
Abstract
Although there is an increase in the development of new cellular therapies, few guidelines have been published to assist in the design of preclinical studies. As no product and therapeutic intention is entirely alike regulators and Contract Research Organizations need to treat each project on a case-by-case basis. One of the most important considerations in study design is to retain all tissues from the study, thereby allowing for further analysis of tissues should unexpected effects be seen in clinical studies. Input from the pathologist at the earliest stages of study design regarding animal selection, cell markers, and phased tissue examination improves the scientific integrity of the study.
Introduction
Cell-based human medicinal products (CBMPs) may contain cells that are autologous, or allogeneic, and may or may not include cells that have been genetically modified, or which may or may not be combined with a device, scaffold or mesh.
Cell-based human medicinal products are aimed at repairing, restoring, replacing, or regenerating the structure and function of a damaged organ in order to ameliorate or cure previously untreatable injury or disease. Viable cells are introduced into the diseased/damaged tissue directly or via an implanted device and are incorporated into and become part of the recipient/targeted tissue, direct/facilitate regeneration by secreting factors, or coordinate intercellular signaling and interactions. There is huge public, scientific, and clinical expectation for stem cell therapeutics as treatments for genetic, degenerative, and inflammatory diseases.
Derivation of Stem Cells
There are 3 main types of stem cells used in CBMPs: (1) human pluripotent stem cells—human embryonic stem cells (hESCs) and induced pluripotent stem cells (iPSCs); (2) adult stem cells (ASC), also referred to as tissue-derived or resident stem cells; and (3) functionally mature/differentiated and structural cells, for example, retinal pigmented epithelium (RPE) cells, cardiomyocytes.
Human embryonic stem cells are pluripotent cells that have the ability to differentiate into derivatives of all 3 germ layers: endoderm, mesoderm, and ectoderm and are potentially able to replicate indefinitely. Induced pluripotent (iPSC) cell lines are derived from a nonpluripotent human adult somatic cell, through the forced expression of specific genes critical for the expression of pluripotency. Various methods exist to revert adult cells to pluripotency including viral-mediated transduction, epigenetic reprogramming, and protein-mediated transduction. 1 -5 The resulting cells have similar biological properties and growth characteristics to embryonic stem cells including long-term self-renewal and the ability to produce other, more mature cell types. Unfortunately, such cells may also retain some less desirable characteristics, such as the ability of iPSCs to develop into teratomas. Induced pluripotent stem cells can be patient-specific, so address some potential issues of cell rejection, however large scale iPSC therapies are likely to be allogeneic so have similar safety concerns to hESCs. There also may be a tendency for these cells to revert to their original cell type with time. 6 Multipotent somatic ASCs are found naturally in differentiated tissue throughout the body, where they function to maintain tissue by replacing damaged or lost cells. Adult stem cells can only differentiate into cell types of the tissue from which they originated and tend to have limited self-renewal. Examples of ASCs include mesenchymal stem cells, hematopoietic stem cells, and endothelial progenitor cells.
Methods: Preclinical Study Design for Cellular Therapeutics
The main challenge in study design is the paucity of guidelines available, and choosing the relevant animal model is key. Regulators should be consulted throughout the program design, and toxicologic pathologists can play a leading role here at the pre-pre-investigational new drug (IND) and pre-IND meetings.
Cell lines to be used to make the CBMP must be well-characterized, as the extent of the preclinical program necessarily depends upon any genetic manipulations and modifications of the cell line. Safety issues to be addressed are similar to those in tumorigenicity studies. The World Health Organization (WHO) guideline on tumorigenicity study design 7 and Food and Drug Administration (FDA) Cellular and Gene Therapy Guidance document 8 are useful starting points when considering the study aims and outcomes.
Program Key End Points
The aims of the therapy are for the cells to either be incorporated into and become part of the recipient/targeted tissue (repair, replace, or restore injured/diseased cells) or the cells direct or facilitate regeneration, by secreting factors or through intercellular signaling and interactions.
