
Abstract
Diverse host factors/phenotypes may exacerbate or diminish biological responses induced by air pollutant exposure. We lack an understanding of biological indicators of environmental exposures that culminate in a physiological response versus those that lead to adversity. Variations in response phenotype might arise centrally and/or at the local tissue level. In addition to genetic differences, the current evidence supports the roles of preexisting cardiopulmonary diseases, diabetes, diet, adverse prenatal environments, neurobehavioral disorders, childhood infections, microbiome, sex, and psychosocial stressors in modifying the susceptibility to air pollutant exposures. Animal models of human diseases, obesity, nutritional inadequacies, and neurobehavioral conditions have been compared with healthy controls to understand the causes of variations in susceptibility. Although psychosocial stressors have been associated with increased susceptibility to air pollutant effects, the contribution of neuroendocrine stress pathways in mediating these effects is just emerging. The new findings of neuroendocrine activation leading to systemic metabolic and immunological effects of air pollutants, and the potential contribution to allostatic load, emphasize the consideration of these mechanisms into susceptibility. Variations in susceptibility to air pollution health effects are likely to underlie host genetic and physiological conditions in concert with disrupted neuroendocrine circuitry that alters physiological stability under the influence of stressors.
Keywords
Introduction
The World Health Organization report of May 2018 shows that ambient (outdoor) and household air pollution causes nearly 7 million annual deaths worldwide. Air pollution accounts for nearly 70% of all deaths attributed to environmental causes. 1 However, for decades, we have recognized that not every individual who is exposed to air pollutants or other environmental stressors will die or develop diseases. 2 –4 For example, only about 20% of smokers develop chronic obstructive pulmonary disease (COPD). 5 Likewise, variations in susceptibility are likely to predispose some individuals to granulomatous diseases after exposure to silica or asbestos. Individuals from the same genetic background will show relatively similar susceptibility for a given disease. 6,7 When discussing susceptibility variations, one also must keep in mind the impact of longevity and the intensity of environmental exposures. This becomes important as chronic exposures to some pollutants may rarely result in active disease. Understanding the causes of susceptibility variations is critical in developing mitigation strategies and therapeutic interventions. In the context of the current article, “susceptibility” is defined as any unhealthy host condition(s) that modify the biological response to environmental or other stressors. Here, we focus on experimental studies of animal models of selected human susceptibilities and how they modify risk of adverse outcomes caused by air pollution.
Targeted hypothesis-driven research has led us to identify and understand some of the biological pathways and key genetic differences or physiological conditions that make one more susceptible to pollutants. 8 –11 In the past 2 decades, the application of a systems approach with high-throughput global gene expression, proteomic, and metabolomic techniques has accelerated the understanding of variations in susceptibilities but has also exposed the complexities of biological networks, cross talks between multiple processes within a cell, dynamics of these processes, and the communication between organ systems. At what step in the biological network, and in what organ system the normal homeostatic processes activated by air pollutants are interrupted due to abnormal host conditions, becomes a critical determinant of the type and the degree of variation in response. Perturbation in any one of the homeostatic processes either due to a genetic defect or from chronic or overstimulated responses will likely lead to cumulative or organ-specific impact after exposure to environmental pollutants. In this article, I will first briefly provide a historical perspective on what we have learned using animal models with defined susceptibilities/diseases by giving examples of air pollutant exposures in these models. Recent work 12 on the role of neuroendocrine stress pathways in mediating response to air pollutants will highlight how these new mechanisms will be critical in identifying variations in individual susceptibilities. Based on this body of work, a new mechanistic paradigm is proposed that will be useful when determining causes of variations in susceptibility to air pollutants and the importance of allostatic load. Experimental studies that provide insights into the biological mechanisms of susceptibility variations (noncommunicable; noncancer effects) will be discussed in this article but will not include models of infections, allergies, or developmental stressors.
Historical Perspective on Susceptibility Variations in Air Pollution Research
A variety of host conditions can modify the response to air pollution exposure that can impact health (Figure 1). It is conceivable that one who is susceptible to developing a disease from unknown or known causes is likely to have increased susceptibility to subsequent air pollutant exposure. Likewise, when an individual is exposed chronically to a given air pollutant, he or she will likely develop exposure-related disease over time. However, the mechanisms by which air pollutants may cause disease, or simply exacerbate the preexisting disease condition may depend on each individual’s collective and individually tailored response for survival. The understanding of variations in each individual’s response is further complicated by the fact that in ambient environmental conditions, individuals are generally exposed to rather low levels of a diverse range of pollutants that produce changes in homeostatic processes through different mechanisms.

Host susceptibility factors that interactively produce variations in individual response to air pollution health effects.
