
Abstract
The Society of Toxicologic Pathology’s Annual Virtual Symposium (2021) included a session on "Regulatory Perspectives on Juvenile Animal Toxicologic Pathology." The following narrative summarizes the key concepts from the four talks included in this symposium session chaired by Drs Deepa Rao and Alan Hoberman. These encompass an overview of various global regulations impacting the conduct of juvenile animal studies in pharmaceutical drug development and chemical toxicity assessments in a talk by Dr Alan Hoberman. Given the numerous regulatory guidances and legal statutes that have covered the conduct of juvenile animal studies and the recent harmonization of these guidances for pharmaceuticals, Dr Paul Brown provided an update on the harmonization of these guidances for pharmaceuticals, in the recently finalized version of the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use S11 guidance document, “Nonclinical Safety Testing in Support of Development of Pediatric Medicines.” The first two talks on regulations were followed by two talks focused on an evaluation of the postnatal development of two major organ systems relevant in juvenile animals. Dr Aurore Varela covered study design and endpoints impacting the skeletal system (bone), while Dr Brad Bolon presented a talk on the study design and conduct of neuropathology evaluations for the developing nervous system.
Keywords
Introduction
Nonclinical testing in juvenile animals is relevant for the toxicity hazard identification and characterization of xenobiotic exposures to infants, children, and adolescents, as well as for the risk assessment of pediatric pharmaceutical formulations. Unlike general toxicology studies in young adult and adult animals, assessment of toxicity in juvenile animals requires evaluation of direct effects, as well as an evaluation of any effects on sexual maturation, growth, and potential effects that might not be evident until later in life after exposure to the chemical has stopped. The time period for juveniles covers premature newborns, neonates, infants, children, and adolescents in humans, reflecting a time period inclusive of postnatal development for multiple organ systems as well as indirect exposures (in utero and/or via milk). Hence, studies to assess toxicity in juvenile animals generally have increased complexity with regard to study design and evaluation.
Numerous guidances or guidelines from regulatory authorities in response to legislative initiatives have existed for safety evaluation of juvenile populations via animal toxicology studies. Because the evaluation of safety for infants including neonates and premature infants is closely associated with reproductive toxicology studies, guidances for reproductive toxicology have traditionally covered some aspects of juvenile toxicity testing. For example, with chemicals, the Organisation for Economic Co-operation and Development (OECD) has two general guidelines that have been globally adopted (OECD 416—Two-Generation Reproduction Toxicity Study 1 and OECD 443—Extended One-Generation Reproductive Toxicity Study 2 ) that typically cover juvenile exposures. In addition, two OECD guidelines directed specifically at the assessment of (developmental) neurotoxicity include OECD 424—Neurotoxicity Study in Rodents 3 and OECD 426—Developmental Neurotoxicity Study. 4 In addition to OECD regulations for chemicals, within the United States, the Environmental Protection Agency (EPA) also has two guidelines for (developmental) neurotoxicity assessment. These include EPA 870.6200—Neurotoxicity Screening Battery 5 and EPA 870.6300—Developmental Neurotoxicity Study 6 . More details on the evaluation of chemical toxicity in developing animals was covered by Dr Wolfgang Kaufmann in session 1 of this Virtual Annual Symposium, and previous publications on this topic include Kaufmann and Gröeters, 7 Bolon et al, 8 Garman et al, 9 and Spencer et al. 10
Pharmaceutical legislation impacting drug development of pediatric formulations was initially driven by the Food and Drug Administration Modernization Act (FDAMA), 11 the Best Pharmaceuticals for Children Act (BPCA) 12 in FDAMA, and the Pediatric Research Equity Act (PREA). 13 Prior to permanent reauthorization of BPCA and PREA in 2012, “more than 80% of the drugs approved for adult use were being used in children, although safety and effectiveness had not been established in children. By 2012, that number had been reduced to 50%.” 14 Older guidances based on legislation include regional documents from the Food and Drug Administration (FDA), 15 the European Medicines Agency (EMA), 16 and the Ministry of Health, Labour and Welfare in Japan (MHLW) 17 that were used by the respective global sites. However, because of the variability in the nonclinical plans proposed by companies within the pharmaceutical industry and because of conflicting recommendations on major aspects of the nonclinical development program among and within regulatory bodies, the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) convened an S11 Expert Working Group 18 in 2014. The result of this ICH Expert Group was the recent finalization (April, 2020) of Nonclinical Safety Testing in Support of Development of Pediatric Pharmaceuticals (ICH S [Safety] 11) 17 guidance that documents nonclinical information impacting pediatric pharmaceuticals. The ICH S11 guidance for nonclinical animal toxicology studies is intended to complement and expand on the existing clinical ICH efficacy guideline (“E11 Clinical Investigation of Medicinal Products in the Pediatric Population” [ICH E11]) 19 released in 2017). The ICH S11 guidance is primarily for small molecule and biologic pharmaceuticals and to some extent, pharmaceuticals for pediatric oncology (in conjunction with ICH S9 20 ). Tissue-engineered products, gene and cellular therapies, or vaccines are not included. However, in addition to the safety of the active ingredient in the formulation, nonclinical studies in juvenile animals may be required to support the safety of excipients based on the history of excipient disasters in children. 21- 23
In general, nonclinical studies in juvenile animals provide supporting data for safety evaluation for the conduct of pediatric clinical trials for new pediatric-only drug products, for drug products approved in adults and to be used in pediatric populations, as well as for drug products already approved in nonpediatric (adult) populations. In addition to general study design guidelines to evaluate toxicity in juvenile animals, attention to specific organ systems is also needed based on the fact that some organ systems undergo significant postnatal development, and for some organs (eg, brain) different regions undergo peak differentiation across different time periods during development. Organ systems with postnatal development include the nervous, reproductive, pulmonary, renal, skeletal system, gastrointestinal, hepatobiliary, and immune systems. Given the variable rates of postnatal development during different periods in childhood, drugs potentially targeting specific organ systems usually require specific assessments in juvenile animal toxicology studies to ensure safety prior to exposure in pediatric populations.
The following narrative sections are summaries that include key highlights from four talks that composed the Symposium Session entitled “Regulatory Perspectives on Juvenile Animal Toxicologic Pathology” chaired by Drs Rao and Hoberman. This half-day session occurred at the Society of Toxicologic Pathology’s Annual Symposium held virtually on Tuesday, June 29, 2021. The first talk by Dr Alan Hoberman covered a history of legislation, an overview of guidances impacting both developmental neurotoxicity studies (DNTS) for chemicals and for juvenile animal studies (JAS) for pharmaceuticals, and general aspects of study design impacting DNTS and JAS. This was followed by a talk from Dr Paul Brown who covered a review of the recently finalized ICH S11 for JAS. Due to particular vulnerabilities unique to developing animals, study designs in juvenile animal toxicology studies typically explore potential test article effects in selected organ systems. This symposium session included talks on two organ systems: Dr Aurore Varela covered an overview of the assessments in the skeletal system (bone), while Dr Brad Bolon covered an overview of neuropathology end points. This manuscript is tailored to be a descriptive narrative on the proceedings and highlights of the symposium session and not meant to provide a detailed resource or even a comprehensive review on this topic.
