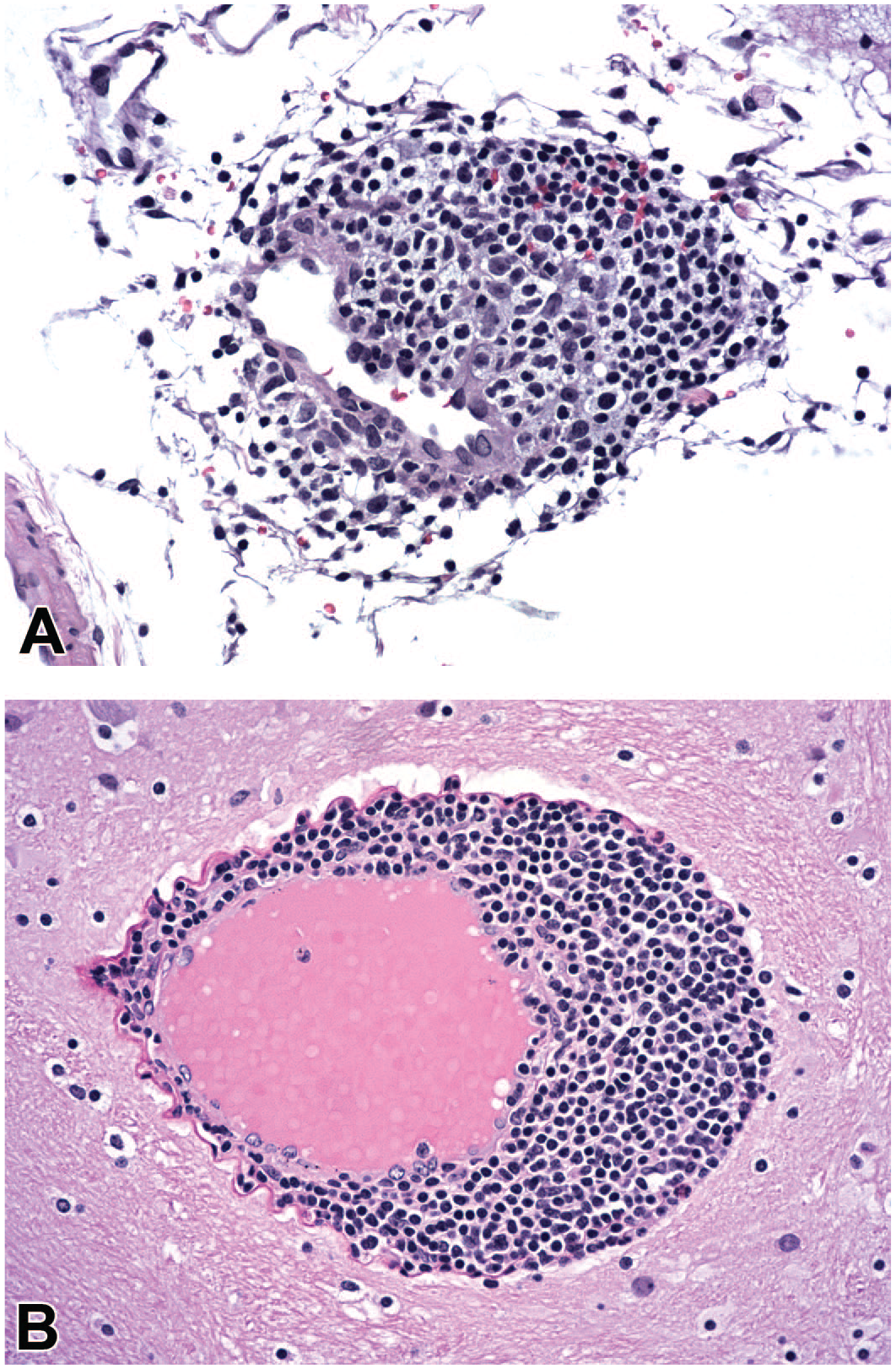
Leukocyte accumulation in the brain following administration of a nucleic acid-based test article. (A) Focal, minimal, leukocyte-mediated vascular disruption (INHAND diagnosis = “inflammation, mononuclear cell”) is characterized by leukocytes within a portion of the blood vessel wall and a few scattered, extravascular, intact erythrocytes. (B) Focal aggregation of leukocytes without parenchymal damage (INHAND diagnosis = “infiltrate, mononuclear cell”) in the perivascular space defined by the intact blood vessel wall and thick, undulating basement membrane. Test system: juvenile cynomolgus macaque. Stain: hematoxylin and eosin. Original objective magnification: 20×. INHAND indicates International Harmonization of Nomenclature and Diagnostic Criteria for Lesions.
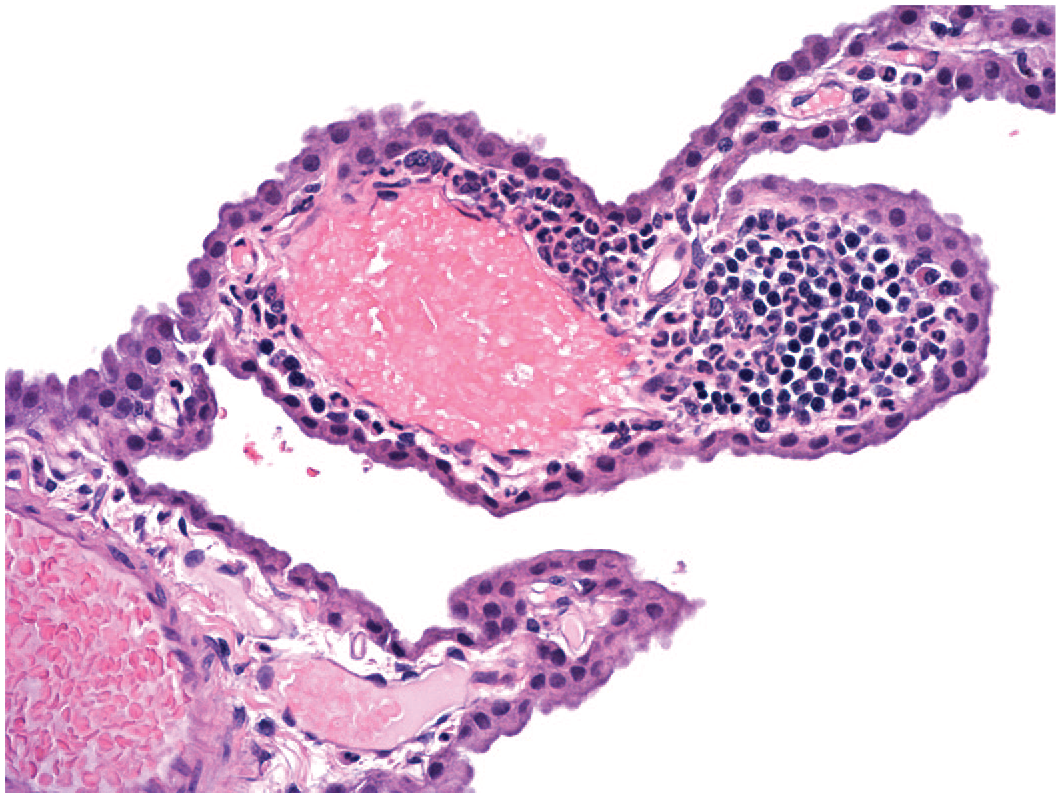
Multi-lineage leukocyte accumulation without damage to the neural tissue (INHAND diagnosis = “infiltrate, mixed cell”) in the choroid plexus following administration of a protein (antibody-based) test article. The focus consists mainly of basophilic granulocytes (neutrophils) with modest numbers of small dark mononuclear cells (lymphocytes). Test system: juvenile Beagle dog. Stain: hematoxylin and eosin. Original objective magnification: 20×. INHAND indicates International Harmonization of Nomenclature and Diagnostic Criteria for Lesions.
