
Abstract
Immune tolerance is defined by an active state of immune system unresponsiveness to foreign and self-antigens. Loss of immune tolerance to self-antigens and the resulting overexpression of autoantibodies can lead to tissue injury and development of various autoimmune diseases. In drug development, the goal of newly emerging immune tolerance therapies is to treat autoimmune disorders by restoring the immunoregulatory capacity of the immune system. Development of immune tolerance targets is initiated with the establishment of pharmacological efficacy in relevant disease animal models, followed by their stepwise translation to humans. This review discusses the major challenges to developing tolerance inducing pharmaceutical drugs, including the selection of appropriate disease models to establish efficacy, adequate, and acceptable in vitro and in vivo safety assessments, relevant biomarkers of human safety and efficacy, and finally, some regulatory guidelines to successfully develop immune tolerance therapeutics.
This is an opinion article submitted to the Toxicologic Pathology Forum. It represents the views of the authors. It does not constitute an official position of the Society of Toxicologic Pathology, British Society of Toxicological Pathology, or European Society of Toxicologic Pathology, and the views expressed might not reflect the best practices recommended by these Societies. This article should not be construed to represent the policies, positions, or opinions of their respective organizations, employers, or regulatory agencies.
Introduction
Therapeutic immune tolerance is defined most simply as the active inhibition of immune responses to specified antigens that would normally induce unwanted inflammatory and pathological responses. For example, feeding Sprague-Dawley rats horse serum or pollen extracts 1 or mice sheep red blood cells 2 induced immune tolerance to these antigens. Immune tolerance includes both central and peripheral immune tolerance (Figure 1). In central immune tolerance, which takes place in the thymus and bone marrow and during lymphocyte development, autoreactive T and B lymphocyte clones are removed by negative selection. 