Most of the earlier research on air pollution nearly 50 years ago focused on occupational exposures to source-enriched pollutants at rather high concentrations. Because of the lack of exposure controls, workers who were exposed to specific source pollutants developed many types of pulmonary diseases based on the nature of inhalants. Due to the dominance of pulmonary effects, the acute and subtle effects on extrapulmonary organs likely went unnoticed. For example, exposure to silica in miners caused acute and chronic silicosis 13 ; coal miners’ exposures were associated with coal worker’s pneumoconiosis 14 ; asbestos mining caused fibrotic lung disease referred to as asbestosis, adenoma, and also pleural diseases with plaques of fibrous tissue, calcification, and mesothelioma 15 ; and exposure of farm dust caused hypersensitivity pneumonitis, often referred to as farmer’s lung. 16 Some of these pollutants were retained in the lungs with little clearance, and the diseases occurred decades later. For example, mesothelioma occurs in susceptible individuals decades after asbestos exposure. 17 The mechanisms of the delay in expression of disease still puzzle researchers and new insights continue to emerge. It was believed that air pollutants affected primarily the lung while the knowledge of peripheral effects was limited or these effects were considered nonspecific and therefore of lower significance when examining pulmonary diseases. Thus, the experimental studies mainly included lung pathologies and functional impairment. Likewise, animal models of susceptibility used for research were primarily of those with lung diseases. 18 –21
The October 1948 Donora Pennsylvania smog incident and the December 1952 Great London Smog incident raised the awareness that ambient-level air pollution can cause mortality and morbidity, and influenced the creation of ambient air quality standards for criteria air pollutants. 22 These episodes also prompted epidemiological and toxicological studies to determine what type of pollutants were responsible for adverse health effects and especially who was susceptible. Human mortality and hospital admissions were primarily associated with pulmonary complications of bronchitis and exacerbation of COPD. Based on these observations, and along with the awareness of cigarette smoke-related, allergic, and infectious lung diseases in the population, the emphasis for research was placed on the animal models of bronchitis, COPD, pneumonia, and asthma. 4,23 –29 The research experiments included understanding of biological factors/host conditions that may increase pulmonary susceptibility to inflammation and obstructive lung diseases.
Host Genetics as the Contributor to Variations in Air Pollution Health Effects
The contribution of genetic differences to the variations in susceptibility to air pollution health effects continues to be examined experimentally. 20,30 –33 Mouse models of different genetic backgrounds have shown differential susceptibilities to pulmonary inflammation after exposure to ozone and other pollutants. 34 –37 Linkage analysis studies have provided insights into the contribution of candidate genes within specific chromosomal loci influencing response to air pollutants. For example, in studies involving genetically diverse mouse strains, the DNA sequences on chromosomes 4, 11, and 17 spanning Toll-like receptor 4 and many inflammatory cytokines were linked to increased ozone-induced lung injury and inflammation. 38 The mutations in Toll-like receptor 4 and its contributions to increased allergen-induced lung injury and inflammation were also identified in mouse models as well as in humans. 9 Although these studies provide insights for the gene targets associated with specific pathological conditions, the understanding of collective contribution of gene networks and the causal factors to observed susceptibility differences remains sparse. The limited genetic diversity of laboratory rodents limits the scope of investigations identifying mechanisms of biological variations among genetically diverse humans.
More recent studies have provided insights into the complex interplay of genetic networks that might govern phenotypic expression of disease or adverse health effects in each strain/species of animals. 39,40 The individual strain/species variability in orchestrating transcriptional response will likely be a major contributing factor to producing variability in ultimate disease phenotype. The newer approach of collaborative cross of mouse models to create highly variable genetic backgrounds, which better represent the diversity seen in the human population, will provide some insights. Their systematic assessment using high-throughput platforms of genetic contribution to each disease phenotype has already begun to provide breakthroughs in understanding how individual gene clusters influence the variability in susceptibility not only to air pollutants but also to other types of environmental stressors. 33,41,42
Use of Animal Models of Human Cardiovascular Diseases in Air Pollution Susceptibility Studies
The robust epidemiological evidence from the 1990s expanding air pollution effects beyond the lung to the cardiovascular system, and how these effects might be important in human mortality and morbidity 43 led to research interest in cardiovascular effects and cardiopulmonary interactions. Likewise, the focus of susceptibility variations expanded from lung disease models to using animal models of cardiovascular diseases for understanding how those with preexisting hypertension or other cardiovascular complications might have exacerbated responses to air pollutants. 4,44 –47 Guided by the epidemiological associations between air pollution and cardiovascular diseases, our introduction of the use of a spontaneously hypertensive (SH) rat model in examining their susceptibility to air pollutants has led to the use of these rats in many air pollution studies. 48 These rats are now used worldwide in understanding of the key susceptibility attributes associated with hypertensive disorder in exacerbating pulmonary and cardiovascular effects of air pollutants. 49 –54
We have shown that relative to healthy normotensive rats, SH rats have greater pulmonary injury and inflammation, especially when air pollution exposures occur at high concentrations. 48,49,55 They have compromised ability to increase lung lining antioxidants when exposed to residual oil fly ash particles. 48 Spontaneously hypertensive rats are not able to respond to inhaled combustion source particles by increasing expression of genes involved in inflammation in the lung and the heart as baseline expression of some of these genes is already high in these rats. 48 When global gene expression in the heart of normotensive control Wistar Kyoto (WKY) rats was compared to SH rats following diesel exhaust inhalation, WKY rats showed remarkable changes in gene clusters reflecting cardiovascular disease, and these expression changes mimicked the expression pattern of the same genes at baseline in SH rats. 56 Diesel exhaust inhalation, however, was not able to further exacerbate the transcriptome expression in SH rats, implying that their ability to induce changes in response to pollutant exposure has been compromised due to disease. 56 When hypertensive rats were made normotensive by treating them with hydralazine, diesel exhaust inhalation–induced lung injury, inflammation, and vascular effects were diminished. 10 These data clearly demonstrated the role of hypertension in exacerbating the effects of inhaled pollutants. However, what brings about hypertension-associated physiological changes becomes an important determinant of the susceptibility of SH rats to pollutant-induced cardiopulmonary effects. These findings led us to understand how susceptibility variations may depend on the phenotypic outcome being selected for in a given genetic background of SH rats and how underlying host conditions might, in a complex and interactive manner, impact the response to air pollution stress. In epidemiological studies, air pollution has been variably linked to increases in blood pressure among healthy and susceptible individuals. 57,58