Pathology-Based Pharmacokinetic and Pharmacodynamic Methods for Biotherapeutics
Histopathologic evaluation is a valuable, often underused tool in pharmacokinetic (PK) investigations of novel bio-derived test articles. In contrast to traditional PK and pharmacodynamic (PD) assays, which are run at the whole organ level, microscopic examinations using special procedures enable cell-specific visualization of test article distribution and activity in fixed, structurally intact tissues. By comparison, molecular-based single-cell assays (e.g., sequencing) require unfixed dissociated tissue, which results in loss of spatial information and may lead to potential artifacts associated with cellular de-differentiation.
Various tools are available to demonstrate test article distribution in sections of fixed tissue. The usual options for toxicologic pathologists are immunohistochemistry (IHC) for detecting cells, non-native nucleic acids, and proteins and in situ hybridization (ISH) for nucleic acids (including transgenes in gene therapies). For IHC, an antibody may be generated by injecting an adjuvant containing the antigen (usually a cell or protein but sometimes a nucleic acid) into a naïve animal (usually rabbits or goats). A specific antibody that detects an antigen that is foreign to the test animal may confirm the distribution of the test article to the target tissue. For more common antigens, short non-native polypeptides such as histidine (His) or DYKDDDDK (FLAG) tags or larger non-native proteins like green fluorescent protein (GFP) may be engineered into the transgene to permit detection.102,106 Molecules with intrinsic fluorescence (e.g., GFP) may be visualized directly in frozen tissue sections or by IHC in standard formalin-fixed, paraffin-embedded (FFPE) tissues. Test article visualization may be coupled with cell-specific IHC or ISH to definitively localize the biotherapeutic to cells of interest. This approach is especially valuable in cells that lack easily distinguishable morphological features in H&E-stained sections, such as immune cells.
In the CNS, PK evaluation of test articles is impacted by the route of administration and flow of CSF. In humans (and thus in most preclinical studies), neuroactive bio-derived test articles that cannot cross the BBB are delivered directly into the lumbar cistern by IT injection. In rats and nonhuman primates (NHPs), the introduction of fluid into the CSF raises intracranial pressure transiently to an extent influenced by the infusion rate and volume. 19 Increased CSF flow associated with transiently altered fluid dynamics (related to the injection procedure) works together with existing pulsatile CSF flow related to cyclic fluctuations in arterial blood pressure 120 to propel the test article cranially from the lumbar cistern up the vertebral canal to reach the cranial vault. In addition, larger IT bolus dose volumes produce greater cranial distribution along the neuraxis. 147 The CSF flow occurs in the subarachnoid space in a spiral pattern, with micro-eddy currents moving the test article into proximity with the surface of the spinal cord (A. Rogers, personal observations). Just prior to entering the cranial vault through the foramen magnum, ascending CSF (and test article) encounters a caudally moving countercurrent related to CSF escaping the fourth ventricle through the two lateral apertures (foramina of Monro) and a median (dorsal) aperture (foramen of Magendie). 163 This dorsolateral countercurrent diverts ascending CSF ventrally so that most of the test articles delivered in the lumbar cistern enter the cranial vault and proceed rostrally along the ventral aspect of the brain to reach the interpeduncular cistern ventral to the midbrain. At this locus, the test article may move dorsally along the caudal aspect of the tentorium cerebelli to encounter the hindbrain, dorsally along the rostral aspect of the tentorium cerebelli to flow over the caudal forebrain, or rostrally to the olfactory bulbs before flowing up and over the frontal cortex. 18 Continued impetus from the pressure of the injected fluid drives some test articles dorsally along the lateral brain surfaces as well. Because the forebrain is divided sagittally by the cerebral crura, separate circuits of flow are established for the left and right cerebral hemispheres. Therefore, drug exposure may not be equal on both sides of the brain.
To penetrate the CNS parenchyma, IT-administered test article transits from the subarachnoid space along perivascular (Virchow-Robin) spaces adjacent to penetrating arterioles. 107 The rate and turbulence of CSF flow are influenced by the tortuosity of radiating vessels and the amount of solid material (i.e., cells and connective tissue) present within the spaces. 93 Arteriole pulsation promotes deeper penetration and movement of the test article from the perivascular spaces into the neuropil following a pressure gradient that moves fluid from the arterioles toward venules. Some investigators posit that neuropil penetration is actively facilitated by aquaporin-4 channels on the endfeet of perivascular astrocytes, 138 but this glial-lymphatic (“glymphatic”) hypothesis is still controversial. 145 Because IT-delivered drugs enter the parenchyma from the subarachnoid space, deeper brain regions such as striatum and thalamus typically show lower drug concentrations than more superficial regions like cerebral cortex. 32 Perivenous fluid is thought to drain to lymphatic vessels associated with the venous sinuses and olfactory nerves, which helps explain why IT-delivered test articles may be visualized in lymph nodes of the head and neck within hours of delivery. 105 Olfactory-related drainage is particularly important in rodents since the olfactory nerves are more prominent in this species compared with NHPs and humans. Finally, test article egress from the CNS also occurs through the arachnoid granulations, which are small leptomeningeal outpouchings that provide CSF recycling into the venous sinuses, especially under conditions of increased intracranial pressure.