3 If the first line of central tolerance fails, peripheral tolerance may be needed to prevent autoimmune diseases. In peripheral immune tolerance, which takes place in secondary lymphoid organs after lymphocyte maturation, autoimmune responses and diseases are inhibited by primary mechanisms such as immune ignorance, anergy (unresponsiveness), deletion (apoptosis), phenotypic skewing, and regulatory T cell-mediated suppression of effector T cells 3,4 (Figure 1). One hypothesized trigger for loss of tolerance is illustrated by the danger or tissue damage model in which cellular remnants called danger-associated signals trigger an immune response, resulting in the breakdown of immune tolerance. The loss of immune tolerance to self-antigens leads to the development of autoantibodies that can trigger various autoimmune diseases including vitiligo, systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), multiple sclerosis (MS), type 1 diabetes (T1D), and Graves’ disease (GD). 4 –6
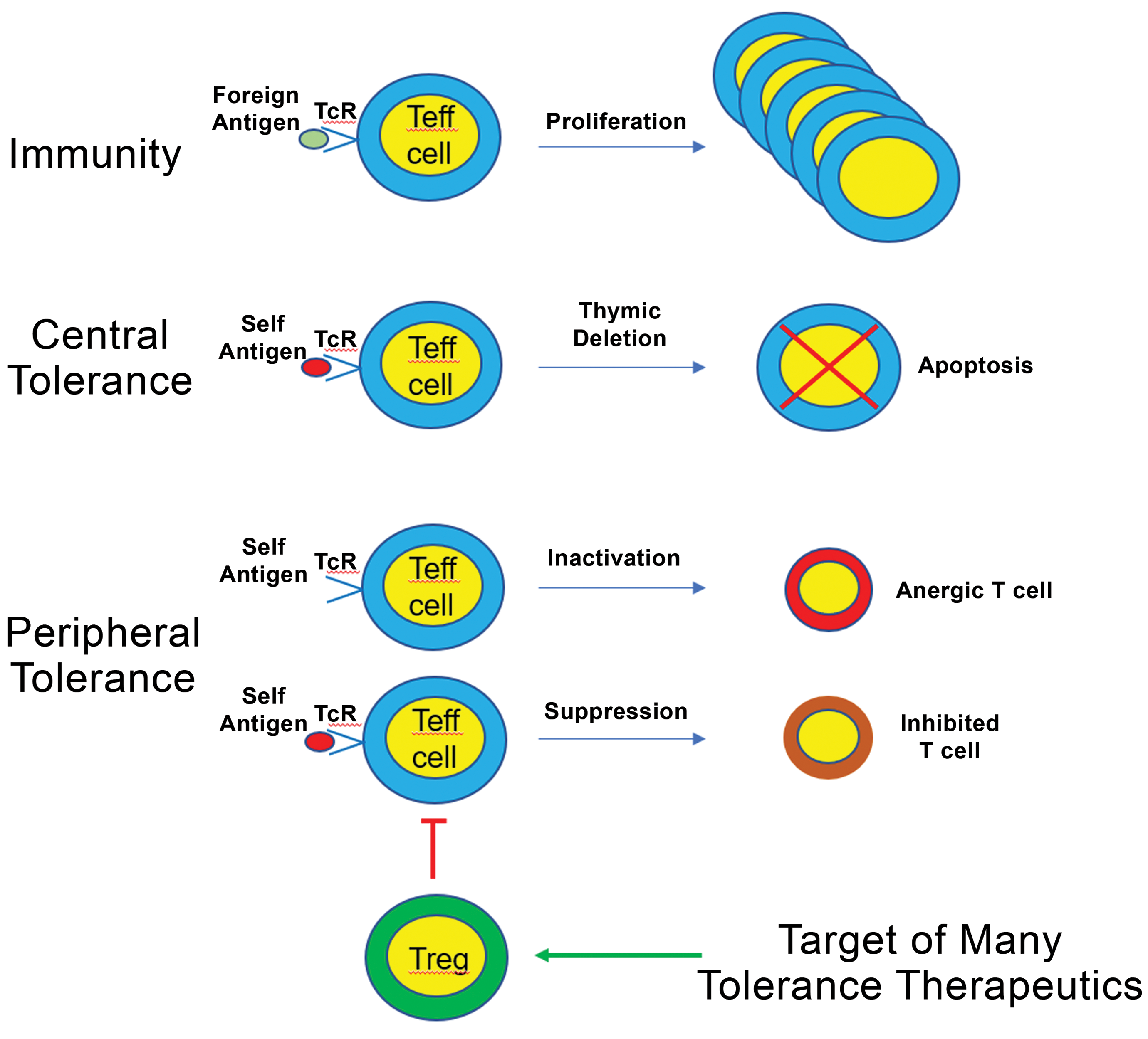
Tolerance inducing therapeutics often target regulatory T-cell expansion and function. Immune tolerance is the state of unresponsiveness to antigens that would normally promote immunity by triggering the proliferation and expansion of effector T cells (Teff). Tolerance to self-antigens is generated by both central and peripheral tolerance mechanisms, as depicted in the figure. Many experimental therapeutic approaches currently in development target peripheral tolerance by inducing the expansion and/or activation of T regulatory cells (Tregs) that actively suppress Teff cells through a variety of mechanisms.
A combination of genetic and epigenetic risk factors and environmental exposures can result in loss of tolerance in individuals with autoimmune disease, and the specific self-antigens to which tolerance is lost often determines the target organs that are affected. Important cellular mediators in the maintenance of antigen-specific tolerance are regulatory T lymphocytes (Treg), which suppress effector T-cell responses. Indeed, deficiencies in Treg cells can lead to autoimmune and inflammatory diseases such as immune dysregulation, polyendocrinopathy, enteropathy, X linked syndrome. 3,4,7 Two of the most important phenotypes of Tregs are those that express the transcription factor forkhead box P3+ (FoxP3+) and FoxP3− type 1 regulatory (TR1) cells. 8,9 Type 1 regulatory cells are memory CD4+ T lymphocytes that express the surface markers CD49b and lymphocyte-activation gene 3 and suppress Th17 and other T effector cell responses by secreting large amounts of IL-10 and transforming growth factor beta (TGF-β) following activation. 8 Consequently, a number of novel autoimmune disease therapies are in various stages of drug development, with numerous preclinical animal efficacy models demonstrating the ability to expand FoxP3+ Treg and/or TR1 cells resulting in substantial efficacy.