Overview of Regulations Impacting JAS
Dr Alan Hoberman provided an overview of various global regulations impacting conduct of JAS in pharmaceutical drug development as well as chemical toxicity assessments including multigenerational and DNTS. The need to protect pediatric populations is recognized in the ICH E11 guideline 19 : “Pediatric patients should be given medicines that have been appropriately evaluated for their use. Safe and effective pharmacotherapy in pediatric patients requires the timely development of information on the proper use of medicinal products in pediatric patients of various ages and, often, the development of pediatric formulations of those products.” The ICH S11 safety guideline for nonclinical testing of pediatric pharmaceuticals was finalized in 2020, harmonizing guidelines issued by various regulatory authorities in the early 2000 and various rules issued from the late 1970s, by regulatory agencies (EPA), and the OECD. In view of poisoning of hundreds of children in the late 1930s from the vehicle used for sulfanilamide, the neural seizures produced by hexachlorophene disinfectants, gray baby syndrome from chloramphenicol, adverse effects of various excipients, and the effects of tetracyclines on bone growth, it remains questionable why guidelines for pediatric testing were not promulgated earlier.
The unintentional exposure of juveniles to chemicals in the environment has been recognized for many years dating back to the early 1970s, and many of the methodologies, especially for evaluation of the function and anatomy of the nervous system that were incorporated into testing guidelines for chemicals, are now being adapted for pediatric testing of pharmaceuticals.
There are several developmental changes and physiologic factors that influence drug disposition in infants, children, and adolescents. These changes are related to age-dependent differences in metabolic capacity, total body water, body fat, integumentary development, gastrointestinal, and renal function. 24 Exposure to juvenile animals covers the gap between exposure of fetuses in utero and/or neonates from milk, until exposure would start in adult toxicity studies (Figure 1).
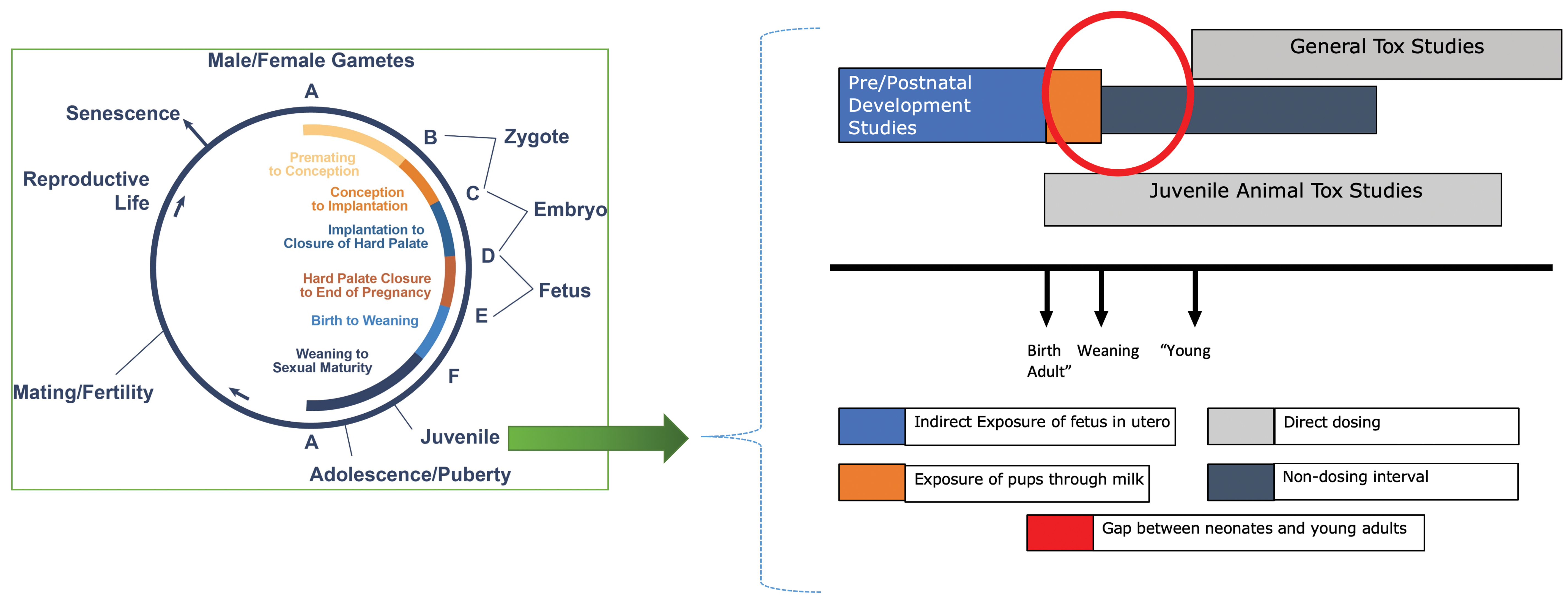
Life cycle stages and dosing periods in juvenile animal toxicology studies. The figure was adapted from chapter 1 page 14 of Pediatric Non-Clinical Drug Testing: Principles, Requirements and Practices, edited by Hoberman and Lewis. 25
The first US guidance was issued for pediatric testing in 2006. This was followed in 2008 with guidance from Europe and in 2012, the guidance from Japan. As previously noted, the harmonization of these guidance documents was accomplished together in 2020 with the ICH S11 guidance.
In addition to guidance, legislation was promulgated in the United States and the European Union. In the United States, the PREA mandated a Pediatric Study Plan (PSP) from drug developers and a review of the PSP by a special FDA committee, the Pediatric Review Committee. In the European Union, a Pediatric Investigation plan (PIP) to be reviewed by the Pediatric Committee was established. Both documents ensure that exposure to juveniles was at least considered during drug development.
The PIP is due at the end of phase 1 clinical trials, while the PSP is due during phase 2 clinical trials. For each plan, the regulatory agency is required to respond, and the documents moved to finalization, to produce an agreed-upon plan for development of the drug for pediatric use. Typically, even for drugs not targeted for pediatric populations, a PSP/PIP is still required to justify a waiver for not conducting any testing in juvenile animals.
The testing designs and evaluation included in a typical juvenile toxicity study can include study use procedures that have been in use for the hazard assessment of chemicals since the early 1990s. Chemical testing has always recognized that exposure can occur at any point in the life cycle. Guidelines including the OECD 416 for multigenerational studies, and the more extensive OECD 443 extended one-generation study (Figure 2), covers direct exposure of animals at human equivalent ages from 2 years to adulthood, with additional indirect exposure to the pups from prior to birth (in utero) and/or through milk. Specifically, for developmental neurotoxicity, the OECD 426 guidance has been covering these evaluations since 2007.