Assessment of PD (i.e., bioactivity) of neuroactive biotherapeutics is enhanced if the CNS target or a known neural cell biomarker is secreted into the CSF or escapes into the serum, thereby permitting intermittent nonterminal sampling to evaluate test article potency and durability. If no such secreted marker exists, PD may possibly be monitored through non-invasive imaging methods. However, in most preclinical settings PD analysis requires tissue collection at necropsy with subsequent evaluation in tissue homogenates or FFPE sections. In homogenates, polymerase chain reaction (PCR) is used for nucleic acids, while enzyme-linked immunosorbent assay (ELISA), western blotting, and sometimes liquid chromatography-mass spectrometry (LC-MS) are employed for proteins and lipids. In tissue sections, ISH for nucleic acids and IHC for proteins may be utilized to directly visualize the pharmacologic target or a surrogate molecule. 32 Multiplex labeling may facilitate dual localization of the test article and its engagement with target in a cell-specific manner. In recent years, multiplexing of either ISH or IHC for dozens of targets in a single section has underpinned the growth of spatial imaging. 122
Limitations associated with histopathologic evaluation of biotherapeutic PK/PD must be factored into preclinical decision-making for novel test articles. Histopathology data are typically qualitative or semi-quantitative, and small differences between treatment groups may be missed. Quantitative image analysis may deliver precise numbers, but the numbers may be impacted by several factors affecting image integrity. Tissue visible in standard 4- to 5-µm-thick sections may not be representative of the entire sample. Differences in staining quality (for H&E as well as IHC and ISH), tissue composition, molecular assay digital analysis algorithms, and instrument performance may impact image quality. 39 In particular, molecular cross-linking associated with tissue fixation excludes many of the antibodies validated for ELISA and western blotting for use with tissue sections. Because of the many potential confounding variables, definitive assessment and interpretation of PK/PD molecular histology assays should ideally be performed in close collaboration with an experienced pathologist(s).
Toxicologic Neuropathology of Cell and Gene Therapies
Product development considerations for cell and gene therapies differ in many respects from those for traditional drugs. In the United States, the Food and Drug Administration (FDA) Center for Biologics Evaluations and Research (CBER) has prepared multiple guidance documents to assist in the preclinical and clinical assessment (efficacy and safety) of cell and gene therapies (https://www.fda.gov/vaccines-blood-biologics/biologics-guidances/cellular-gene-therapy-guidances). Guidance for preclinical testing of cell therapies and gene therapies is provided in a common document. 55 This guidance states that animal test systems for evaluating the safety of cell and gene therapies must be selected with several considerations in mind: “a) comparability of physiology and anatomy to that of humans; b) permissiveness/susceptibility to infection by, and replication of, viral vectors or microbial vectors for gene therapy; c) immune tolerance to a human CT product or human transgene expressed by a GT product; and d) feasibility of using the planned clinical delivery system/procedure.” Preclinical work may utilize animal models of disease to permit a simultaneous assessment of efficacy and safety. 55 Endpoints relevant to toxicologic neuropathology are evaluated using in vivo models, but in vitro studies are recommended before initiating definitive in vivo preclinical studies. 55 Similar guidance is available for cell therapies and gene therapies in other parts of the globe.49,50,129
Stem Cells
Therapeutic stem cells may be autologous (obtained from the patient and then returned to that same person), allogeneic (collected from a healthy human donor and given to a different individual with an illness), or xenogeneic (harvested from an animal for transfer into a human). The cells may or may not have been genetically modified and may or may not be combined with a medical device, mesh, or scaffold. Common variants under investigation include embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs), which can differentiate into multiple cell types, and somatic-origin adult stem cells, which can differentiate into fewer cell types. For clinical products, stem cells typically undergo a complex, multi-step process in vitro to induce differentiation into a desired cell lineage. 74
Platform effects reported variously in preclinical studies for stem cell test articles include neoplastic transformation (i.e., formation of teratomas or other neoplasms), ectopic tissue generation especially following cell migration out of the engraftment site, host immune responses, and transfer of infectious diseases.45,53 Clinical manifestations of neurotoxicity associated with stem cell therapy are limited and may be related to co-treatments (e.g., busulfan chemotherapy given alongside autologous stem cells to treat multiple sclerosis 152 ) or contaminants (e.g., dimethyl sulfoxide as a cryo-protectant. 14 Neoplasia within the CNS has been described in human patients following the administration of neural stem cells.6,22
Test systems for preclinical safety testing of cell therapies include wild-type rodents (especially neonates), minipigs, and NHPs; immunocompromised rodent models; and induced or spontaneous models of disease. Animal disease models are often preferred since both efficacy and safety can be tested in a single, well-designed study. A further advantage of immunocompromised models is that engrafted cells are less likely to be rejected, which removes one confounder to data interpretation.
For preclinical testing, stem cells generally are administered to animals by orthotopic injection (i.e., into the parenchyma at the site that is intended as the administration site in human patients). Key considerations in neuropathology evaluation include examining the size of the injection track, screening for migration of engrafted cells away from the injection site, assessing for aberrant differentiation (e.g., non-neoplastic cartilaginous nodules in meninges following engraftment of neural stem cells), evaluating any tissue response (e.g., glial or leukocyte) to the stem cells, and determining the existence and extent of any neoplasm formation. Suitable control groups are
Initial screening of engrafted stem cells typically starts with H&E-stained sections, but detection of cells often requires IHC labeling to demonstrate markers specific for the implanted cell type. 30 For example, human cells injected into animal organs may be highlighted using human leukocyte antigen (HLA) 144 ; STEM121 (a cytoplasmic protein found in brain, pancreas, and liver) 92 ; human nuclear antigen (usually HNA or HuNu) 10 ; Ku80 (another human nuclear antigen) 88 ; or Clone 113-1 (an antibody against a human mitochondrial protein). 139 Alternatively, ISH for the human Alu sequence may also serve as a marker for human-origin stem cells. 11 Assessment of Ki67 or a similar marker may be helpful in characterizing proliferation in the implanted cell population, especially if stem cells have formed a mass rather than integrating into the tissues at the injection site.