Numerous epidemiological studies have linked air pollution to exacerbation of cardiovascular diseases. Since autonomic imbalance can mediate cardiovascular effects of air pollutants, 59,60 the individuals with underlying cardiovascular diseases might be impacted through effects on sensory and central autonomic pathways. Using WKY, SH, and 6 other healthy and cardiovascular and/or metabolically compromised rat models, 46,61 we obtained insights into how underlying host genetics and disease phenotype may influence the response to inhaled ozone. Measures of breathing, 62 injury, and inflammation varied greatly even between healthy strains. 63 Furthermore, alveolar lining levels of antioxidants, namely ascorbate and glutathione, at baseline 64 were correlated with lung physiological changes, protein leakage, and inflammation when the theoretical ozone dose was plotted against levels of ascorbate in the lung lavage fluid. 11 It was apparent that strains with highest levels of ascorbate in the lavage fluid were minimally affected by ozone in terms of injury and inflammation. 63 For example, obese and insulin-resistant James C. Russell (JCR) rats, which have a propensity to develop vascular disease, each having received low ozone effective dose (based on theoretical calculation) and high levels of ascorbate at baseline in the lung lining fluid, were least affected by ozone-induced lung vascular leakage and inflammation. On the contrary, the stroke-prone SH rats, which had the lowest level of lung lining ascorbate and received the highest effective ozone dose, were highly susceptible to lung injury and inflammation from ozone exposure. 11,63 These data collectively demonstrated that ascorbate and glutathione, the first line of defense in the lung lining, 65 are at least one of the critical determinants of susceptibility variation in how ozone may induce lung injury and inflammation. However, the causal mechanisms responsible for achieving differential lung lining antioxidant levels and the connectivity to other cellular physiological processes require further investigations.
When examining the global gene expression patterns of the lung in 5 genetically related strains which included healthy WKY, SH, stroke-prone SH, obese spontaneously hypertensive heart failure (SHHF), and obese insulin resistant JCR rats with vascular impairment, 61 we observed marked strain-related differences in transcriptome at baseline. We noted that the baseline pulmonary expression patterns of genetically closer strains (SH, SH-stroke-prone, and SHHF), which were cardiovascular compromised, were similar and reflected more severe cardiac complications. 40 However, the lung gene expression pattern of JCR rats (genetically somewhat distinct than other strains, derived by crossing LA/N-cp and SH obese rats 46 ) was generally the opposite of the other strains, implying a strain-specific variation in the transcriptome. We noted that 2 obese strains due to their leptin receptor mutation (JCR and SHHF) had opposing patterns of expression for most gene clusters in the lung including the genes involved in metabolic processes. 39 When exposed to ozone, these 5 strains shifted their lung expression in the same direction, albeit at different levels. 39 These changes in different rat strains suggest that while inhaled ozone induces similar effects on the lung, there are quantitative differences in expression of specific gene clusters in each strain based on their underlying condition. 39 Ozone effects on genes in the lung were primarily noted in SHR and stoke-prone SHR, but not in severely compromised SHHF and JCR. 39 The whole-transcriptome analysis of WKY and SH after ozone exposure, 39 unlike few inflammatory gene changes examined in these strains after fly ash particle exposure, 48 provided a different picture and insights into greater effects in SH rats of ozone than in WKY rats.
As in the lung, when baseline expression of genes in the heart tissue was determined, it was further apparent that JCR and SHHF had opposing expression profiles, whereas those genetically closer strains SHR, stoke-prone SHR, and SHHF showed similar expression of genes. 40 These data clearly emphasized that the phenotypic expression of disease might be similar in 2 strains of rats; however, the mechanisms that bring about these phenotypes depend on the underlying genetic differences and individual variability in the transcriptional response of regulatory networks. Because of these complexities, the therapeutic interventions targeted for a given disease phenotype might not be similarly effective in all individuals. These studies emphasize that the susceptibility variations are complex, and a systems-based approach with continued use of high-throughput technologies and identification of patterns of changes in gene expression, epigenetic regulators, and phenotypes collectively will provide increased understanding of mechanisms and guide personalized therapeutic interventions that will be effective.
Use of Rodent Models of Metabolic Alterations in Air Pollution Susceptibility Studies
Research on air pollution further expanded to include metabolic diseases as a risk factor for exacerbated air pollution effects, as new epidemiological evidence began to emerge linking air pollution to these diseases (ie,. diabetes, metabolic syndrome, obesity). 66,67 The interest in how diet could modify health effects of pollutants prompted studies that included unhealthy diets as modifiers of air pollution health effects. Since air pollution was linked to a wide array of systemic effects, the quest to identify circulating mediators that produce systemic effects was also apparent. 68 –70 Likewise, the choice of animal models used for understanding mechanisms of susceptibility variations began to include diet-induced predisposition to metabolic diseases, such as obesity and diabetes. A number of studies have been published examining how ozone-induced inflammation may be associated with greater production of interleukin-33 in obese mice and the modulation through interleukin-17A and the influence of gut microbiota in exacerbating ozone-induced airway hyperresponsiveness. 71 –73 Although these studies have shown increases in ozone-induced inflammatory response in obese mice, our use of 2 rat models of obesity with genetic predisposition to cardiovascular diseases showed no exacerbation of net ozone–induced injury and inflammation relative to healthy rats. 63 This suggests that multiple host factors including genetic background might be critical in the exacerbation of the inflammatory response in obese models. Moreover, the regulatory mechanisms that control obesity might influence susceptibility to lung injury and inflammation differently in individuals.