Development of immune tolerance therapeutics begins with the establishment of pharmacologically relevant efficacy in disease animal models followed by their translation to humans. Consequently, to successfully develop tolerance-inducing therapeutics, scientists, pharmaceutical drug developers, and clinicians must use a weight-of-evidence approach based on scientific data and select the most appropriate disease models, most appropriate and more sensitive toxicology species for in vivo assessment, in vitro assays, and safety and efficacy biomarkers and follow regulatory guidelines. In this review, we discuss these important issues to help clarify the major obstacles that must be overcome to better inform the path forward for tolerance-inducing therapeutics.
Commonly Used Animal Models of Immune Tolerance
Immune tolerance approaches have been tested in several animal models including T1D, MS, RA, GD, and celiac disease, in which pathology is caused by autoantibodies or T cells targeting autoantigen(s). For example, systemic administration, via intravenous infusion, of nanoparticles (NPs) coated with disease-relevant peptide major histocompatibility complex (MHC class) II molecules resulted in antigen-specific tolerance and signs of efficacy in spontaneous diabetes and experimental autoimmune encephalomyelitis. 10,11 Celiac disease is an immune-mediated intestinal disease in which individuals are sensitive to antigenic-specific (ie, gluten) peptide exposure and subsequent activation of CD4+ T cells. Pro-inflammatory cytokines (eg, IL-21 and interferon gamma [IFN-γ]) are secreted following activation of gluten-specific T cells leading to intestinal pathology. 12 If the immune response to gluten is eliminated via the induction of immune tolerance to relevant antigens, dietary restrictions may be eliminated for some individuals, thus providing substantial relief from the most severe disease manifestations. Therapies employing the intravenous delivery of soluble peptide antigens have also been used to induce peripheral tolerance. For example, tolerogenic immune modifying NPs [TIMP GLIA] encapsulated with gliadin protein were administered to mice intravenously, resulting in the restoration of peripheral tolerance to gluten and protection from celiac disease. 13 The treatment significantly reduced the histopathological changes observed in the duodenum and reversed the effects of dietary gluten challenge when compared with the gluten positive control. 13
The nonobese diabetic mouse is a commonly used animal model to investigate T1D. In this model, pathological autoimmune T cells mediate the damage of insulin-producing β cells in the pancreas. The 2 dominant epitope antigens in this model that are implicated in T1D immunopathogenesis are proinsulin and islet-specific glucose-6-phosphatase catalytic subunit-related protein (IGRP). 14 Interestingly, however, development of immune tolerance to insulin and prevention of T1D was induced following genetic overexpression of proinsulin but not by IGRP. 14
The Aryl hydrocarbon receptor (AhR) is a transcription factor that plays an important role in immunity as the activation of AhR maintains peripheral tolerance and prevents autoimmune pathologies. 15 Aryl hydrocarbon receptor deficiency in mice leads to the development of the autoimmune disease, SLE, and human SLE has been associated with an altered AhR transcriptional signature. 15 It was demonstrated, using in vitro model of bone marrow–derived macrophages or bone marrow–derived dendritic cells cocultured with apoptotic thymocytes, that apoptosis and phagocytic removal of apoptotic cells (efferocytosis) is required for AhR activation. 15 Efferocytosis triggered AhR activation in macrophages in turn was associated with reduced inflammatory responses and increased IL-10. 15 Therefore, to prevent the development of autoimmune diseases, tolerance triggered by apoptotic cell–mediated AhR activation may be a critical determinant.
The examples presented above were conducted in what is considered a pharmacologically relevant efficacy disease animal model. Also, animal models might be used on a case-by-case to collect select safety end points. However, although several mouse models show promise (eg, TIMP-GLIA mouse model in celiac disease), these models also pose several challenges and limitations when developing immune tolerance therapeutics. For example, it is challenging to use animal models to accurately predict the precise antigen dose needed to achieve the desired therapeutic benefit in humans (ie, scaling up directly from animal to human). 16 In addition, considering the vast genetic diversity of humans, in vitro studies and/or experiments in genetically diverse animal models may be needed to better predict potential adverse effects and toxicities in humans. 16 Furthermore, various target organs are collected and analyzed depending on the animal model (eg, pancreatic islet cells, pancreatic draining lymph node, and spleen in T1D models), but these tissues are not easily accessible in humans. In addition, findings from human peripheral blood rarely replicate the findings observed in mouse spleen and lymph nodes, and this is particularly true when it comes to phenotyping Treg responses in patients with autoimmune diseases. 16 Thus, although animal models are useful tools to test specific hypotheses, they are not always useful to predict outcomes of immune tolerance-inducing protocols in humans.