Chimeric Antigen Receptor (CAR) T-Cells
Engineered CAR T-cells are T-lymphocytes that express recombinant receptors that recognize and destroy neoplastic cells. These immunotherapies are especially effective in treating relapsed and refractory B-cell malignancies. 67 Target engagement of CAR T-cells leads to CAR T-cell proliferation and cytokine secretion, resulting in lysis of the targeted tumor cells. 68
Clinically, target engagement by CAR T-cells may be associated with cytokine release syndrome (CRS) and immune effector cell-associated neurotoxicity syndrome (or ICANS).68,110 The incidence of neurotoxicity varies from 20% to 64% of clinical trials, and pediatric and young adult patients may be at higher risk.67,82 After CAR T-cell administration, CRS usually precedes ICANS by about 2 weeks though the lag phase varies among clinical trials.67,82,110 ICANS can also be associated with lymphodepletion regimens. 67
Clinical presentation of CAR T-cell neurotoxicity includes various combinations of cognitive dysfunction, aphasia, memory loss, seizures, headache, ataxia, coma, and cerebral edema, among others.67,68,82,153 In one patient with cerebral edema symptoms, microscopic changes included microglial activation, astrocyte clasmatodendrosis (i.e., disintegration and beading of astrocyte processes seen on GFAP IHC), and perivascular edema 153 ; in this patient, CAR T-cells were not present in the brain parenchyma, suggesting that neurotoxicity may have resulted from dysfunction of the BBB. Other microscopic changes associated with CAR T-cell neurotoxicity include microhemorrhages, perivascular aggregation of CD8+ T-cells, endothelial activation, microthrombi, and occasionally vascular necrosis.68,82 Fatalities due to neurotoxicity may necessitate that a clinical trial be discontinued. 153
Mechanisms that initiate and drive ICANS are not well understood. 67 In some cases, neurotoxicity may be associated with on-target effects of CD19 CAR-T cells because brain pericytes and vascular smooth muscle cells express CD19.110,123 Other CAR T-cell varieties such as those expressing CD20, CD22, or BCMA/TNFRSF17 are less likely to cause neurotoxicity. 110 Preclinical models of CAR T-cell neurotoxicity are lacking at present, but innovation in animal model development is progressing.51,67,82 CAR T-cell neurotoxicity has been modeled in mice and rhesus monkeys.64,116,134,149 Humanized (xenotolerant) mouse models show meningeal infiltration by human macrophages, 82 while rhesus models exhibit IPa infiltrates of CAR T-cells in the brain as well as meningitis and perivascular edema. 149 Animal models may not effectively predict human responses due to species-specific differences in hematopoiesis and development of graft-versus-host disease.82,110
Neuropathology evaluation of preclinical CAR T-cell models may include a tailored screening battery in which routine methods (H&E, GFAP, and Iba1) are supplemented by IHC for CD3 (a T-cell marker) to detect subtle T-cell accumulation. 153 In addition, histochemical staining (e.g., phosphotungstic acid hematoxylin [PTAH]) may be helpful for highlighting intravascular (i.e., microthrombi) and perivascular fibrin deposits. Selection of time points for neuropathology evaluation during preclinical studies may be problematic since the variable onset of neurotoxicity symptoms observed during clinical trials suggests that lesion development does not follow a consistent pattern in vivo. One proposed approach to preclinicial study design would be to necropsy animals exhibiting clinical signs to capture acute, often subtle changes associated with CAR T-cell neural damage. 149
Gene Therapies
Nearly 3000 clinical trials of GTx test articles have been launched around the world in the past three decades to alter the expression of genes or repair/replace defective or missing genes. Many of these trials are for severe neurodegenerative or neuromuscular diseases. Platforms now under development include viral vectors (e.g., adenovirus [AdV], AAV, herpesvirus, lentivirus [and other retroviruses], poxvirus); non-viral vectors (e.g., naked DNA/plasmid DNA delivered via liposomes, nanoparticles, or exosomes); and various hybrid systems (e.g., CRISPR-based genome editing and virosomes).12,55,57,168 The choice of GTx vector depends on several factors including persistence and biodistribution patterns of transgene expression and the extent of the host immune response. Viral vectors are GTx delivery systems of choice for in vivo applications based on their superior ability to efficiently introduce foreign DNA into a host cell; non-viral vectors are of interest due to their reduced cytotoxicity and immunogenicity relative to viral vectors; and hybrid systems are attractive options due to their manufacturing simplicity and versatility.101,130,165 Genome-integrating viral vectors (e.g., lentivirus and other retroviruses) offer long-term transgene expression, but their predisposition for integration into existing genes (i.e., insertional mutagenesis) does pose a risk for test article-related mutagenesis or oncogenesis.69,80,100,103,108,109,162 Non-integrating vectors (e.g., AAV and AdV) generally remain episomal and thus have a lower likelihood of insertional mutagenesis,15,65,66 but the potential for integration of such vectors to induce neoplasms remains a concern.37,133 Moreover, the therapeutic efficacy of non-integrating vectors may decline over time. 72 Genome-integrating vectors are typically used ex vivo to modify gene activity in a patient’s cells while non-integrating vectors are commonly utilized for in vivo applications. Ex vivo GTx permits greater control and confirmation of appropriate gene modification prior to infusion of the engineered autologous cells back into the patient, while in vivo GTx provides both a higher likelihood of systemic exposure and a greater potential for inadvertent modification of non-target cells.
Due to the innovative nature of bio-derived GTx products, regulatory guidance for this therapeutic modality is still evolving. Preclinical safety studies for GTx products are defined on a case-by-case basis that is determined by the disease indication and test article attributes.50,54,55 A plethora of safety considerations need to be addressed for GTx biotherapeutics,54-57 including the following principal considerations.
“Pharmacologic” properties Biodistribution to target organs and major non-target tissues Expression levels and persistence Extent of target cell activation Test article composition (e.g., ratio of full to empty capsids)
Procedural effects related to the route of administration
Toxicity profile Anticipated and unexpected findings (on-target and off-target) attributed to the GTx test article Extent of the anti-viral host response Potential for insertional mutagenesis and oncogenicity Toxicity of final formulation components (intended and contaminating)
Transmission Germline Horizontal (shedding)
Each viral platform is associated with particular safety concerns (FDA, 2013). For the purpose of this discussion, we are using AAV vectors as a prototypic GTx biotherapeutic as (1) these vectors are widely used as the basis for current GTx test articles destined to treat neurological diseases and (2) AAV-based GTX vectors exhibit a well-known pattern of neuropathology related to their effects on ganglionic sensory neurons.