Diet supplementations and nutrient deficiencies have been examined as modifying factors of air pollution–induced cardiopulmonary health impairments. 74 –77 Feeding of a high-fat diet in healthy rats has been linked to increased metabolic impairment and insulin resistance after repeated particulate matter instillation. 78 In a mouse model, high-fat diet has been shown to increase insulin resistance after repeated exposure to particulate matter, which correlated with increased plasma to tissue ratio of heat shock protein-70 that has been shown to be a biomarker of systemic changes associated with type 2 diabetes. 79 High-fat diet in a mouse model has also been shown to exacerbate changes in blood–brain barrier integrity after traffic-related motor vehicle exhaust exposures as evidenced by increased expression of oxidized low density lipoprotein signaling in the vasculature. 80 High-fat or high-carbohydrate/fructose diets in male and female Brown Norway rats, which are resistant to developing diet-induced obesity, showed increased body fat percentage relative to lean mass in males, 81 but when exposed to ozone, diet did not significantly exacerbate lung injury or inflammation. These studies imply that that diet-induced susceptibility differences are dependent on model selection and biological end point.
Individuals consuming healthy dietary fats might have reduced responsiveness to air pollutants. Based on the health benefits of omega-6 and omega-3 fatty acids in a clinical study showing decreases in particulate air pollution–induced vasoconstriction, 82 we demonstrated that rats fed an omega-3-rich diet, similar to humans, had attenuated vascular contractility response to ozone. 75 However, these rats developed foamy macrophage accumulation in the lung with impaired lipid transporters expression, suggesting inhibition of lipid surfactant clearance. 75 Although it is likely that the type and the concentration of dietary supplements might play a role in this adverse effect, lipid-rich diets with several unsaturated binding sites are likely to have impact on the lung as lipids can accumulate in the form of surfactant. And constant exposure to oxygen and oxidant pollutants also makes the lipids more susceptible to oxidation within the lung. Thus, not only host factors but also the type of intervention can impact the effects of air pollutants.
Air Pollution and Neurobehavioral Impact: Use of Animal Models of Neural Diseases
In the past decade, significant epidemiological and experimental evidence has been accumulated pertaining to the effects of air pollutants on various brain areas and neurobehavioral alterations. 83 In epidemiological studies, exposure to air pollutants has been linked to Alzheimer disease, 84 depression and suicide, 85 cognitive dysfunction, 86 Parkinson disease, 87 autism spectrum disorder, 88 and neurodegenerative diseases. 89 Recently, short-term exposure to air pollution has also been associated with increases in criminal activities. 90 However, to date, only limited experimental studies have used animal models of brain disorders to determine whether and how air pollutants interactively exacerbate the disease phenotype. In one study, exposure to diesel exhaust for 5 to 13 weeks in female 5X familial AD mice prone to Alzheimer disease has been shown to accelerate amyloid-β plaque formation in the brain without having significant systemic inflammation. 91 In a genetic stress-sensitive rat model of depression, the Flinders Sensitive Line rat, repeated ozone exposure is associated with exacerbated behavioral alterations and oxidative stress in the hippocampus and frontal cortex, relative to control animals. 92 These studies are beginning to emphasize the role of neural regulation in health effects of air pollutants and how central mechanisms may contribute to human susceptibility variations.
Evidence of the Role of Neuroendocrine System in Mediating Air Pollutant Health Effects and Its Implications in Susceptibility Variations
It is well-documented that when a stress is encountered, a highly conserved host response will involve the activation of 2 major survival mechanisms, that is (1) mobilization of energy sources from storage sites and channeling of metabolite precursors to organs where they are needed the most and (2) recognizing the location of stress and mobilizing immune cells to the site of injury to fight invading organisms or creating an innate immune response that removes unwanted or damaged tissues/cells to initiate healing process in case of an injury or tissue damage. 93 The degree of host response to a stressor will depend on the underlying physiological condition and genetic variability. The peripheral autonomic sensory nerves recognize stress in the body and relay the information to the brain centers through the activation of stress responsive regions. 94 The sympathetic and parasympathetic arms nervous system are activated. This leads to the activation of neuroendocrine stress response pathways involving sympathetic–adrenal–medullary (SAM) and hypothalamus–pituitary–adrenal (HPA) axes, which release adrenal-derived stress hormones, such as epinephrine and corticosterone/cortisol, respectively. 95,96 These are the hormones, which through their receptors in various tissues, mediate cellular metabolic and immune changes to protect the host from stress. When stress is no longer a threat, these same hormones reverse acute stressor–induced effects through feedback inhibition. 97 Because of selective distribution of the stress hormone receptor subtypes in the brain centers, these hormones also regulate learning and memory that is likely linked to an adaptation/habituation response. 95,98,99 These host survival mechanisms, well characterized for bodily injuries and infections, are tailored to the type of stressor encountered and the location of the injury in the body (Figure 2). Any variations in these mechanisms 100 might impact how individuals differ in their susceptibility to stressors.