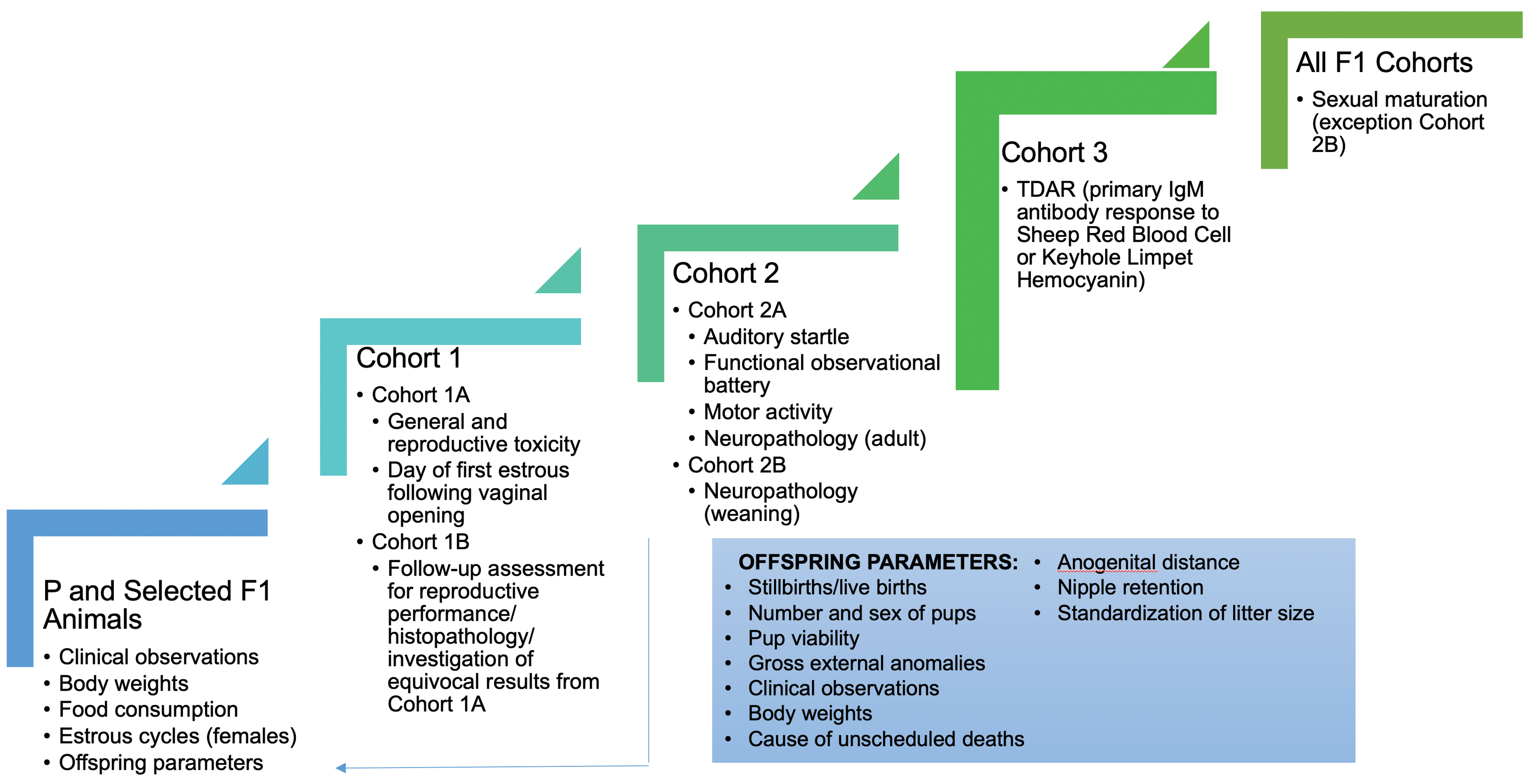
End points collected for F1 generation in an extended one-generation reproductive study Organisation for Economic Co-operation and Development (OECD) guideline 443.
Prior to conducting a juvenile toxicity study, one must determine the key factors that impact study design. These include the need for testing (will juveniles be treated), number and type of species (usually one when adult nonclinical data already exist), the duration of treatment (several weeks, or chronic), potential translatable end points relevant to the clinical trials, and the target organs from adult toxicity studies. Each juvenile study design is unique varying by species, strain, age at dosing initiation, and stage of organ system development. One should also keep in mind that the pediatric formulation for children may be different from the adult formulations or tablets, thereby necessitating the potential safety evaluation of excipients. Figure 3 illustrates comparable timing in ages of development across species.
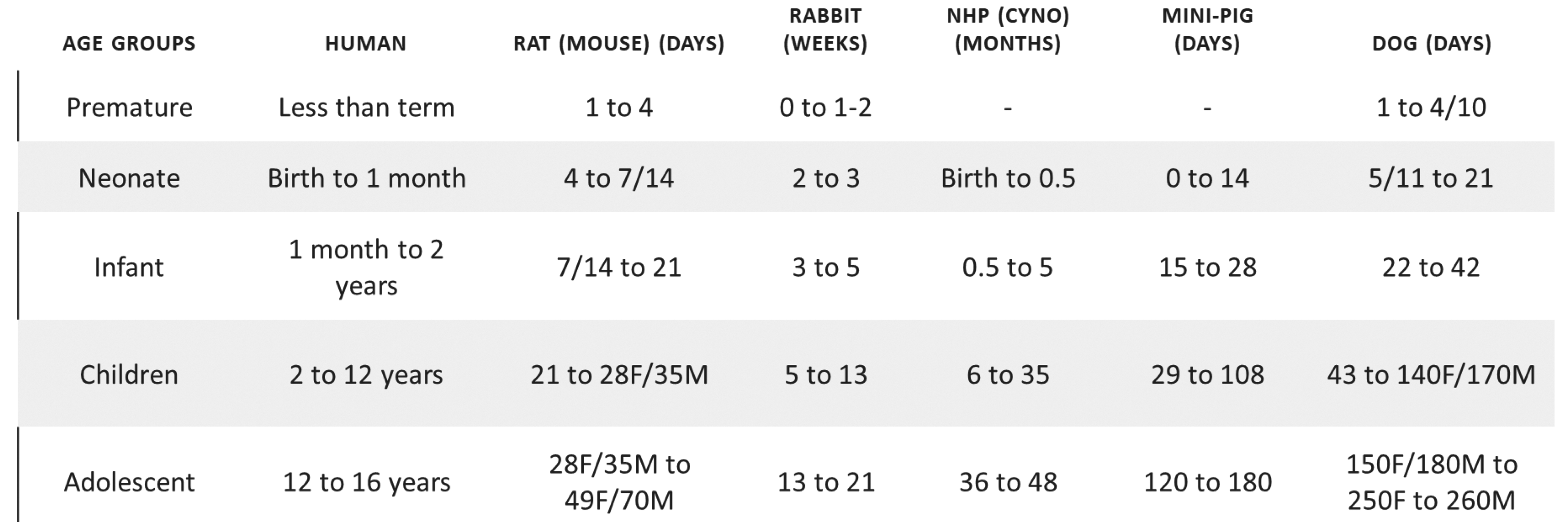
Dosing guide: matching ages between animals and humans through postnatal development.
Common to many juvenile toxicity designs, neurobehavioral measures reflecting end points of function are included. Specific testing paradigms are not included in the ICH guidelines. The ICH S11 guidance includes reference to a functional observational battery (FOB), assessment of locomotor activity, evaluation of coordination and reflexes, acoustic startle response, and learning and memory. No specific testing paradigms are noted, and numerous tests and test methodologies are available. A testing laboratory should use methods that have been validated with positive controls, have trained personnel with standard operating procedures in place, and be able to evaluate data using a priori statistical analyses. Most testing paradigms have been developed for rodents, and these are the preferred species. However, if other species are used, there are some tests that can be conducted in rabbits (FOB, eye blink), dogs (modified FOB), and nonhuman primates (modified human pediatric screens and neurological examinations).