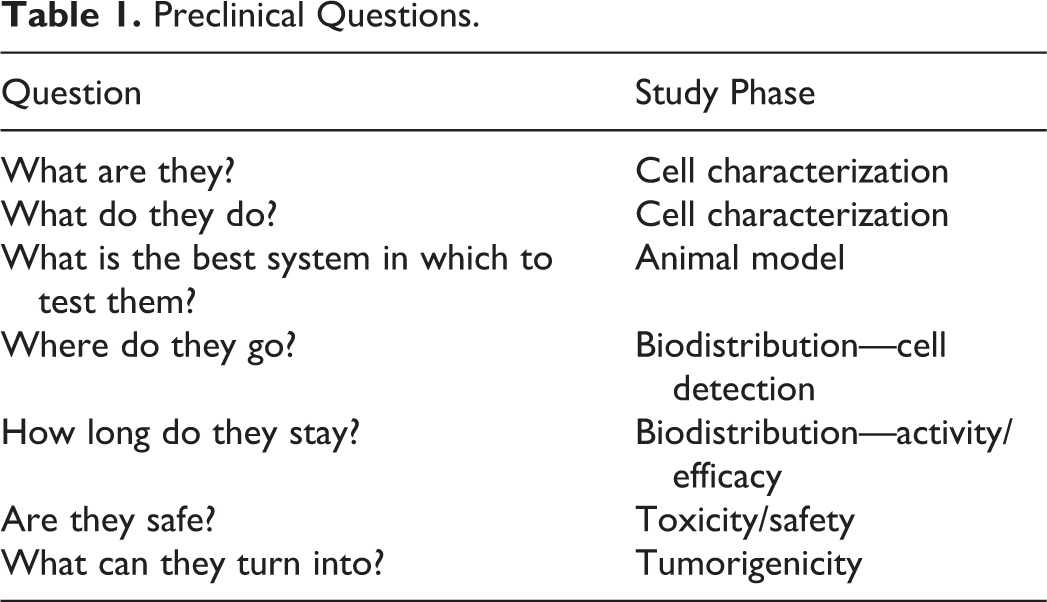
There are 7 key questions to be asked and answered which will guide the study and/or program design as listed in Table 1.
Preclinical Questions.
Program design should include discussions between preclinical and discovery scientists, pathologists, and regulatory specialists to produce a testing regimen to show that the cell therapy is safe and effective (Figure 1). The program must support clinical trials and use a consistent and well characterized product in a biocompatible formulation. The program should be customized to the particular cell, its actions, and the intended clinical use, at each step. Sponsors and contract research organization scientists should communicate with regulatory agencies on the preclinical program designs and animal model selections before studies are initiated. Proof of concept studies may be needed to aid in the selection of animal model(s).
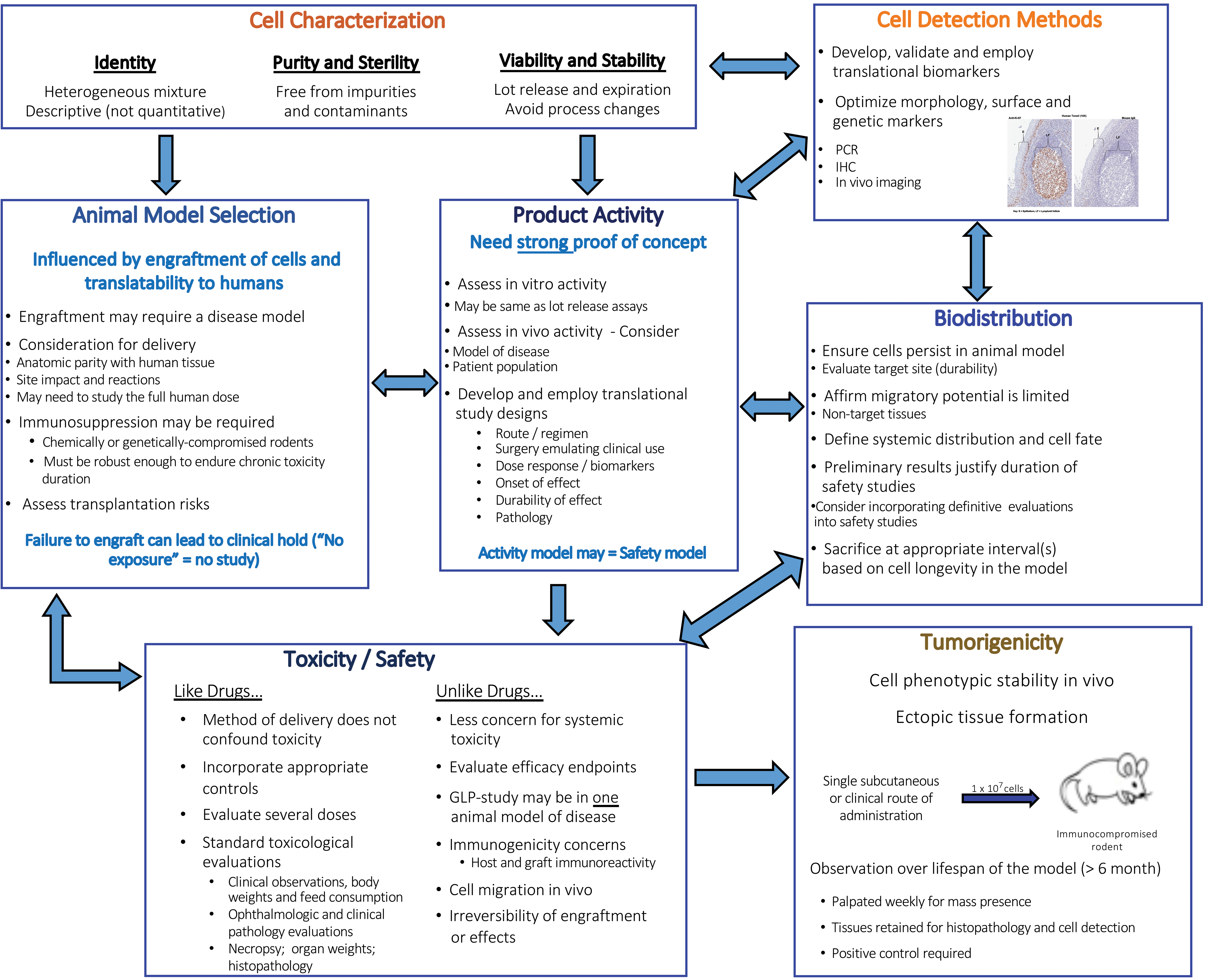
Summary of cell therapy study program.
Non-GLP pilot studies can be undertaken to demonstrate that persistence is associated with efficacy (biodistribution assays), determine dose levels and time points (if cells do not persist then shorter time study times may be possible), and ascertain the timing of administration in relationship to progression of disease (eg, acute or subacute phase of disease models). The main preclinical studies should be to Good Laboratory Practice (GLP) with a test item manufactured under Good Manufacturing Practice (GMP).
For the GLP/GMP studies, the route of administration and dosing regimen should mimic the proposed clinical program, using the intended clinical product by the intended clinical route administered with the intended delivery device.