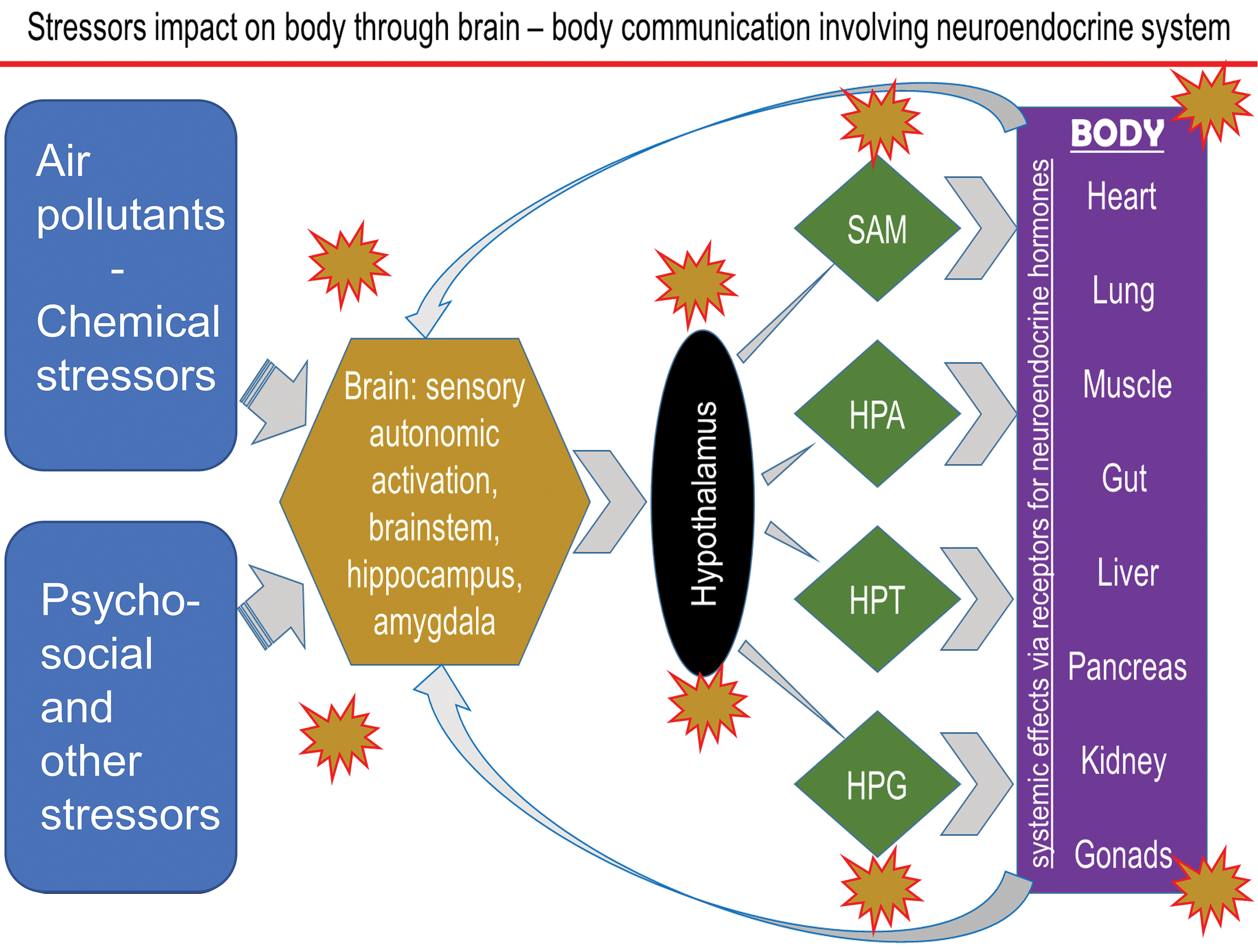
The proposed schematic of the mechanisms by which chemical and nonchemical stressors affect multiple organs through the activation/suppression of neuroendocrine axes. HPA indicates hypothalamic-pituitary-adrenal; HPG, hypothalamus–pituitary–gonadal axes; HPT, hypothalamus–pituitary–thyroid; SAM, sympathetic–adrenal–medullary.
Although the neuroendocrine-mediated activation of SAM and HPA stress axes in maintaining homeostatic balance are well characterized, prior air pollution studies neither systematically examined their involvement in peripheral and local health effects nor assessed their potential significance in disease causation or susceptibility variations. In inhalation toxicology studies, these acute reversible physiological changes have been often recognized but not characterized for their systemic and pulmonary consequences. 101 Air pollution effects on autonomic nervous system, and sympathetic and parasympathetic reflex cardiopulmonary changes, however, have been well characterized 102,103 such that air pollution regulations, in part, rely on evidence of autonomically mediated cardiovascular changes in heart rate variability. 104,105
The research effort for identifying circulating mediators responsible for extrapulmonary effects of air pollutants prompted us to examine a wide array of circulating cytokines, acute phase proteins, and metabolic hormones in our experimental studies. The lack of increases in circulating cytokines, in general, but striking increases in leptin and acute phase proteins in many of our studies 106 –108 led us to consider neural effects and subsequent peripheral changes. The Gackière et al 109 paper of 2011 showing the activation of hypothalamic stress responsive regions in rats acutely exposed to ozone served as a catalyst for us to consider neural mechanisms, especially the neuroendocrine stress pathways in examining air pollution health effects and how this may be important in susceptibility variations.