Neurobehavioral tests can be divided into several categories including reflex ontogeny/sensory motor function (reflex tests, eg, auditory startle ontogeny, components of the FOB), motor activity (beam break, video monitoring), reactivity (auditory startle, components of FOB), and learning and memory (water maze, avoidance paradigms). As noted previously, a laboratory should use the tests that they have the most familiarity with, but the test must also be appropriate for the mechanism of action of the drug. Illustrative of this is the difference in types of learning that are evaluated in the Morris water maze versus the Biel or Cincinnati water maze. Water mazes are appropriate for juvenile studies because they can be conducted without food deprivation that can affect the growth and development.
A Morris maze is a large circular maze with an escape platform placed just under the water, so the escape is invisible to the animal. This maze tests allocentric or spatial learning. Rodents must learn the location of the platform based on clues throughout the testing room or position of objects in relation to the escape platform. Biel or Cincinnati mazes measure egocentric learning and are composed of a series of connected alleys that require the animal to learn the length of each alley and remember turns to take to find the submerged escape platform. Animals do not have to key off objects in the room for this test. Morris mazes use the hippocampal subregions (dentate gyrus, cornu ammonis CA3 and CA1). Each of these areas are known to be important in learning and memory based on object recognition. Specifically, the dentate gyrus is important for identifying different patterns with overlapping information (eg, the visual cues used for navigation). The CA3 and CA1 regions are known for retrieving previously stored information and remembering corresponding associations (such as where the visual cues were in the prior day’s test session).
A Biel or Cincinnati maze utilizes the striatum that is part of the motor circuits relayed through the basal nuclei. This circuit is an important circuit from motivation to movement. It starts with the limbic loop that involves brain regions associated with motivation, then transitions into the prefrontal loop that involves brain regions associated with cognition and decision-making, then to the oculomotor loop where it is about orienting in the direction of the decision, and finally to the body movement loop where the action is executed. This circuit is integral in executing each directional choice involved in the Biel or Cincinnati water maze task.
A relatively new test for safety evaluations is the social interaction test. The test is conducted in two sessions. Session 1 evaluates the inclination of the subject rat to socialize or “affiliate” with an unfamiliar rat (ie, not a littermate and not a cagemate). The rats are observed for typical social behaviors (eg, sniffing, grooming, following). Session 2 evaluates the preference of the subject rat to socialize with the “affiliate” rat from session 1, as opposed to a third rat (ie, also not a littermate and not a cagemate) that is considered “novel” to the test. The two-session social interaction test has been used in research to investigate disorders such as autism, bipolar disorder, depression, and schizophrenia. It has been requested by the FDA in the evaluation of drugs affecting the prefrontal cortex.
Following neurobehavioral testing, neuropathology can be conducted to look for anatomical changes in the central nervous system (CNS) and peripheral nervous system (PNS). These techniques were discussed by Dr Bolon. In conclusion, the take home messages from this presentation are:
− Children are not small adults; physiological differences are present that change over time.
− Although juvenile toxicity testing guidelines were not issued until the 2000, the susceptibility of infants and children to agents that were recognized as safe in adults has been known since the 1930s.
− Juvenile toxicity study designs need to consider comparative species development.
− Rodent models allow evaluation of both neurobehavior and neuropathology.
Update on ICH S11: Nonclinical Safety Testing in Support of Development of Pediatric Medicines
Dr Paul Brown, who served on the ICH S11 Expert Working Group, provided the background, update, and an overview of the recently finalized ICH S11 guideline impacting the conduct of JAS for pharmaceutical products. An increased emphasis on pediatric drug development in recent years and the long-standing recognition of the differences in response to drugs between adult and pediatric subjects has led to a greater conduct of clinical trials in pediatric subjects and the nonclinical JAS to support such trials. The ICH endorsed the concept of developing a guidance on the nonclinical safety studies important to support a pediatric development program in 2014. Specific problems identified at that time included: Lack of harmonized criteria for determining when all previous animal data and human safety data are considered insufficient to support pediatric studies. Lack of harmonization of specific aspects of study design of the JAS where it is determined that studies are appropriate. Lack of guidelines describing in detail the studies that need to be performed to support a pediatric-only development program with no indication in adults.
In addition to the existing three regional guidances (FDA, 15 EMA, 16 and Japanese MHLW 17 ) that provided a foundation for the new ICH S11 guidance, the ICH Working Group conducted surveys of the pharmaceutical industry and regulatory submissions to better understand the current practices and the utility and impact of the nonclinical studies conducted to support pediatric drug development programs. The draft guidance was published for public comment in 2018. After the Working Group addressed the comments, the ICH Assembly signed off on the final ICH S11 26 guidance in April 2020.
The final guidance describes a weight of evidence (WoE) approach (Figure 4) to determine when nonclinical toxicity studies may be recommended in juvenile animals to support development of medicines to be used in pediatric patients. The potential nonclinical support needed for pediatric drug development should be considered early in an overall drug development program. It may be appropriate to consider the WoE approach several times throughout the development of a drug as additional data are accumulated and/or if the intended use of the drug changes. Major factors, although not necessarily all factors, to consider in the WoE include the clinical context (indication, age group, etc), the pharmacology and pharmacokinetics (PK) of the drug, the existing clinical and nonclinical information about the drug, and the feasibility of conducting nonclinical studies in juvenile animals. If the WoE conclusion suggests that additional nonclinical studies are recommended, the guidance also provides appropriate study designs.