Conventional AAV vectors targeting the nervous system may be delivered either directly into the CNS (by ICM, ICV, IP, or IT administration) or systemically (usually by intravenous [IV] injection).29,112 Upon reaching the target tissue, AAV capsid proteins bind to cell surface receptors to initiate endocytosis, leading eventually to transcription of the transgene payload.42,46,112 Certain AAV serotypes and differences in expression cassette engineering (such as a tissue- or cellular-directed promoter) have an enhanced affinity for neural expression.42,81,127,159 The durability of AAV activity can be limited in some organs, particularly those with cellular replication and replacement, but clinical trial data suggest that CNS administration of AAV often leads to durable transgene expression. 143
In clinical trials, neurotoxicity is among the most common undesired effects of AAV administration, often being flagged as a serious adverse event. 143 The clinical presentation, as reported in a patient with amyotrophic lateral sclerosis (ALS), involves enhanced pain sensations (like “electrical shocks”); this sensory disturbance was associated with DRG neuronal loss and spinal nerve root inflammation (i.e., meningoradiculitis) at autopsy. 111 Other organs frequently linked to AAV-related toxicity include the coagulation system (as thrombotic microangiopathy), heart, and liver.
Suggestions for design of a comprehensive preclinical safety assessment for AAV-based GTx test articles have been detailed elsewhere.13,83 The known targeted neural organs in animals indicate that tissue sampling for studies with AAV test articles should be more extensive than current industrial best practices,27,28 chiefly by more systematic collection of multiple ganglia and nerves. In preclinical studies, the cardinal feature of AAV-related neurotoxicity in test species (especially NHPs) is primary damage to sensory neurons in the DRG, trigeminal, and/or autonomic ganglia with secondary effects in tissues containing sensory neuron processes (including brainstem, spinal cord, spinal nerve roots, and nerves).12,20,76,83,118 Key morphologic changes in affected ganglia include neuron degeneration, necrosis, and sometimes loss (depending on the time point); widespread but typically transient leukocyte infiltration or inflammation; and increased satellite glial cellularity (to support injured neurons and ultimately fill gaps left by necrotic neurons).20,76 Secondary findings related to degeneration of ganglionic neuron-derived cell processes include vacuolation and gliosis (mainly microgliosis) of the spinal cord white matter (dorsal > lateral funiculi) and less often the spinal cord gray matter (dorsal > lateral and ventral horns).20,76 Similar degeneration of cell processes may be seen in spinal nerve roots and somatic nerves.20,76 Preclinical safety data (even when obtained using NHPs) do not always correlate with effects reported in humans.79,81,143 The reason for this divergence is that AAV-associated neural findings in animals (related to modest morphological changes in sensory ganglia, nerve, and spinal cord) typically induce no routinely detectable or, rarely, very subtle clinical signs when severity extends to affecting proprioceptive parameters20,33,75,76,154; importantly, assays to investigate altered sensation (e.g., electrophysiological tests like nerve conduction velocity) are not included in routine preclinical studies. Neurotoxicity may be related to the viral dose,20,76,118 but in many instances the incidence and severity of neural findings does not exhibit a dose-response relationship.
In addition to the potential for neurotoxicity, AAV test articles have been suggested to have a very low risk of genome integration, 113 which could serve as the launch point for neoplastic transformation of transfected cells.55,136,137 While AAV generally retain an episomal location in transduced cells, 58 AAV rarely integrate into host DNA. For AAV vectors encoding CRISPR gene editing machinery, rates of integration may be higher. 71 AAV-associated oncogenesis has not been described in humans to date. 137
Conventional preclinical safety studies for AAV-based test articles are not designed to assess the underlying pathogenesis of neurotoxicity. Potential mechanisms of neurotoxicity include immunogenicity of the transgene or viral proteins,53,143 cell exhaustion resulting from protein overexpression, 77 and toxicity of the transgene product. 140 Non-mammalian reporter genes such as GFP are highly immunogenic, so the safety profile of an exploratory construct containing GFP may be very different from that of the final clinical product.8,33,95,140 Accordingly, preclinical assessment of an AAV-based test article bearing a reporter gene may have little relevance in determining the safety profile of a given AAV vector.
Neuropathology evaluation of AAV-based biotherapeutics must be designed with the knowledge that AAV effects typically impact only a subset of DRG (and thus a limited number of contiguous spinal nerve roots and nerves). Therefore, capturing DRG changes depends mainly on the sampling strategy, which can be controlled, with a sprinkling of serendipity. In general, multiple ganglia should be collected and evaluated, emphasizing caudal (lumbar and sacral) DRG that appear to be more sensitive than more cranial DRG regardless of how the AAV agent is administered. 20 The sampled DRG should include the dorsal and ventral spinal nerve roots as changes in the DRG do not always align completely with changes in the dorsal roots,20,21 and ventral roots do contain a modest complement of sensory fibers originating from DRG neurons. 41 In instances where white matter changes are present in the spinal cord yet the DRG appear to be unaffected (potentially skewed by the sampling approach), the presence of effects on DRG may be inferred (or extrapolated) as findings in the white matter of the dorsal funiculi depict the sum of axonal changes for all DRG caudal to the spinal cord level present in the tissue section. Additional confidence in interpreting such spinal cord findings may be gained where bilateral symmetry exists for such dorsal funiculus effects although frequently the apparently random variations in the severity of neuronal necrosis in individually affected DRG will result in less than perfect symmetry of lesions. Findings in the DRG (including nerve roots), spinal cord, and nerves are often limited at early time points (14 days or earlier) due to the still-building transgene expression and immune response, variably robust as DRG inflammation and sometimes neuronal degeneration and necrosis rise at intermediate time points (1-3 months), and often limited to minimal mononuclear cell infiltrates and/or glial cell responses (especially satellite glial cellularity in DRG and microgliosis [identified by Iba1 IHC] in spinal cord white matter) at late time points (6 or more months).20,76,118
Infrequently, other neural findings may be seen in preclinical studies of AAV-based test articles. Across multiple constructs, neuronal necrosis sometimes associated with gliosis may be seen in the cerebral cortex, deep cerebellar nuclei, or spinal cord motor neurons. In general, this change affects rare individual neurons, though in exceptional cases the change may impact an entire region (e.g., a cerebrocortical layer or a large brain nucleus). These neuronal changes occur at a distance from the administration site; a possible explanation for this pattern is toxicity to the neuron following anterograde or retrograde transport of the transgene. Small molecule test articles typically initiate neuron degeneration soon after administration, leading to necrotic neurons in a narrow time span of 2 to 4 days post-dosing. 148 In the authors’ collective experience, AAV-induced neuron necrosis occurs later, commonly in the time frame 2 weeks to 3 months post-dosing, in association with the later onset and persistence of transgene expression.