Preclinical studies need to assess the risks of the injection/implantation procedure, any potential inflammatory/immune response to the injected/implanted cellular product, and indicate the minimally active dose and no-adverse-effect-level dose. Biodistribution studies need to check for cell migration to nontarget tissues. Robust, reliable, and sensitive cell detection methods (polymerase chain reaction [PCR], immunohistochemical [IHC]) are crucial for this stage. Implantation sites need to be assessed for inappropriate cell proliferation (ie, tumor formation) and inappropriate cell differentiation (ie, ectopic tissue formation). Toxicity studies should evaluate local and chronic toxicity, including tissue reactivity and off-target effects, and may also be used to predict interactions with concomitant therapies.
The Pathologist’s Contribution
Cell characterization (identity, purity, viability, sterility, and stability) should have been carried out long before the start of any animal studies. The starting hypothesis is that cell-based therapeutics are a heterogeneous mixture of cells. Pathologists can assist with determining identity and composition by evaluating morphology, phenotype-specific cell surface antigens, and validated cell identification markers. Purity and sterility can be assessed by looking for infectious agent-derived cytopathic effects on cell sheets in vitro and detection of biomarkers for undifferentiated stem cells.
Histopathological evaluation accounts for a significant portion of the data in preclinical studies of cellular therapeutics. Toxicologic pathologists evaluate cell biodistribution; determine engraftment and/or persistence within the host; assess local tissue reaction, safety, toxicity, and efficacy.
Therefore involvement of toxicologic pathologists in early stages of study design is important for the choice of tissues to be collected and evaluated. Toxicologic pathologists can assist with species and strain selection, development of specific cell markers, and triaging of tissue examination on a time point-specific basis.