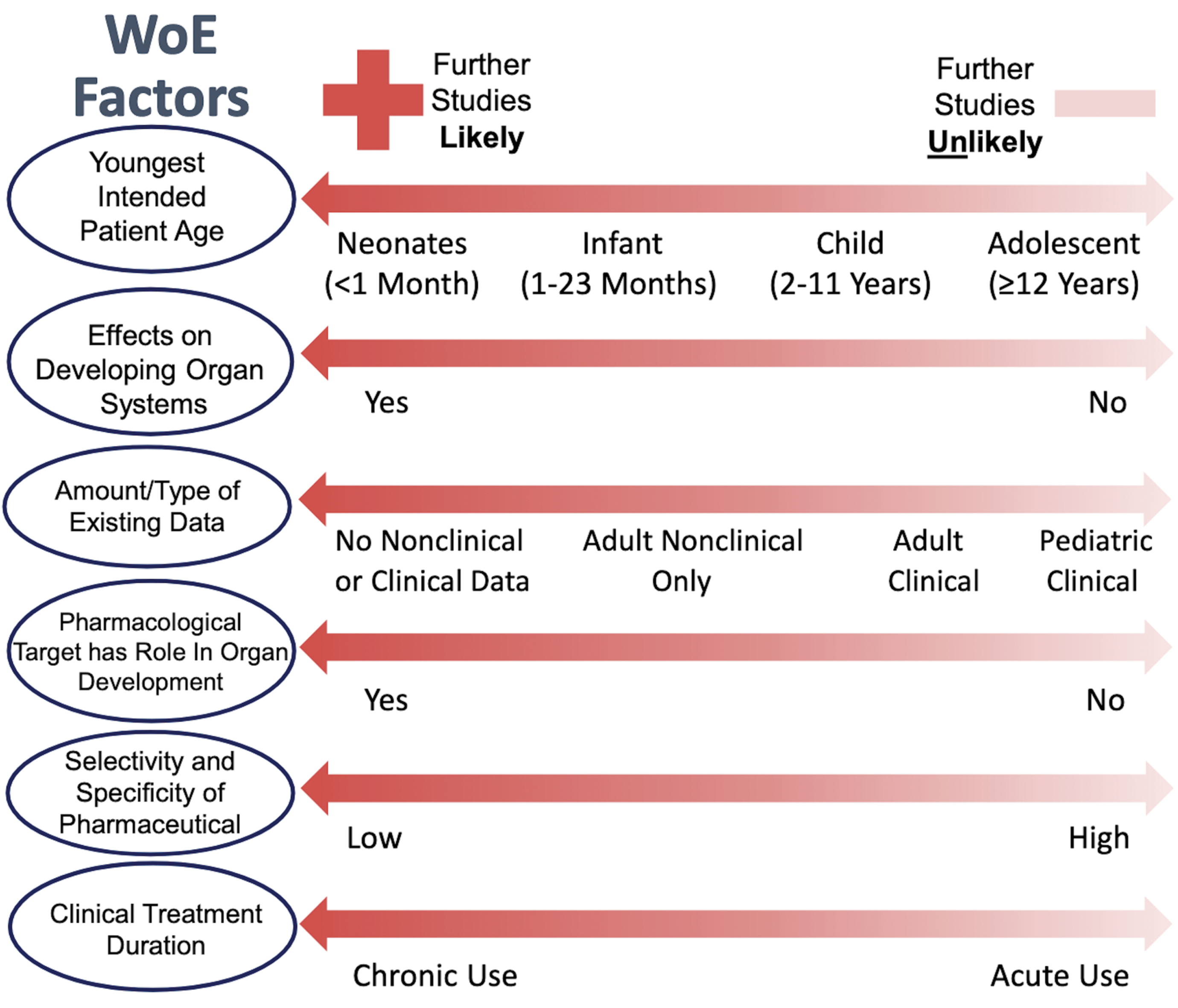
Key weight of evidence (WoE) factors to be considered in determining if nonclinical studies are warranted. The most important factors that should be highly weighted (listed first) are the youngest intended patient age and whether there are suspected adverse effects on developing organ systems of patients during the conduct of the pediatric trial. The other factors are not listed in order of importance (excerpted from ICH S11 guideline).
The guidance describes study designs as consisting of a core set of end points that can be supplemented by additional end points depending on the concerns identified in the WoE approach. Core end points include:
Mortality and clinical observations,
Growth (eg, body weight and long bone length),
Postweaning food consumption,
Sexual development,
Clinical pathology,
Anatomic pathology, and
Toxicokinetics.
Additional end points can include:
Other growth end points,
Bone assessments,
Expanded clinical pathology,
Expanded anatomic pathology,
Ophthalmologic examinations,
CNS assessments,
Reproductive assessments, and
Immunologic assessments.
Dose range finding (DRF) studies for juvenile animals are recommended to assist in the appropriate design of definitive studies including considering whether dose adjustments may be appropriate during the study to maintain adequate drug exposure.
An appendix was included in the ICH S11 guidance with tables of the age-dependent development of various organ systems in human, rat, dog, minipig, and cynomolgus monkey. These tables can aid in selecting the appropriate animal species and age to represent the human developmental period and organ system of concern.
The guidance notes that the dosing period used in a JAS may not always correspond to the proposed clinical treatment duration in humans. Rather, the safety concern and the relevant period of organ development need to be considered. In some cases, a relatively short period of treatment in juvenile animals may be sufficient to address an issue (eg, kidney development in the rat), whereas in other cases a longer duration of treatment in the juvenile animal may be appropriate to cover all vulnerable developmental time periods even if the clinical treatment duration is short (eg, CNS concerns).
Post-treatment (recovery) assessments are recommended for JAS to address the potential reversibility, persistence, or progression of any findings and to assess the possibility of any delayed onset of effects.
When selecting doses for JAS, the guidance notes that it is desirable to obtain a dose response and to establish a no observed adverse effect level. Some overlap with doses/exposures in adult animal studies is also desirable to allow comparison between juvenile and adult studies. Ideally, the high dose used in a JAS should not result in marked toxicity that could confound growth and development end points and complicate study interpretation. In some cases, it is appropriate to consider dose adjustments during a JAS to compensate for changes in drug exposure which can be related to the development of the animals during the study.
The ICH S11 guidance also describes the potential approaches for the nonclinical support of drugs that will be developed only for use in pediatric patients or that will be first tested in pediatric patients. When no adult clinical trials are conducted prior to pediatric trials, JAS in two species are recommended. In general, the conduct of JAS in primates, while potentially appropriate in some situations, should be carefully considered and alternatives used when possible.
The ICH S11 guidance provides a few pointers on data interpretation. For example, the value of having age-matched concurrent control data to enable distinguishing drug-related effects from normal age-dependent organ changes is noted. Once results of a JAS are obtained, the results should be integrated with all other available data on the drug so the findings can be related to the intended pediatric use.
The guidance recommends that the WoE approach be used also for assessing excipients that will be used in pediatric formulations. If insufficient data are available to support the safe use of an excipient in a pediatric formulation, then further safety evaluation is warranted.
The guidance notes that the principles related to combination drug products described in the ICH M3(R2) guidance (“Nonclinical Safety Studies for the Conduct of Human Clinical Trials and Marketing Authorization for Pharmaceuticals”) also apply to pediatric drugs. In those cases where previous information on the combination is not considered sufficient to support the pediatric use, a JAS could be appropriate. It may be possible to study only the combination or to include a combination arm in another JAS of one of the individual drugs if such a study is already being conducted.
As noted in the guidance, early consideration of nonclinical support for pediatric pharmaceutical development as well as interaction with regulatory authorities to seek more specific advice is recommended. The guidance is intended to promote harmonization of recommendations for the nonclinical support of pediatric drug development and should facilitate the timely conduct of pediatric clinical trials and reduce the use of animals in accordance with the 3R (replace/reduce/refine) principles.
Study Design and Focus on Bone End Points in Juvenile Toxicology Studies
Dr Aurore Varela discussed the end points impacting the developing skeletal system in juvenile toxicology studies. Bone evaluations in juvenile studies are important end points in evaluating pharmaceutical effects, given that the skeletal system has significant postnatal growth and that numerous pharmaceuticals have been demonstrated to affect the skeletal system. Drugs impacting bone structure and/or function include bisphosphonates and denosumab (bone antiresorptive agents), parathyroid hormone and antisclerostin antibodies (bone anabolic agents), peroxisome proliferator-activated receptor-γ agonists, fibroblast growth factor agonists (antidiabetics), tenofovir (antiviral), clenbuterol (β2-adrenergic agonist), and omeprazole (proton pump inhibitor) to name a few. 27- 32 Evaluation of bone in developing animals can initially include comparatively simple measurements of growth that are easily incorporated into standard toxicology study designs. This section covers various aspects of the evaluation of the skeletal system in JAS.