To date, neural neoplasia has not been reported with AAV-based vectors. However, AAV-associated neoplasia has been described in hepatocellular carcinoma in wild-type mice. 47 A 10-year study in dogs treated with a hepatotropic AAV for hemophilia A showed potential effects of integration in hepatocytes beginning at 4 years post-dosing. 115 Current FDA guidance suggests clinical monitoring for 5 years following AAV exposure. 58
Toxicologic Neuropathology of Nucleic Acid Therapies
Since the ASO approach to gene regulation was first proposed in 1978, the field of nucleic acid-based biotherapeutics has experienced a mix of successes and challenges. Fortunately for patients, dedicated scientists have made substantial advances even in times when the technology was nearly abandoned. As a result, at present more than a dozen marketed nucleic acid-based therapies (ASOs, mRNAs, and siRNAs) are available for treating or preventing rare and common diseases in many organs. The pipeline of nucleic acid therapies for severe and life-threatening neural and neuromuscular diseases (e.g., Alzheimer’s disease, ALS, and various muscular dystrophies) has increased exponentially in the past decade. The remainder of this section focuses on nucleic acid therapies targeting neural or neuromuscular diseases. All of the currently marketed nucleic acid agents for neural and neuromuscular diseases are ASOs (both DNA and RNA variants).
In most applications, nucleic acid therapies generally are administered systemically by either IV infusion or subcutaneous (SC) injection. Nucleic acids given by these two routes typically have limited biodistribution, so local delivery systems and conjugation strategies have been employed to more effectively target nucleic acids to the intended site of action. In preclinical testing, mRNAs and siRNAs are still administered either IV or SC, but ASOs have been successfully delivered via the digestive tract (pH-dependent slow-release microparticles), eye (intravitreal injection), nervous system (IT injection), and skin (topical application). The first approved nucleic acid-based therapy (fomivirsen, an ASO) is administered by intravitreal injection to treat cytomegalovirus retinitis. The use of ASOs for CNS diseases was first investigated in the early 2000s, leading to the 2016 approval of nusinersin to treat spinal muscular atrophy. Importantly, the long half-life of nusinersin in the CNS permits a regimen in which the ASO is given three times yearly once steady-state levels have been reached. In the future, additional chemical modifications to the nusinersin backbone could realistically extend the half-life of this ASO in the nervous system and support once-yearly dosing. 24 A pharmacologic advantage of nucleic acid therapies over gene therapies is that nucleic acids will be cleared in time, allowing for potential reversal of adverse effects if they occur.
Upon administration, ASOs and siRNAs enter the cell where they interact with the target mRNA sequence, primarily resulting in degradation in the cytoplasm. ASOs can also enter the nucleus where they can be involved with splice modulation. Unmodified nucleic acids are degraded rapidly in biological matrices, so current-generation test articles undergo chemical modification to increase their RNA-binding affinity and alter their protein binding, thereby increasing the half-life, as well as decrease their pro-inflammatory properties. 96 In general, modifications are made to the phosphate backbone and/or sugar residues of the oligonucleotide. For example, the phosphorothioate (PS) modification is employed in all marketed ASO and siRNA products to stabilize the phosphate backbone, while 2’-O-methyl (abbreviated 2’OMe) or 2’-O-methoxyethyl (abbreviated 2’MOE) conjugation for ASOs or 2’F modifications for siRNAs are used to protect sugar residues from enzymatic degradation.44,142 Such chemical modifications dramatically alter the PK, PD, and toxicology profiles of nucleic acid therapies. The resulting properties can be similar for molecules in a given chemical class but can also be quite variable for molecules in different chemical classes. Moreover, some sequence-specific effects can affect the profile of molecules within a given chemical class. Therefore, generalizations regarding the likely preclinical profiles of nucleic acid test articles should be avoided.
Nucleic acids are classified as small molecules for the purposes of drug development. Therefore, the preclinical development packages for marketed ASOs for neurologic and neuromuscular diseases follow the international harmonised ICH M3(R2) guidance. 84 The medicinal chemistry and drug discovery processes for nucleic acid therapeutics are more efficient than for small molecules because of our comprehensive understanding of RNA biology and how nucleic acid therapies interact with their nucleic acid target and because our past experiences with this platform inform the development of new molecules. When a promising target is identified, thousands of potential oligonucleotide sequences may be generated. The first step toward selecting a candidate molecule is in silico screening to eliminate molecules with known problematic sequences that have unfavorable off-target binding properties. Next, high-throughput in vitro screens in cultured cell lines are utilized to select sequences that are potent and not inflammatory. These sequences are then evaluated in vivo in rodent tolerability screens. The best sequence or sequences are then moved into an in vivo NHP study that, depending on the study objectives, may or may not comply with Good Laboratory Practice (GLP). Finally, the single best sequence is advanced into GLP-compliant toxicity studies in both rodents and NHPs. One major difference in the preclinical development of ASOs compared with traditional small molecules is that short-term studies are generally not performed because oligonucleotides have much longer half-lives than small molecules, so studies of at least 3 months in duration are generally performed to allow steady-state kinetics to be reached. Preclinical packages for nucleic acid test articles generally include in vitro and in vivo mutagenicity assessments, subchronic and chronic toxicity studies in wild-type mice and wild-type NHPs, development and reproductive toxicity (DART) assessments in mice and rabbits, and carcinogenicity bioassays in 1 or 2 rodent species. Safety pharmacology assessments are generally incorporated into an NHP GLP toxicity study but occasionally are performed as a standalone study in NHPs.