Robust Detection Methods
There is usually significant cell death within the implant after transplantation, with limited engraftment and differentiation of transplanted stem cells. At early time points, it is possible to detect a bolus of administered cells within a tissue using hematoxylin and eosin (H&E) stained tissue sections, however in later time points, detection of small numbers of migrating cells using H&E alone may not be feasible. Cell products administered to direct and reprogram host cells will require analysis of biomarkers of safety and efficacy and any off-target cellular toxicities associated with cellular implantation. Immunohistochemical staining can be used to detect administered cells and determine whether or not the cells proliferate and/or persist. Potential tumorigenicity is a major public concern regarding stem cell-derived therapies. Pluripotent stem cells have the potential to develop a phenotype very different from the original cells. Both H&E and Immunohistochemistry (IHC) can be used to assess the administered cells’ ability to differentiate into the intended cell type or into a non-intended cell. Various markers, including human nuclear antigen and human mitochondrial antigen, have been used to demonstrate administered cells of human origin. Proliferating cell nuclear antigen or Ki67 may be used to look for evidence of proliferation. Assessment of gene expression by PCR, using markers of transcriptional activation, proliferation, and differentiation should also be used as necessary. Once the intended product has been formulated, studies should be carried out to GLP and GMP.
Regulatory Issues
In the United States, stem cells/stem cell-derived products are regulated by the Office of Tissues and Advanced Therapies (OTAT). Sponsors should arrange nonbinding informal scientific discussions between the Center for Biologics Evaluation and Research (CBER)/OTAT in “INTERACT”—Initial Targeted Engagement for Regulatory Advice on CBER products meetings (previously pre-pre-IND meeting).
In Japan, stem cells/stem cell-derived products are regulated by the Ministry of Health, Labour and Welfare. Guidelines on ensuring the quality and safety of pharmaceuticals and medical devices derived from processing human stem cells from different origin were issued in 2012, which include English translations of the Japanese notifications (notifications 0907-2 to 6).
In the European Union, cell therapies are assessed by the Nominated Regulatory Authority according to Regulation (EC) No 1394/2007 of the European Parliament and of the Council of November 13, 2007, on Advanced Therapy Medicinal Products and amending Directive 2001/83/EC and Regulation (EC) No 726/2004. For European purposes, cell therapies are defined as medicinal products when there is more than minimal manipulation of any cell type destined for clinical application or where the intended use of the cells is different to their normal function in the body. For both regulators, there is no “one size fits all” approach. The data needed for a submission depend on the characteristics of the product. Dossiers can include GLP and GMP studies, reference data in peer-reviewed journals, cross references to similar products, and clinical trial reports.
Species Selection
The behavior of stem cells is strongly influenced by local environmental factors, and accurate administration to the intended target site is crucial for proper assessment of cell safety. 8 There is no default species. Sponsors should provide scientific justification for the species/model(s) used. 7 The overall design of the toxicology studies should mimic the proposed clinical trial design as closely as possible. The animal test system needs to be a ‘progressive host’ allowing the cellular therapeutic to thrive, and consideration needs to be given to the intended clinical delivery device and route of administration when choosing a rodent or nonrodent species. Rats (including neonates), mice, nonhuman primates, pigs, surgically altered rodents, and immunocompromised animal models have been used. The use of an immunocompromised animal model reduces the likelihood of rejection of the cells. The nature of cell therapeutics allows the evaluation of product safety in combination with efficacy studies, using animal models of disease. Organ and tissue size are important when using the intended human dose levels using the targeted delivery device, formulation volume, and location. For example, an RPE cell scaffold implant to be used in human eyes is inherently large and so an animal with an eye similar in size to humans, such as pigs, would be the most suitable animal model 9 . Adequate numbers of animals per gender, appropriately randomized to each group, are necessary to provide statistically sound group sizes. Appropriate control groups are essential, while taking into consideration the principles of the “3Rs,” (reducing, refining, and replacing animal use). Examples include animals that are left untreated, receive sham surgery, or are administered formulation vehicle only, adjuvant alone, null vector, or scaffold alone. Justification should be provided for the number of individuals and type of specific control group(s) selected. Disease models should include littermates or a parental unaffected wild-type line for knock-out/knock-in.
Standard safety end points monitored include mortality (with cause of death determined, clinical observations, body weights, physical examinations, food consumption/appetite, water consumption (as applicable), clinical pathology (serum chemistry, hematology, coagulation, urinalysis), organ weights, gross pathology, and histopathology. No recovery groups are necessary as cells cannot be removed, but sufficient duration time points should be designed to assess chronic and off-target effects.