Linking Peripheral Metabolic and Immune Effects of Irritant Air Pollutants to Neuroendocrine Stress Pathways
Our first few studies on neuroendocrine pathways focused on circulating stress hormones and characterization of resultant peripheral effects. We demonstrated that ozone exposure increased circuiting epinephrine, corticosterone/cortisol, and acute phase response proteins. 70,107,108 A single exposure to ozone also resulted in a series of metabolic effects together with depletion of circulating lymphocytes in a reversible manner. These effects were not associated with the release of cytokines from the lung into the circulation as metabolic effects occurred before detectable lung inflammation. 107,108 These effects were characterized by the release of numerous free fatty acids into the circulation, likely through ozone-induced adipose lipolysis, increases in circulating branched-chain amino acids indicating muscle protein catabolism, increased gluconeogenesis, inhibition of lipid synthesis as indicated by downregulated gene expressions in the liver, and inhibition of bile acids release into the circulation with increased cholesterol, indicating inhibition of cholesterol breakdown. 108 Ozone exposure produced hyperglycemia, glucose intolerance, and diminished glucose-mediated insulin secretion from pancreatic β cells. 70,107,108 Subsequently, published epidemiological studies incorporating metabolic homeostasis markers have reported association between air pollution and indices of altered glucose homeostasis. 110,111
Animal studies involving exposure to another irritant air pollutant, acrolein, which deposits primarily in nasal airways, also exhibited similar peripheral metabolic changes. 112 Acrolein-induced nasal injury/inflammation and also lung inflammation were associated with increases in circulating stress hormones, with diabetic rats being more susceptible to metabolic effects. 112 In a human clinical study where ozone exposure occurred during intermittent exercise, similar increases in a variety of free fatty acids in the circulation and metabolites of membrane lipid breakdown together with increases in cortisol/corticosterone were shown. 113 Virtually, all pathways associated with lipid metabolism were altered as evident by changes in circulating lipid metabolites in humans exposed to ozone. 113 Thus, we demonstrated that this stress response to ozone was coherent between species. There are a few other recent studies that have also examined the activation of the neuroendocrine system, especially the HPA in mediating ozone effects. 114 –116 One earlier study examining the effects of concentrated ambient particles in rats with allergic airways disease showed changes in the stress response pathway. 117 All these effects observed in rat models and in humans indicated that SAM and HPA axes were stimulated by ozone and acrolein exposures and caused peripheral effects. 118 This fight-or-flight response, induced by other host stressors or injuries, likely contributes to variations in susceptibility to effects of air pollution. Epidemiological studies have begun examining the biomarkers of HPA activation and subsequent associations with circulating stress hormones as well as metabolic alterations after exposure to increased levels of air pollutants. 119
The confirmation of the role of circulating stress hormones in mediating systemic and pulmonary effects of ozone was initially made using the intervention of adrenalectomy, since adrenals are the main source of adrenaline and glucocorticoids. As expected, bilateral total adrenalectomy depleted circulating corticosterone and epinephrine nearly completely, but norepinephrine levels did not change as it is produced by nerve terminals and acts locally in a paracrine manner. 120 To our surprise, adrenalectomy resulted in near elimination of all ozone-mediated systemic metabolic, immunological, and pulmonary effects emphasizing the role of circulating adrenal-derived stress hormones in mediating these effects. 120,121 In follow-up studies, we demonstrated that if the receptors of these stress hormones (ie, adrenergic and glucocorticoid) are selectively blocked using pharmacological treatments, ozone-induced lung injury and inflammation were inhibited in an antagonist-specific manner. 122 It was evident that the effect of blocking glucocorticoid receptors was more central and reduced all metabolic effects and systemic immunosuppression effects of ozone (ie, depletion of circulating lymphocytes). On the other hand, as β-adrenergic receptors are densely distributed in the lung and regulate respiratory muscle tone, 123 the blockade of these receptors was associated with reduced local lung vascular leakage and extravasation of neutrophils at the site of stress. 122 These data implied that the tissue distribution for adrenergic and glucocorticoid receptors is a critical determinant of the organ-specific effects of air pollution–induced stress response. The roles of β-adrenergic and glucocorticoid receptors in mediating ozone-induced lung injury/inflammation and systemic effects were further established by a gain-of-function experiment where adrenalectomized rats were treated with β-adrenergic and glucocorticoid receptor agonists and exposed to ozone. 124 Thus, any variations in these receptors distribution in specific organs or the alterations in receptors sensitivity can contribute to variability in host response to environmental stressors in peripheral organs.