The pharmacologic properties of nucleic acids depend on their chemical structure. In preclinical studies, unconjugated oligonucleotides distribute primarily to the kidney (concentrating in the tubular epithelium following glomerular excretion) and the liver (accumulating in sinusoidal macrophages [Kupffer cells] with only a small fraction delivered to hepatocytes). Nucleic acids conjugated to N-acetylgalactosamine (GalNAc), a sugar moiety that strongly binds to a surface receptor found exclusively on hepatocytes, are delivered almost entirely to hepatocytes via receptor-mediated endocytosis. Selective uptake of GalNAc-linked ASOs and siRNAs drastically reduces the administered drug dose needed to obtain the desired pharmacologic effect and, for siRNAs, obviates the need for lipid encapsulation and pre-medication to avoid the infusion reactions that commonly accompany lipid nanoparticle therapeutics.
In addition to chemical structure, the toxicity profile of nucleic acid therapeutics in preclinical studies depends on the route of administration. For ASOs marketed to treat neuromuscular indications, the primary target organ after systemic administration is the kidney, where the oligonucleotides may be visualized in H&E-stained sections within proximal tubule epithelial cells as basophilic cytoplasmic granules. 62 As renal ASO accumulation increases, tubular epithelial cells become progressively vacuolated. At very high doses, tubular epithelial cells undergo degeneration and necrosis, and serum concentrations of renal damage markers (e.g., blood urea nitrogen [BUN] and creatinine) may be elevated. Renal tubular epithelial injury is a tolerable risk because it is monitorable and generally occurs in preclinical studies only at doses far above the clinically efficacious dose in humans. Other findings induced by nucleic acid test articles following systemic administration include altered coagulation, systemic inflammation, and thrombocytopenia.62,73 Neuropathological findings are not observed following systemic ASO administration.
For preclinical studies utilizing IT administration, ASOs are widely distributed in the brain and spinal cord and are taken up by all major cell types including neurons and glial cells. Delivery of ASOs directly to the CNS produces a robust PD effect as shown by substantial mRNA repression throughout the brain and spinal cord. Neurobehavioral observations seen in NHPs following IT injection are generally limited to transient (reversible within 24 hr of dosing) absence of reflexes (especially the patellar reflex), which is common in both control and treated animals (and thus represents in part a procedural effect), but may be of increased incidence with ASO treatment. 97 A possible explanation for this finding is that the lumbar cistern in NHPs (located in the subdural space located at lumbar vertebrae L3 to L6) is quite narrow, which may lead to inadvertent physical trauma to the spinal cord and spinal nerve roots. (In comparison, IT delivery to humans is routine and has a high success rate with a low incidence of side effects.) Neurobehavioral observations in NHPs that have been attributed to IT administration of ASOs and that are not reported to occur in control animals include muscle tremors, hind limb paresis or paralysis, and ataxia. 97 These latter findings are often considered adverse, while transient loss of reflexes is usually interpreted as non-adverse. The most common microscopic findings associated with IT-administered ASOs are granulated or vacuolated macrophages in the meninges (brain and spinal cord) and perivascular (Virchow-Robin) spaces (usually brain) as well as cytoplasmic vacuolation of neurons, both reflective of ASO accumulation, as well as mononuclear cell infiltrates in the meninges of the spinal cord and spinal nerve roots.17,98 These findings are dose- and dose frequency-dependent in severity, may be reversible depending on the dose frequency, and thus are interpreted as non-adverse. Nerve fiber degeneration (minimal to occasionally mild) in the spinal cord and spinal nerve roots may accompany large leukocyte aggregates, especially in rodents, but this finding typically is uncommon and is more frequent near the IT injection site; therefore, it may be caused in part by trauma related to the injection procedure. Other findings often observed in rodents but seldom in NHPs include meningeal fibrosis and spinal cord gliosis. These rodent findings may be of increased incidence and severity in ASO-treated animals.
Investigator-initiated development of nucleic acid therapeutics for single patient (i.e., “N of 1”) clinical trials has rapidly gained momentum in recent years since individuals suffering from neural and neuromuscular diseases may have rare or even unique genetic mutations. A draft FDA guidance for the preclinical development of oligonucleotides for “N of 1” indications has been published recently for test articles with well-characterized chemical properties (including PS, mixed backbone, 2’MOE-conjugated, and phosphorodiamidate morpholino [PMO] variants). 59 The guidance states that general toxicity data with safety pharmacology endpoints in a single species (rodent) are acceptable for a First-in-Human (FIH) trial for a rapidly progressing disease. As little as 2 weeks of in-life data may be necessary to open an investigational new drug (IND) application, with submission of the final report when available.
Toxicologic Neuropathology of Proteins Designed to Cross the Blood-Brain Barrier
Protein-based therapies including monoclonal antibodies, antibody fragments, and fusion proteins exhibit such desirable properties as high specificity, thereby maximizing on-target while limiting off-target effects, and generally long half-lives. 99 Such molecules have the potential to be effective in treating neural diseases, but only if they can cross the BBB in sufficient quantities to achieve therapeutic concentrations. 166
In lieu of direct delivery to the CNS, engineered carrier proteins have shown promise as a platform to facilitate systemically delivered nucleic acid-based and protein-based drugs to cross the BBB. 70 Native carrier proteins tightly regulate nutrient transport required for CNS health and are found on brain endothelial cells, especially in capillaries. A number of transporters have been identified,1,2,151 and several have been explored in animal models.43,121,167 By far the most-studied carrier protein, and the one currently being evaluated in multiple clinical trials, is the transferrin 1 receptor (TfR1). 151 This transporter carries iron via ferritin into cells through receptor-mediated transcytosis, and it is ubiquitously expressed on metabolically active cells that require iron including brain endothelial cells, erythroid precursors, and placental trophoblasts.23,87,126 Compared with standard antibodies, carrier proteins enable the delivery of approximately 10-fold more protein-based drugs through the BBB into the brain.91,160 The remainder of this section discusses the toxicologic neuropathology of carrier proteins as model protein-based biotherapeutics.