Study Duration and Dosing Regimen
Duration of the study depends on the predicted persistence of the cell therapy product, for example, 4 to 12 weeks for transient products to confirm clearance; 3 to 12 months for multidose or products expected to engraft, dependent on product and life span of test species; for example, 1 month for acute disease models which need to be evaluated during early stages of disease, not after the animals start to be debilitated. Therefore, sacrifice time point 1 should be 48 hours or 1 week postdose; sacrifice time point 2—2 weeks or 3 months postdose; sacrifice time point 3—4 weeks or 6 months postdose. The toxicologic pathologist should fully evaluate 1 time point before the laboratory processes the next as this may allow a less aggressive sampling protocol.
Study Design
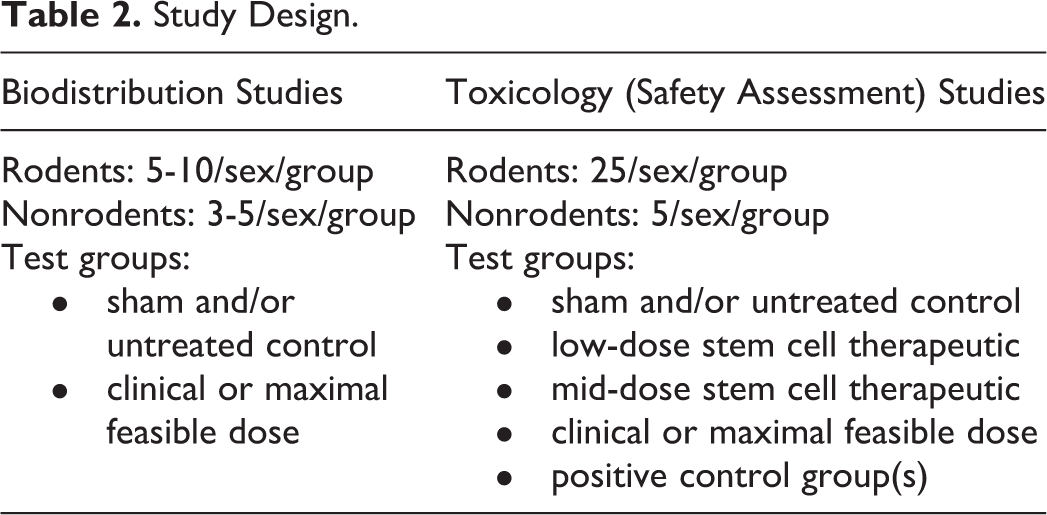
Design of biodistribution and safety assessment studies are dependent on the animal species to be used but should be GLP compliant. Example study parameters are listed in Table 2.
Study Design.
Efficacy studies can be combined with the toxicology study. Necropsies should be performed blinded to treatment however histopathology should not be blinded. The pathologist needs full access to treatment groups to fully assess the test item. Blinded evaluation of tissues by the pathologist should only be done as a secondary step to address specific morphologic parameters.
Pathology should be undertaken on all animals and a full tissue set retained at necropsy. The H&E assessment of the inoculation site and regional lymph nodes, major organs (liver, lung, spleen, heart, kidney, brain, bone marrow, gonads), gross lesions, and other therapy specific targets based on mode of action and likely distribution of the test item should be evaluated in the first instance. The inoculation site and the associated draining lymph nodes are most likely to demonstrate efficacy and safety parameters. Evaluation of liver, spleen, kidney, and lung will assess the potential for hematogenous spread. Gonads should be evaluated if reproductive toxicity is expected due to stem cell-released factors. Any gross lesions, especially masses, should be screened to exclude the possibility of toxicity and stem cell tumorigenicity. Systemic toxicity is less of a concern for many cellular therapeutic products, for example, stem cells implanted in the retina, an immune privileged site.
Quantitative PCR (Q-PCR) can screen a larger tissue volume of nontarget tissues for the presence of cells and reduce the need for the examination of IHC-stained sections of the nontarget tissues. If any positives are found with Q-PCR, then they should be confirmed with IHC or in situ hybridization. If no cells have been injected into a control group, tissue collection can be modified and minimized to include only sufficient tissue to demonstrate both background findings and the expected negative staining pattern for IHC markers.
Use of Phased Tissue Examination
Multiple sacrifice time points are recommended to capture potential acute, chronic, and/or delayed-onset toxicities, as well as the potential for resolution of toxicities. Although there may be additional cost incurred by processing and examining tissues at each time point, this is justified as findings from an early time point can be used to direct a reduced slide production protocol at later time points. Negative results from Q-PCR used to screen the nontarget tissues at 1 time point may be used to justify a reduced tissue list at the intermediate time points.