The autonomic activation by air pollutants likely involves stimulation of sensory vagus nerves in the lung and brain regions including brain stem and hypothalamus, 109 leading to the activation of SAM and HPA axes. The vagus nerve innervating the lung originates from jugular and nodose ganglion, which receives projections from the nucleus tractus solitarius (NTS) of the brain stem. 125 The neural connection from NTS to hypothalamus involves norepinephrine and glucagon-like peptide-1 neurons. 126 Although ozone has been shown to activate these stress responsive regions, it is still not clear if ozone-induced changes in the lung initiates changes in the brain through vagal sensory nerves or if there are circulating mediators which reach and cross the blood–brain barrier to induce discrete effects on different brain regions including the hypothalamus. The key initiating event for systemic changes is currently being actively investigated in the field of air pollution health effects. 127
Our recent study has shown that acute ozone exposure induces a wide array of gene expression changes in the brain stem and hypothalamus that are similar to those induced during hypoxia (ie, interferon signaling and the activation of stress pathways). 128 These ozone-induced gene expression changes in rats are associated with increases in adrenaline and corticosterone while depleting prolactin, luteinizing hormone, and also thyroid-stimulating hormone, suggesting the concomitant inhibition of hypothalamus–pituitary–gonadal (HPG) and hypothalamus–pituitary–thyroid (HPT) axes. The activation of SAM and HPA and associated inhibition of HPG and HPT axes have been reported after application of other stressors. 129 These data suggest that air pollutants, especially those that induce lung irritation, impact all neuroendocrine axes leading to suppression of thyroid and reproductive pathways along with the activation of adrenal axes (Figure 2). Importantly, most of the ozone-induced changes in hormones are not evident in adrenalectomized rats, implying the contribution of circulating adrenal-derived stress hormones in regulating brain effects of ozone. Circulating stress hormones are needed to produce lung effects of ozone, and if no lung effects occur due to lack of stress hormones, no changes in brain regions are noted. 128 Glucocorticoids in conjunction with mineralocorticoids and catecholamines have been shown to interactively regulate the process of adaptation and habituation centrally. 99 The regulatory roles of adrenergic and glucocorticoid receptors systems and feedback inhibition need to be further examined in relation to health effects of acute and chronic air pollution exposures and more importantly how neuroendocrine response variations may lead to variations in human susceptibility to air pollution health effects.
The Potential Contribution of Stress Hormones in Adaptation/Plasticity and Susceptibility Variations
Once released into the circulation, stress hormones exert their cellular effects through adrenergic and glucocorticoid receptors. 130,131 Subtypes of adrenergic receptors regulate cardiovascular functionality, smooth muscle tone, and other homeostatic processes. 130 Impaired adrenergic receptor function in the brain has been shown to contribute to chronic stress in the central and peripheral organs. 132 –135 Glucocorticoid receptors are important in homeostatic cellular metabolic and immunological response of peripheral organs and also in regulating stress, memory, learning, and habituation centrally. 98,136 Impaired regulation of adrenergic and glucocorticoid receptors in the brain has been linked to chronic stress and a number of neurological disorders including depression and posttraumatic stress disorder. For example, glucocorticoid resistance has been noted in a variety of neuropathies and inflammatory conditions. 131,137 There is evidence that the selective distribution of mineralocorticoid and glucocorticoids receptors in limbic areas, which regulate stress response, is involved in habituation and learning. 99 However, the impacts of air pollutants on these receptor pathways have been poorly examined.
Reversibility of air pollution–induced changes and the loss of effects upon repeated daily exposure (often referred to as adaptation, habituation, or tolerance) to some pollutants, specifically ozone, has been the area of research interest with limited understanding of the mechanisms. 138,139 Based on the role of adrenergic and glucocorticoid regulation of habituation, learning, and memory 99,130,131 and the activation of SAM and HPA axes after a single air pollution exposure, it is conceivable that the adaptation that occurs upon repeated exposure to ozone will involve the loss of neuroendocrine activation through adrenergic and glucocorticoid mechanisms. The importance of the presence of catecholamines and glucocorticoids in the peripheral circulation, their increases following stress, and in mediation of systemic, pulmonary, and even brain effects of air pollutants 120,121,124,128 emphasizes their role in regulation of adaptive, homeostatic, and pathological processes. 131,132 It has been shown that after applying psychological stress, the changes in peripheral stress hormones is necessary for inducing inflammatory changes in the brain, 135 implying the regulatory role of peripheral stress hormones in orchestrating brain response. Our recent study showing the lack of ozone effects on brain in adrenalectomized rats corroborates with these findings. 128 Thus, the potential interactions between underlying host neurobehavioral disorders and air pollutant modification of susceptibility through neuroendocrine system are likely (Figure 3). The role of developmental reprogramming of neuroendocrine pathways has been proposed to increase susceptibility of children to allergic respiratory diseases in epidemiological studies. 140,141 The use of animal models with impaired activation of neuroendocrine stress response will prove useful in understanding how loss of adaptation can impact chronic diseases after exposure to air pollutants.