Some key features of tolerogenic peptides for autoimmune diseases would include the following: (1) high solubility to minimize destruction at injection site, (2) mimicking the naturally processed antigen when bound to MHC II, (3) inducing apoptosis or anergy, (4) suppressing the secretion of inflammatory cytokines and promoting the secretion of anti-inflammatory cytokines, and (5) safe administration to patients. 17
Toxicology Species Selection for Immune Tolerance Therapies
There are no regulatory guidelines specific to species selection for immune tolerance targets. Appropriate toxicology species selection is an important factor in nonclinical safety and risk assessment and should be scientifically justified. Some of the general principles and factors used to select the most appropriate and more sensitive species in other modalities might also apply to immune tolerance programs. These factors include comparative cross-species amino acid and protein sequence homology, pharmacology, and/or physiology; adequate in vitro screening for target tissue expression, binding, and function in animals and humans; pharmacodynamics and pharmacokinetics; using only a single rodent species (rats or mice) without conducting toxicity studies in a nonrodent species; principles of 3Rs (Replacement, Reduction and Refinement). Furthermore, the Food and Drug Administration (FDA) guidance suggests prior to the initiation of definitive preclinical studies, in vitro studies should be conducted, including functional assays, immunophenotyping, and morphologic evaluation, followed by experimental in vivo studies to establish the biological relevance of a specific animal species to the investigational product(s). 18 Comparative cross-species sequence homology and physiology can be the starting point in immune tolerance program species selection. For immune tolerance approaches using NP coated peptide MHC (pMHC) class II complexes, the most appropriate preclinical toxicology species needs to be carefully considered. For example, Sponsors may consider using a nonrodent species such as nonhuman primates or minipigs in toxicity studies evaluation due to physiological and anatomical similarities with humans for a specific immune tolerance target but only if scientifically justified and supported by a weight-of-evidence approach. The FDA guidance recommends Sponsors consider in species selection criteria such as “comparability of physiology and anatomy to that of humans.” 18 Sponsors should also consider whether the use of preclinical disease models for efficacy and safety, adequate in vitro screening assays, and in vivo toxicity studies in a single rodent species is a sufficient preclinical safety data package for an immune tolerance drug. It is known that MHC class II molecules are highly polymorphic. 19 Since anaphylactic responses are a potential safety concern following intravenous administration of soluble peptides 20 as are immune responses to foreign pMHC class II molecules, investigators might attempt to minimize these concerns by performing some studies in a single healthy toxicology preclinical species such as rats or mice typically used in routine safety assessment without needing to conduct in a nonrodent species unless scientific data support nonrodent is most appropriate and more sensitive species.
Although there are few specific guidelines that can be applied to species selection for immune tolerance programs, some regulatory guidance documents contain useful information and concepts (eg, sequence homologies, functional activities in cell-based systems, binding avidity, and animal disease models) that may help guide toxicology species selection. This is particularly relevant for immune tolerance antigens that are only present in disease models but not in healthy animals typically used in toxicity studies and is consistent with the principles of 3Rs. For example, ICH S6 addendum states that “When the target is expressed at very low levels in typical healthy preclinical species (eg, inflammatory cytokines or tumor antigens), binding affinity and activity in cell-based systems can be sufficient to guide species selection,” and “when animal models of disease are used to evaluate proof of principle, a safety assessment can be included to provide information on potential target-associated safety aspects.” 