Tumorigenic Potential
Undifferentiated stem cells may have the risk of unregulated growth and have the capacity to differentiate or dedifferentiate into teratomas or other neoplasms. Tumorigenicity is defined as “the capacity of a cell population inoculated into an animal model to produce a tumor by proliferation at the site of inoculation and/or at a distant site by metastasis.” 7 A separate tumorigenicity study may be set up following the FDA notes for guidance on in vivo tumorigenicity studies and/or the WHO Technical Report Series 878. These studies assess cell phenotypic stability, and tumor or ectopic tissue formation in an immunocompromised rodent. As assessing the tumorigenic phenotype of a cell substrate requires the inoculation of xenogeneic or allogeneic cells, the test animal should be rendered deficient in cytotoxic T-lymphocyte activity. This is accomplished either by the use of rodents that are genetically immunocompromised or by inactivating the T-cell function with antithymocyte globulin, antithymocyte serum, or antilymphocyte serum. The animal models most often used are athymic nude mice (Nu/Nu genotype) and severe combined immunodeficient mice with a lack of functional T and B lymphocytes. There is a single subcutaneous or other intended clinical route inoculation with at least 3 dose groups including positive control cells from a reliable source. HeLa cells from the WHO cell bank are recommended as the positive control reference preparation, stored at the American Type Culture Collection (Manassas, Virginia) and National Institute for Biological Standards and Control (South Mimms, Hertfordshire, United Kingdom). The test item cell line and the control cells are each injected as 107 viable cells, suspended in a volume of 0.1 mL of phosphate buffered saline, into separate groups of 10 athymic female mice 4 to 7 weeks old at the start of the study. The purpose of the positive control is to assure that an individual test is valid by demonstrating that the animal model has the capacity to develop tumors from inoculated cells (ie, a negative result is unlikely to be due to a problem with the in vivo model). Negative controls are not recommended because the rates of spontaneous neoplastic disease in nude mice are low, and small numbers of negative control animals are unlikely to provide meaningful data. The mice are palpated weekly for a minimum of 16 weeks for the presence of masses at the inoculation site. If a nodule appears, it is measured in 2 perpendicular dimensions, the measurements being recorded weekly to determine whether the nodule grows progressively, remains stable, or decreases in size over time. Animals bearing nodules that appear to be regressing should not be killed until the end of the observation period. Cell lines that produce nodules that fail to grow progressively are not considered to be tumorigenic. At the end of the observation period, or at an earlier time if required due to the death of an animal or other justifiable circumstances, all animals are killed and examined for gross and microscopic evidence of the proliferation of inoculated cells at the site of inoculation and in other sites such as the heart, lungs, liver, spleen, kidneys, brain, and regional lymph nodes. Some cell lines may give rise to tumors at distant sites without evidence of tumor at the sampled inoculation site. The tissues are fixed in 10% formalin and sections are stained with H&E. Toxicologic pathologists assess anatomical location, size, incidence, morphological type, and cell origin of any tumors, that is, whether the tumors that arise at the inoculation site contain cells derived from the inoculated cells. If there is no evidence of a progressively growing nodule at the end of the observation period, the cell line may be considered to be nontumorigenic. 7
Results—The Pathology Report
The toxicologic pathologist’s safety assessment should include the following questions: Were there findings at the inoculation or distant sites? Did the disease model prove relevant? Is there any historical control data available? Were there adequate and enough control animals? Could the cell surface antigens and/or secreted products induce a polyclonal response? How immune privileged are the stem cells at the site of inoculation? Does the maturation status of the cells alter with time points and/or an inflammatory response? Will there be a need for prolonged immune suppression? What is the effect of an aging immune system?
Conclusion
The field of regenerative medicine is buoyant with many cellular products being developed for multiple therapeutic targets. Safety assessment requires a comprehensive and integrated approach at the molecular, cellular, and tissue levels. The toxicologic pathologist possesses training and expertise in systems biology, pathology mechanisms, and preclinical study design and as such are best placed to aid in program design and ensure the translatability of preclinical studies into clinical trials. The toxicologic pathologist can make invaluable contributions to species and strain selection, design of biomarkers, evaluation of host immunological response, and characterization of safety and efficacy end points.
Footnotes
Authors’ Note
This article was presented at both the IATP Session at the Annual STP meeting in 2018, and the Annual SFPT meeting in 2019.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.