Identifying bone as a target tissue in drug development to address any potential for liability is important as data emerge to show that signaling pathways may positively or negatively affect the skeletal system. Regulatory guidances for juvenile toxicology studies include those from the FDA, 15 EMA, 16 Japanese MHLW, 17 and the recent ICH S11. 26 These guidances indicate that the skeletal system is an “organ system of concern” in juvenile animals and that growth should be assessed by body weights in conjunction with long bone length.
In general, if there is no specific concern related to a bone liability, minimal requirements include simple physical measurements such as long bone length measurements along with body weight and a crown-to-rump measurement. Integration of these data into toxicology studies, including in the pre- and postnatal reproductive toxicology studies, should be routinely performed as part of nonclinical drug development. However, when there is an identified concern about bone metabolism or structure, the measurements of bone-related biomarkers and/or expanded histopathology (eg, histomorphometry) should be considered. Additional assessment of bone mineral density (BMD; eg, microcomputed tomography [micro-CT], dual-energy X-ray absorptiometry [DXA], peripheral quantitative computed tomography [pQCT]), bone structure (eg, micro-CT), or bone quality (eg, biomechanical strength testing) can also be conducted as appropriate. 33
Factors impacting study design include species, sex, and study duration. Species selection is an important point to consider, and rodents are a frequently accepted and recommended species for testing bone compounds. Although rodent models do not completely reflect human bone responses, they provide a detection of a signal in bone with wellpowered studies where bone end points are well validated. Dogs and nonhuman primates are similar to humans with respect to intracortical bone remodeling but have a more rapid turnover. Males of all species are often more sensitive than females as they have a higher growth rate on a longer time frame. Study duration might vary depending on the species and its duration of the bone turnover cycle. In rodent juvenile studies, the study duration should at least cover the rapid phase of growth (ie, up to 13 weeks of age).
Although, in most cases, qualitative histopathology evaluation of decalcified hematoxylin and eosin (H&E)-stained bone tissue sections has sufficient sensitivity to detect major pathological changes, the standard microscopic evaluation of decalcified H&E stained bone sections is not a very sensitive technique to evaluate changes in bone mass and geometry. There are several established imaging modalities that can be used to evaluate bone end points, but each modality has its own advantages and disadvantages. For example, in vivo evaluations of the skeletal system include radiology to identify abnormalities (focal lesions, abnormal shape, and other radiological findings including growth plate assessment); biochemical markers of bone turnover; and measures of bone mass, density, and geometry. Ex vivo measurements include bone architecture, bone dynamics, and bone strength as needed. More detailed information regarding these advanced techniques may be found elsewhere. 33,34
Growth and development of the skeleton can be assessed through important postnatal milestones including the appearance/fusion of secondary ossification centers, longitudinal bone growth, diametric bone growth (bone expansion), and bone accrual. 35 Growth is completed when adult longitudinal size (ie, length and diameter) is attained, while the skeletal maturity is achieved by the closure of the physes (“growth plates”). Peak bone mass assessed by bone mineral content (BMC) and/or BMD is a determinant of bone strength when associated with bone geometry and bone material properties. In many nonrodent species including humans, skeletal maturity occurs after the growth plate closure and the end of longitudinal bone growth. Generally, peak bone mass occurs after the closure of the epiphyseal growth plates and varies by sex and skeletal site. These sex differences for the timing of growth and development of the skeleton are also encountered in animals, and marked differences are present among species. 36 The sequence of growth plate closure is generally similar in mammals. Given the variability among sites, bone end points in animals commonly are evaluated in one long bone from the appendicular skeleton (femur or tibia) and bones from the axial skeleton (usually lumbar vertebrae).
Radiology remains an essential tool of skeleton evaluation and provides a simple and commonly available technology, although it requires a skilled and experienced veterinarian or veterinary radiologist for interpretation. Bench-top radiography units are a cost-effective means of screening the entire skeleton of rodents and isolated intact bones of larger nonrodents. Radiology is a straightforward means for examining the size, shape, skeletal abnormalities, and overall density of the entire skeleton or large subdivisions thereof when the skeletal elements are positioned in a stereotypical orientation (eg, dorsoventral and lateral views). Longitudinal evaluations on skeletal growth (by direct measurements of length and width of specific bones) and development of bones and growth plate assessments are also key in juvenile toxicology studies. Radiological diagnosis can effectively support histopathological diagnosis and often is helpful in deciding which bone locations should be examined microscopically to best characterize the disease process or treatment effect. 37
Bone densitometry techniques (DXA, pQCT, and micro-CT) provide information on BMC, BMD, and bone geometry. Data derived from bone biomarkers and bone densitometry can be used to trigger more in-depth analyses using histomorphometry to derive quantitative data on bone cell function. 33 If appropriate, bone strength can also be measured. In vivo body composition assessments of lean mass and/or fat mass can be captured simultaneously and further characterized histopathologically or quantitatively using histomorphometric techniques. These studies can be run using all standard and specialized routes of administration in the major rodent and nonrodent laboratory species. The DXA limitations are important to understand especially in the context of juvenile studies: two-dimensional projections of a three-dimensional structure (effect of bone size and shape) are good but not perfect approximations of the true values, geometrical evaluations and effects on trabecular and cortical bone density and dimensions cannot be made, and body size and composition can impact BMD measurement. The BMD data are influenced by bone size and the composition of surrounding soft tissue. Therefore, DXA data can be influenced by treatment effects on body weight where growth may affect bone size, or treatment may affect body composition (lean mass vs fat mass). However, DXA data from nonclinical animal studies provide clinically relevant data regarding bone density effects. Other techniques, primarily pQCT, can compensate for DXA limitations and are recommended for use in nonclinical JAS in addition to, or instead of, DXA. For example, pQCT scans provide information on bone mass (BMC), volumetric BMD, bone size and geometry (total, trabecular and cortical bone areas, cortical thickness, periosteal, and endosteal circumferences), and cross-sectional moment of inertia, an indicator of bone mass and its distribution in the bone used to derive surrogate measures of bone strength. Peripheral quantitative computed tomography allows differentiation between trabecular and cortical bone and allows assessment of changes in size and geometry along with bone mass and density which are critical to assess and understand changes in relation to all aspects of bone growth and development.