21 Similarly, European Medicines Agency guideline states “in certain cases, studies performed in animal models of disease may be used as an acceptable alternative to toxicity studies in normal animals. The scientific rationale for the use of these animal models of disease to support safety should be provided.” 22 Some specific examples of species selection include the TIMP-GLIA program in which mice that are MHC II deficient but express the human leukocyte antigen DQ8 risk allele (HLA-DQ8) human CD4 transgenes were used to obtain the optimal mouse dose thus allowing for determination of the human equivalent dose. No drug-related toxicity was noted in rodent species, mice and rats, which were used to conduct toxicokinetic and repeat-dose good laboratory practice toxicity studies to establish the no observed adverse effect level. 13 In phase 2 clinical trials, celiac disease subjects needed to be positive for HLA-DQ2 or HLA-DQ2/DQ8 as inclusion criteria. In another example, first in human doses for an antigen-specific immunotherapy using a combination of 2 thyrotropin receptor peptides (termed ATX-GD-59) for the treatment of GD were determined based on safety margins from repeat-dose toxicity studies using only a rodent model. 6
Screening Considerations to Establish Immune Tolerance Safety
To safely develop antigen-specific immune tolerance regimens to treat autoimmune diseases, in vitro testing should be used prior to conducting in vivo toxicity studies in a single rodent species. Also, animal models might be used on a case-by-case to collect select safety end points (ie, combined efficacy and safety study). These in vitro and in vivo safety studies are used to better understand the overall safety risk and provide the necessary evidence to avoid potential off-target adverse effects, including disease exacerbation. This is especially important for immune tolerance therapies that employ NPs. The in vitro NP preclinical and/or human safety characterization might include assays for complement activation (eg, C3a, C5a), inflammatory cytokine (eg, IL-6, IL-8, TNF, IFN-γ, IL-10,) release measures, hemolysis, and/or platelet activation or aggregation. 23 The panel of in vitro assessments can be followed up later with in vivo assessments that include repeat-dose toxicity studies, which provide clinical pathology evaluations such as effects on extrinsic, intrinsic, and common coagulation pathways (ie, prothrombin time, partial thromboplastin time, thrombin time, and D dimers). In addition, the size of the NP is an important feature in the development of organ-specific toxicity profiles. For example, NPs having 10-nm size can easily distribute to blood, liver, spleen, kidney, testis, thymus, heart, lung, and brain, while larger particles may have a more limited distribution, such as the spleen, liver, and blood after intravenous administration, as demonstrated in rats. 24 Thus, in vitro cytotoxicity and cellular uptake assessments may be considered for early screening and profiling of NPs.
Systemic administration of peptide-coated NP can lead to complement activation and subsequent hypersensitivity reactions and anaphylaxis. 20,23 In addition, complement activation (eg, C4d, C3b) can affect a pharmaceutical drug biodistribution (ie, if the drug has high clearance) from the systemic circulation via mononuclear cells phagocytosis. 23 In addition, species physiological differences can have a significant impact on NPs distribution and phagocytosis. For example, pulmonary intravascular macrophages, which are lung phagocytic cells, are more abundant in the lung of pigs than in rats, mice, monkeys, or humans. 25 Pulmonary intravascular macrophages can clear NPs quickly after their injection and such phagocytosis of NPs might elicit the release of inflammatory mediators such as thromboxane A2, and prostaglandins leading to adverse events (eg, vasoconstriction, pulmonary hypertension). 26 Hemolysis assays can help in those cases where NPs might affect erythrocyte membrane integrity, especially considering some of the unique surface properties of NPs, including charge. 23 For TIMP-GLIA, in vitro safety assays using human blood from healthy donors demonstrated that TIMP-GLIA did not cause complement activation, hemolysis, or platelet aggregation. 13 Using organoid technology might have future applications specific to investigating immune tolerance.