Although a variety of TfR1-targeting molecules are in development, antibodies serve as the scaffold for most. The target epitope for these molecules must not compete with or inhibit transferrin binding. One region of the molecule (e.g., the Fc or Fab [“fragment, antigen-binding”] domain) binds TfR1, while another region may bind a target receptor (e.g., TREM2 [triggering receptor expressed on myeloid cells] 156 ) or carry a therapeutic protein (e.g., a recombinant human enzyme 9 ). Within the general framework of the immunoglobulin amino acid sequence (commonly human-derived IgG1), the TfR1 binding domain can be attached to an antibody via a linker, 48 located in one or both Fab regions, 160 or can be provided as a moiety embedded within the Fc domain. 91
To date, neuropathology evaluation has found few effects related to CNS delivery of therapeutic proteins. Accordingly, protein therapeutics have generally been considered to have an excellent safety profile based on the specificity of target binding and lack of off-target effects. Interestingly, conventional anti-amyloid β antibodies have been shown to have a risk of amyloid-related imaging abnormalities (ARIA), which is thought to result from weakened blood vessel walls leading to edema and/or hemorrhage due to on-target efficacy related to the removal of β-amyloid from vascular walls.4,16 With regard to TfR1-based transporter therapies, 89 theoretical concerns associated with TfR1 binding include unintentional down-regulation or degradation of the receptor that would result in iron deprivation to cells 125 and damage to brain endothelial cells resulting from IgG binding (i.e., engagement of the TfR-binding therapeutic) to Fcγ receptors on the effector cells. Neither of these hypothetical phenomena has been identified in vivo.
One carrier protein has been linked to glial changes in a preclinical study. 121 Reactive microglia (indicated by enhanced Iba1 immunolabeling) and astrocytosis (shown by increased GFAP immunolabeling) were reported in rhesus monkeys administered a high-affinity, effector-competent TfR monoclonal antibody (directed against a human TfR1) that also cross-reacted with the primate TfR. 121 The authors speculate that these findings represent either a direct result of the TfR binding or a secondary outcome of effector function. The study included only 1 male and 1 female per dose group, so the conclusions should be interpreted with caution. In one author’s (RM) experience with TfR-binding Transport Vehicles, 91 neither gliosis nor microglial activation have been observed in nonclinical studies as a consequence of TfR binding. In the same study, minimal to moderate nerve fiber degeneration was observed in the sciatic nerve. 121 The cause of the degeneration was not determined.
Similar leukocyte infiltrates may be observed in the CNS of animals treated with TfR-binding test articles. Generally, the incidence of perivascular leukocyte infiltrates is higher in the choroid plexus, meninges, and perivascular spaces in the brain parenchyma of cynomolgus monkeys administered transport vehicles compared with vehicle-treated control animals. Perivascular leukocyte infiltrates are a common background finding in many organs, including neural tissues, in NHPs.35,36,141 Perivascular and intramural leukocyte infiltrates are also a common microscopic change associated with immune complex deposition in blood vessels of many tissues, especially small- to medium-size arteries and arterioles, following exposure to protein test articles.135,155 The incidence and severity of leukocyte infiltrates increase in sites known to have preexisting (i.e., background) infiltrates such as the choroid plexus and meninges as a result of biotherapeutic administration and anti-drug antibody (ADA) formation; the presence of scattered eosinophils and plasma cells is a characteristic feature of these therapeutic-associated leukocyte infiltrates. 114 The novelty of systemically delivered, CNS-penetrating biomolecule therapeutics for neurological indications indicates that reviewing health authorities may not be familiar with the reactions of NHPs to human-derived protein therapeutics. 114 Accordingly, toxicologic pathologists should meticulously articulate the locations and appearances of leukocyte infiltrates in neural tissues and conduct additional assays as needed to provide a strong weight of evidence for interpreting the mechanism and implications of such findings in making adversity decisions and assessing human risk.
Summary
Novel test articles of biological origin including entities of human derivation (e.g., cells, engineered proteins, and transgenes in gene therapy vectors) and modified nucleic acids exhibit considerable promise as exquisitely targeted, long-lasting therapeutics to ameliorate or prevent severe and life-threatening neural diseases. The unique features of novel biotherapeutics generally warrant a case-by-case approach to designing preclinical safety studies, but some common themes should be familiar to all toxicologic pathologists. Bio-derived test articles typically must be administered directly to the CNS to ensure entry in therapeutically relevant levels although certain platforms now allow systemically delivered biomolecules to circumvent the BBB. Biotherapeutics of all classes commonly induce leukocyte infiltration (usually mononuclear cells, mainly lymphocytes and macrophages) in the choroid plexus, meninges, and perivascular spaces in the CNS and less often PNS organs. In some cases, biotherapeutic test articles also damage the neural parenchyma in a consistent fashion, such as scattered neuron degeneration and necrosis of sensory neurons in ganglia following the administration of AAV-based gene therapy vectors. More importantly, direct CNS delivery (especially into the parenchyma) may elicit locally extensive injury at the injection site, and such procedure-related effects will need to be differentiated (if possible) from test article-related effects. Primary damage to neurons, neuronal cell processes, and myelin in the CNS typically is interpreted as adverse in the context of the particular preclinical study since such effects are not subject to effective repair, 117 but such injuries may be acceptable on a case-by-case basis in the context of an overall risk-benefit analysis. 94 For severe and/or life-threatening neural diseases, an “acceptable” (or manageable) level of test article-related harm is implied by neuropathological findings that are modest (minimal [even if multifocal] or localized and mild), not progressive, and unaccompanied by consequential (especially persistent) in-life behavioral or neurological deficits. Glial reactions to direct CNS delivery of bio-derived test articles often occur in the absence of visible damage to the neural parenchyma and therefore may be interpreted to reflect a non-adverse adaptive response to altered microenvironmental conditions rather than as evidence of neurotoxicity. 25 Pathologists and toxicologists who participate in the evaluation of novel bio-derived therapeutics should be prepared to play a leading role in the interpretation and communication of research data to properly position the main messages for regulatory reviewers.
Footnotes
Acknowledgements
The authors gratefully thank the STP Annual Symposium Committee (specifically the Continuing Education course subcommittee) for allowing the authors to present a course covering the points reiterated in this article and Ms. Beth Mahler for her invaluable assistance in optimizing the annotation and resolution of the figures.
Author’s Note
This synopsis paper summarizes the contents of a ½-day continuing education course organized by the Special Interest Group for Neuropathology (SIGN) of the Society of Toxicologic Pathology (STP). The course was presented at the annual STP symposium in Summerlin, NV, on June 25, 2023.
Author Contribution
The analysis, conclusions, and opinions expressed in this article are solely those of the authors.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.