Bone histomorphometry is an important tool for assessing changes in bone tissue and to characterize the mechanisms by which a test article acts on bone. Bone histomorphometry provides valuable information about bone structure, bone formation, bone resorption, bone mineralization, and bone modeling/remodeling activity. Bone histomorphometry nomenclature has been standardized. 38,39 Fluorochrome labeling with calcium chelators can be employed during a study if effects upon bone formation are suspected. Dynamic bone histomorphometry is critical to assess bone turnover and mineralization in trabecular or cortical bone. Injections of fluorochrome label are usually performed on at least 2 occasions prior to bone harvesting. Intervals for injecting labels depend not only on species but also of the age of the animals as bone cycle can be shorter in juvenile animals. Although formalin-fixed tissues are typical for histopathologic evaluation and bone histomorphometry, the decalcification used in preparing routine paraffin-embedded bone sections for routine qualitative histopathology cannot be employed for histomorphometry as sections for the latter quantitative technique must be undecalcified. Accordingly, formalin-fixed bones slated for histomorphometry are embedded in plastic (eg, methyl methacrylate) as detailed in Varela and Jolette. 33
Bone strength constitutes a critical end point in skeletal assessment, serving as the gold standard of bone quality. 40,41 Biomechanical testing of bone constitutes the unique opportunity to evaluate the functional impact of alterations in bone turnover, bone mass, and bone geometry. Statistical correlations between biomechanical measurements and bone densitometry parameters provide important insights into the effects of drug treatment on bone quality. Bones are scanned (DXA, pQCT) to obtain information on bone geometry to normalize strength parameters for bone size. Bone density and biomechanics parameters are used to assess the relationship between bone mass and strength. If an “apparent” effect on the skeleton is only related to body weight, normalization of data could help in the data interpretation: pQCT will reliably provide measures of BMC and BMD as well as bone geometry that will reflect size, while biomechanical testing showing parameters unaffected when corrected for bone size will support those skeletal changes related to body weight but those that did not affect bone quality.
As with any technique, standardization procedures are key for data reproducibility including technical training, animal or specimen positioning, and anatomical landmarks (particularly critical in growing skeleton where morphology can vary very rapidly). Site selection is also critical due to differential growth rate and sequences at different sites and even at proximal versus distal aspects within the same bone. More specifically for imaging, it is important to highlight the scanner calibration and maintenance, scan settings, image processing, and procedure precision, along with Good Laboratory Practice validation of technologies, software, and procedures.
The choice of end points for bone assessment will depend on the mechanism of action of the drug, type of changes expected, objectives/questions asked, duration, and type of study. It is important to understand the techniques, their applications, and limitations, in order to fully integrate the generated data in the interpretation in nonclinical studies. Where there are concerns for off-target effects or when pharmacological end points are required to assess postnatal musculoskeletal development, as well as bone quality, more comprehensive evaluations are required.
Study Design and Conduct of Neuropathology Evaluations in Juvenile Toxicity Studies
Dr Brad Bolon provided an overview on the design of neuropathology evaluation in juvenile toxicity studies. Neuropathology evaluation is a core component in examining the effects of test articles in juvenile animals, but the design of the analysis depends on the study design (and especially the study objectives and timing of exposure). Neurobiological factors that often underpin the decision to perform a juvenile toxicity study typically are related to developmental aspects of at least the CNS, especially the brain. The evolution of the vertebrate brain is divided into discrete stages based on the waxing and waning of morphological landmarks and the unfolding of biochemical and/or molecular signals over time. These events follow a consistent sequence across species, but the timing of events in a given species varies with the lengths of both gestation and the life span. 42- 47 The brain as a whole is susceptible to neurotoxic agents for an extended time during prenatal and postnatal stages of development, while specific sites and cells in the CNS and PNS possess more limited critical periods of vulnerability.
A JAS is likely warranted if damage is anticipated in one or many developing organs, 26,48 of which the nervous system is a principal target. Current globally standardized guidance for JAS 26 recommends a WoE approach (see previous section by Dr Paul Brown on ICH S11) in considering whether or not a nonclinical JAS is needed to support clinical trials. The neurotoxic potential of a pharmaceutical candidate is a major consideration in making such WoE decisions. 49,50 Guidance for pharmaceutical development by regulatory agencies (eg, the FDA Redbook 51 ) or global consortia (eg, ICH S11) are applied to standardize safety assessment methods for new medical products. For example, the ICH suggests that several CNS organs (multiple regions of the brain and several divisions of the spinal cord) and one PNS component (usually the sciatic or sometimes the tibial nerve) should be evaluated. 52 The neuropathology evaluation of JAS differs in design depending on the neural-oriented (especially CNS neuroactive) effects of the test article. 26,53,54
The design of the neuropathology component of juvenile toxicity studies varies with the study objective and especially with the dosing window. In rat JAS with test articles having unknown potential for developmental neurotoxicity, the approach to neuropathology evaluation is generally comparable to that performed for a general toxicity study in adult animals (eg, as recommended in the FDA Redbook, 51 ICH M3(R2), 55 or similar guidances). At necropsy, CNS and PNS tissues are removed and assessed macroscopically, brains are weighed, and then all samples are fixed by immersion in conventional neutral-buffered 10% formalin (NBF). Some pathologists or sponsors may choose to examine and weigh brains after fixation to reduce artifactual microscopic changes (eg, “dark neurons” in certain cerebrocortical, hippocampal, and cerebellar regions 56 ) associated with handling-related microtrauma of unfixed neural tissue. Neural organs (typically brain, spinal cord, and sciatic nerve, in keeping with “best practice” recommendations for nervous system sampling) 57,58 are processed routinely into paraffin, and the sections are stained with H&E for light microscopic evaluation.
In contrast, in rat JAS for CNS-neuroactive test articles or test articles with potential off-target neurotoxicity, the neuropathology evaluation is more extensive by design, though the extent of this “expanded neurohistopathology” evaluation is varied to fit the particular questions raised by the nature of the test article. 49,54 At necropsy, CNS and PNS tissues are removed for macroscopic evaluation, brains are weighed, and all samples are fixed by immersion in NBF. Although the CNS is anticipated to be a primary target tissue in such situations, perfusion fixation is not employed since this technique may affect the organ weights and tissue architecture of highly vascular organs (eg, lung, spleen) that are also important target organs for JAS. This means of fixation is distinctly different from the perfusion fixation recommended for another juvenile toxicity test: the DNTS, described in EPA 870.6300 6 and OECD 426 4 guidances. Although perfusion fixation is ideal for the evaluation of neural tissues, the reason for this divergence in fixation procedures between guidances is based on the fact that the nervous system is the only focus in a DNTS and as such allows the inclusion of special preparation to preserve this one organ system. On the other hand, in JAS, the tissue preparation must be suitable for all organ systems, including the CNS and PNS, thereby allowing for some balanced accommodation regarding fixation procedures. The routine neural tissue list (brain, spinal cord, and sciatic nerve), which is used in typical JAS and DNTS, may be expanded to include additional brain sections as described in the study by Bolon 59 (based on the studies of Rao et al 60 for rodents and Pardo et al 61 for monkeys), and sometimes more PNS tissues (eg, cervical and lumbar dorsal root ganglia, tibial and fibular nerves). In addition to H&E, expanded neurohistopathology for JAS (and the standard stain battery for DNTS) may incorporate one or more special neurohistological stains, commonly Luxol fast blue (for myelin integrity), anti-glial fibrillary acidic protein (to demonstrate astrocytes), anti-ionized calcium-binding adaptor molecule 1 (to highlight microglia), and sometimes Fluoro-Jade B (to detect necrotic neurons). 8,9,57,62 Plastic (epoxy resin) embedding of nerves is appropriate if in-life neurological signs and/or light microscopic evaluation of nerves suggest that the PNS is impacted, 58 but in practice this medium is used routinely in DNTS (based on its inclusion in the EPA 870.6300 guidance) but infrequently in JAS. Quantitative analysis—typically performed as areal or linear morphometry of various brain regions 8 —as recommended for a DNTS is seldom performed in JAS because the major structures subject to measurement (eg, cerebral cortex, hippocampus, and cerebellum) have already attained approximately adult contours and sizes when exposure begins. As noted above, the resulting neuropathology data set will be integrated in the study report with the results of neurobehavioral (cognitive, motor, and sensory) testing via collaborative discussions between the toxicologist and toxicologic pathologist. A recent FDA review notes that JAS data for CNS-active drugs are often used in labeling for pediatric drugs, and that neurobehavioral tests are more commonly affected relative to neuropathology end points. 49
Symposium Session Platform Discussion
In summary, this 4-hour symposium session highlighted several important aspects where the practicing toxicologic pathologist encounters regulatory considerations during the design, analysis, and interpretation of juvenile toxicity studies. A short engagement between speakers and the audience (via online queries) generated a lively discussion across all topics. The following narrative summarizes these interactions.