Efficacy and Safety Biomarkers of Immune Tolerance
Unlike traditional small or large molecules therapeutics modalities, one of the key challenges in the development of immune tolerance therapies is having the appropriate in vitro and in vivo efficacy and safety biomarkers, especially given that the goal of these approaches is to establish long-term or durable tolerance while avoiding the risk of exacerbating disease or increasing the risk of opportunistic infection(s) resulting from systemic immune suppression. However, infection risk would be expected to be much less likely with antigen-specific tolerance mechanisms. Possible biomarkers of immune tolerance induction include: (1) reduced expression of costimulatory molecules (eg, CD80 and CD86), (2) increased expression of coinhibitory molecules (eg, inhibitory Ig-like transcripts), (3) increased production of immunosuppressive cytokines (eg, IL-10, TGF-β), and (4) expansion and/or activation of Tregs (eg, Foxp3+, CD8+ Tregs). 27
dnaJP1 is a peptide that contributes to RA autoimmune pathology, 27 and induction of immune tolerance to dnaJP1 leads to a shift from a pro-inflammatory phenotype to a tolerogenic phenotype. 28 In a phase 2 clinical trial, oral administration of 25 mg of dnaJP1 for 6 months to induce tolerance in RA subjects was safe and well-tolerated. 28 In this study, in vitro assessment of the pro-inflammatory cytokine TNFα and the tolerogenic cytokine IL-10 were measured as biomarkers of efficacy, showing significant decreases in TNFα and a trend toward increased IL-10 production. In addition, peripheral blood mononuclear cells (PBMCs) obtained from dnaJP1-treated subjects were stimulated with dnaJP1 and demonstrated significant increases in FoxP3 expression. 28
In another example, the ability of a multiepitope, filaggrin, β-fibrinogen, vimentin, and collagen type II, citrullinated peptide (Cit-ME) to upregulate anti-inflammatory mediators (Treg) and downregulate proinflammatory cells (Th17) to restore homeostasis in RA was investigated in vitro. 29 Specifically, PBMCs from RA subjects were incubated with Cit-ME and upregulation of TGF-β and CD4+FoxP3+ Treg and downregulation of TNFα and IL-1β were noted. 29 In a related phase 1 clinical safety study, T1D subjects were administered intradermally 10 million autologous tolerogenic dendritic cells once every 2 weeks for a total of 4 administrations and the tolerability, efficacy, and safety were evaluated. 30 Safety and efficacy end points included serum cytokine detection, peripheral blood immune phenotyping (total and activated CD4+ and CD8+ T cells, total B cells, and peripheral blood CD11c+ dendritic cells), antinuclear antibodies, de novo thyroid-specific autoantibodies, and in vitro cell (eg, T-cell) proliferation assays. 30 In a GD transgenic mouse model, immunogenicity was investigated by detecting antigen-specific T-cell proliferation or by measuring IL-2 secretion. 17 No safety issues were noted including exacerbation of autoimmunity, which included clinical evidence of fever, hypersensitivity, malaise, chills, pains, or cardiac or ventilation abnormalities. 30 In summary, there are a number of different in vitro and in vivo tools and biomarkers available that are available to help characterize and better understand the efficacy and safety of immune tolerance therapies.
Regulatory Guidelines
Immune tolerance poses some challenges not only for pharmaceutical companies but also for regulatory agencies reviewing drug exposures, efficacy, and safety. Immune tolerance therapies are reviewed by the Center for Biologics Evaluation and Research (CBER) division of the FDA. Cell and tissue therapies, which use cells/tissues to treat a disease, fall under CBER. Regulatory guidances for Investigational New Drugs (INDs) and early clinical development in CEBER that are relevant to immune tolerance include: Guidance for Industry: Preclinical Assessment of Investigational Cellular and Gene Therapy Products, 18 Guidance for Industry: Considerations for the Design of Early-Phase Clinical Trials of Cellular and Gene Therapy Products, 31 and Guidance for FDA Reviewers and Sponsors: Content and Review of Chemistry, Manufacturing, and Control (CMC) Information for Human Somatic Cell Therapy Investigational New Drug Applications (INDs). 32
Other regulatory guidance documents include ICH M3(R2) guidance on nonclinical safety studies for the conduct of human clinical trials and marketing authorization for pharmaceuticals which give recommendations on the nonclinical safety studies “to support human clinical trials of a given scope and duration as well as marketing authorization for pharmaceuticals.” 33 The ICH S6(R1) guidance on preclinical safety evaluation of biotechnology-derived pharmaceuticals gives some concepts and recommendations that can be applicable to immune tolerance therapies and are “intended primarily to recommend a basic framework for the preclinical safety evaluation of biotechnology-derived pharmaceuticals.” 21
Conclusions
The successful development of tolerance inducing therapeutics will depend of most appropriate disease model, toxicology species selection, in vitro safety assays, safety and efficacy biomarkers, and following applicable regulatory guidelines. Animal models are useful tools to test specific hypotheses and might be used on a case-by-case to supplement safety assessment. Adequate in vitro safety testing screening can be used prior to conducting in vivo toxicity studies. Some in vivo toxicity studies in only a single rodent species such as rats or mice without the need for conducting toxicity studies in nonrodent species with the principles of 3Rs in mind might be considered as part of an acceptable overall weight-of-evidence approach based on scientific data and preclinical safety assessment package. Knowledge of some of existing regulatory guidelines can be applied to immune tolerance drug development.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.