Specific queries on regulatory aspects included one on how the factors influencing WoE are weighted by regulators, and whether absorption, distribution, metabolism, and excretion (ADME) factors were also used in the WoE. This query was addressed by Drs Brown and Hoberman. Dr Brown reiterated that certainly PK data are used to determine whether a JAS is needed and also allows for appropriate selection of species. He mentioned that available data/knowledge on the ontogeny of CYP 450 in humans and even animals may also be factored in toward selection of species. Dr Hoberman also agreed that the PK and ADME data are usually used to drive species selection. In response to weighing out individual factors, Dr Brown stated that the ICH S11 Working Group had considered options to potentially prioritizing the factors; that in the end, the consensus as stated in the finalized ICH S11 is providing slightly higher importance to the age of the intended pediatric population and effects on developing organs (with the caveat that much is contingent on the variables in question for the specific drug and situation that drives the contextual decision). Another question for Drs Hoberman and Brown included solicitation of comments on readily implemented modifications to the pre- and postnatal development (PPND) reproductive toxicology study to expedite the JAS. The underlying rationale was to provide a better use of the first progeny (F1) generation animals in the PPND study as many pups are not evaluated following culling on postnatal day 4 or postweaning. Dr Hoberman states two possible approaches: the first option would be to designate the pups from the control group into a DRF study for a full-fledged JAS (for rats and mice, this would be the equivalent of dosing a child at 2 years of age); the second option is to allow dosing pups from each dose group postweaning into adulthood for evaluation. Although the latter is possible, Dr Hoberman did not recommend the approach because the exposure in utero or via milk that might have occurred cannot be accurately accounted for.
Queries regarding bone end points included solicitation of a recommended collection method for supplemental bone sampling that would maximize use for multiple evaluations/end points (ex vivo, CT, imaging, etc). Dr Varela acknowledged that there is no one catch-all method but can generally be viewed as two main approaches. First, frozen samples that allow the bone to remain intact should be reserved for potential future bone densitometry or ex vivo biomechanical strength testing, while fixation in formalin is typical for histopathology evaluation, while specific additional requirements are important for histomorphometry. Another bone-focused question focused on differentiating treatment-related effects from physiological changes during development. For a drug that impacts organ weight and body weight, is there a way to normalize femur length measurements that will allow one to distinguish general effects compared to specific effects on bone? Dr Varela responded the approach would be similar to a WoE approach used during routine study interpretation where absolute and relative organ weight data are considered along with data for other bone end points to tease out whether these effects are general versus specific. Yet another query on bone end points focused on whether a set number of “bone cycles” should be considered over which animals should be dosed or recovered to actively determine whether a test item has an effect on bone and/or whether the effect is reversible? Dr Varela responded that the timing varies with the animal species in adults, and that specifically, for juvenile studies, the study duration should allow for at least 13 weeks to cover the rapid phase of growth for the skeletal system in rodents.
An initial question on neuropathology evaluation in JAS sought to define the importance and relevance of strain differences impacting developmental findings noted following treatment with agrochemicals in two-generation studies. Dr Bolon responded that strain differences in anatomic and functional neurodevelopment as well as responsiveness to xenobiotics are not uncommon in mice and rats, so an effort should be made to keep strains consistent across studies for a regulatory submission, and that reliance on concurrent controls in interpreting pathology data is the default approach. Drs Bolon and Hoberman both iterated that outbred strains would be preferable compared to the inbred strains that are inherently predisposed to spontaneous developmental anomalies. Dr Rao concurred and added that when such effects are noted in regulatory toxicology studies, the potential issue should be addressed in the narrative where justification is provided to demonstrate that the finding is or is not related to strain-specific background congenital/developmental anomalies so as not to confound risk assessment.
Another neuropathology query regarding the number of sections to be evaluated included whether the 8-section approach for DNTS per Bolon et al 8 was comparable to the 7-section approach for general toxicology studies per Bolon et al 57 (based on studies of Rao et al 60 for rodents and Pardo et al 61 for monkeys). Although specific comparisons between the two approaches were not discussed, both Drs Bolon and Rao agreed that these two methods are comparable. Both also emphasized that the 7- or 8-section approach (whole sections for rodents and hemisections in dogs/nonhuman primates) was meant for a baseline (“Tier 1”) screening of test articles with unknown neurotoxic potential. If neurotoxicity is suspected (direct or off-target), or if the test article targets the nervous system, then additional sections and/or specific neuroanatomical areas should be examined in an advanced (“Tier 2”) assessment (see Bolon et al 54 on neuropathology for juvenile toxicity studies in this symposium issue).
A final neuropathology query focused on the use of results from neurobehavioral assessments to inform the scope of expanded neurohistopathology in a juvenile toxicology study. A number of factors impacting neuropathology evaluations were mentioned, including the location and timing that would be important especially for comparative assessments between end points. Specifically, for JAS, Dr Bolon underscored the importance of comparisons between neuropathology and neurobehavioral assessments, especially if these end points are evaluated in a separate satellite group of animals. Different animals may be used for neurobehavioral assessments because the handling and activity encountered in behavioral testing serves as an enrichment variable that cannot be ignored when evaluating some neuropathology end points (eg, quantitative measurements gathered for synapse-rich neural domains such as the cerebral cortex and hippocampus). Given inherent study design factors that can potentially impact interpretation, close coordination between the toxicologist and the pathologist continues to remain an important aspect in nonclinical animal toxicology studies.
Supplemental Material
Supplemental Material, sj-docx-1-tpx-10.1177_01926233211046869 - Regulatory Perspectives on Juvenile Animal Toxicologic Pathology
Supplemental Material, sj-docx-1-tpx-10.1177_01926233211046869 for Regulatory Perspectives on Juvenile Animal Toxicologic Pathology by Deepa B. Rao, Alan M. Hoberman, Paul C. Brown, Aurore Varela and Brad Bolon in Toxicologic Pathology
Footnotes
Authors’ Note
All authors contributed toward the writing of this manuscript. This document has been reviewed in accordance with policies of the authors’ respective institutions of employment. The opinions expressed in this article are solely those of the authors should not be construed as representing views or policies of their employer and/or any government agency.
Acknowledgment
The authors gratefully acknowledge Ms Beth Mahler for assistance in optimizing the figures for publication.
Declaration of Conflicting Interests
The author(s) declared no potential, real, or perceived conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.