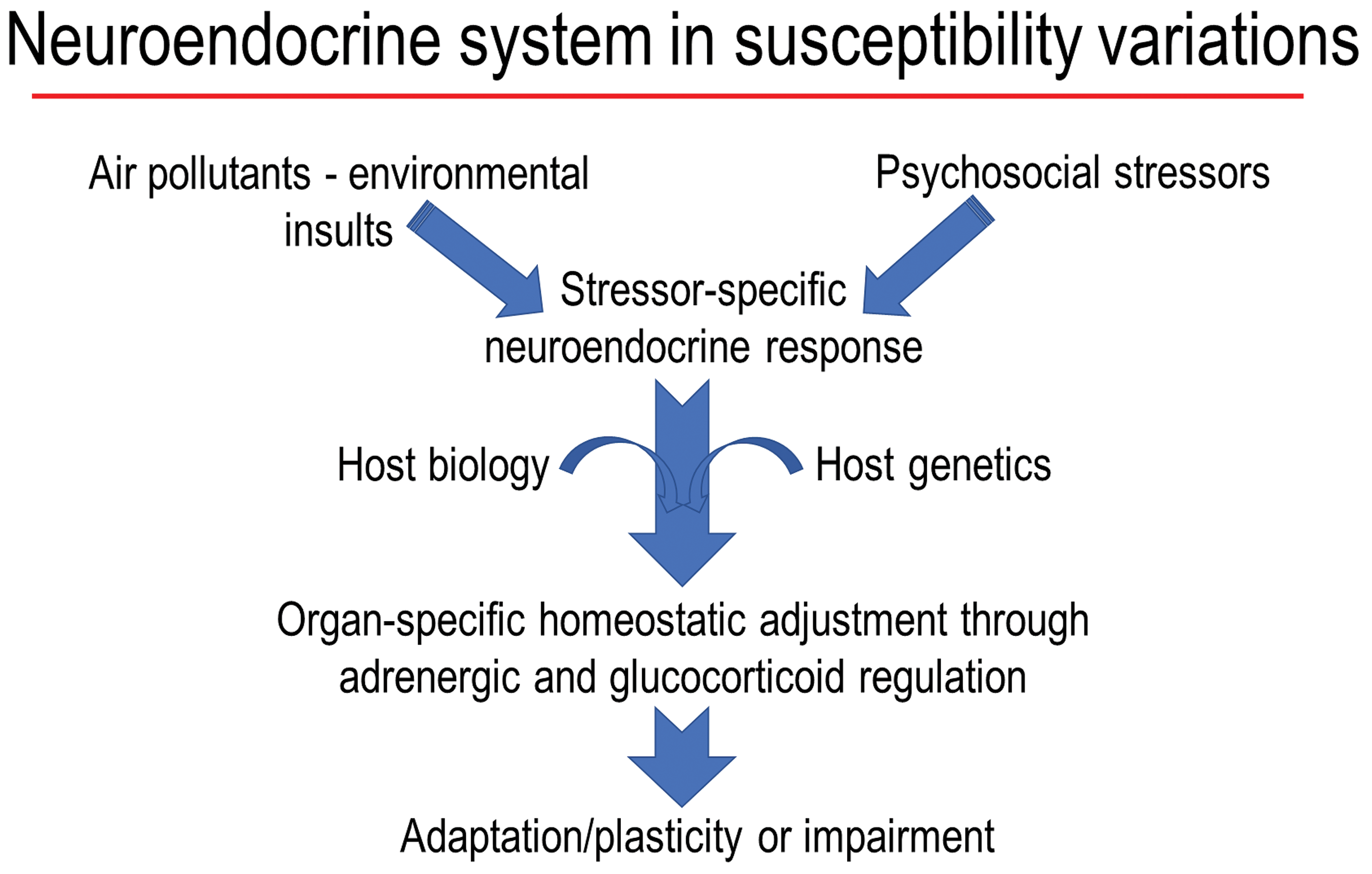
A flowchart of interactive effects of air pollutants and nonchemical stressors. Air pollutant–induced neuroendocrine activation producing a stress response, which when combined with the impact of other stressors and host susceptibilities may still maintain plasticity and lead to adaptation through adrenergic and glucocorticoid receptors or produce an impairment.
Neuroendocrine Activation and Allostatic Load in Susceptibility Variations
The concepts of neuroendocrine-mediated allostasis, habituation/adaptation, homeostasis, and allostatic load are not new and have been examined for decades in relation to the plasticity or the lack thereof of neuroendocrine system to accelerate disease. 95,126,142 –145 How poor socioeconomic conditions, diets, environmental exposures, and psychosocial stressors together with genetic predisposition can push the homeostatic system to lose plasticity and increase the susceptibility to chronic diseases, including acceleration of aging, has been studied and incorporated in modeling approaches to quantify health burden from stressful life conditions. However, this has not been done for air pollution as an environmental stressor. The new epidemiological 119,146 and experimental 12 evidence of the involvement of neuroendocrine system in mediating air pollution effects necessitates its incorporation in allostatic load modeling and assessing the contribution to human mortality and morbidity.
Air pollutants are physicochemically diverse, and therefore, their deposition, solubility, and retention can influence the extent to which acute neuroendocrine stress response is produced after inhalation. When exposures occur repeatedly over a long period of time or when chronic exposure leads to retention of inhaled pollutants, local lung irritation can result in induction of fibrotic processes ultimately causing proliferative changes or even cancer with some exposures (Figure 4). The pollutants that activate central stress responsive regions and the neuroendocrine axes will, based on host susceptibilities or the presence of other stressors, induce systemic metabolic and immunological changes to counteract stress. When overwhelmed by repeated encounters of stressors or cumulative impacts, the adrenergic and glucocorticoid functions can be impaired. This can contribute to allostatic load, causing neural dysregulation, chronic inflammation, and peripheral metabolic disorders based on the preexisting conditions (Figure 4). Thus, when examining susceptibility variations from exposure to air pollutants, multiple host conditions, the contribution of neuroendocrine pathways, and chronicity of air pollution exposure should be incorporated into study designs.
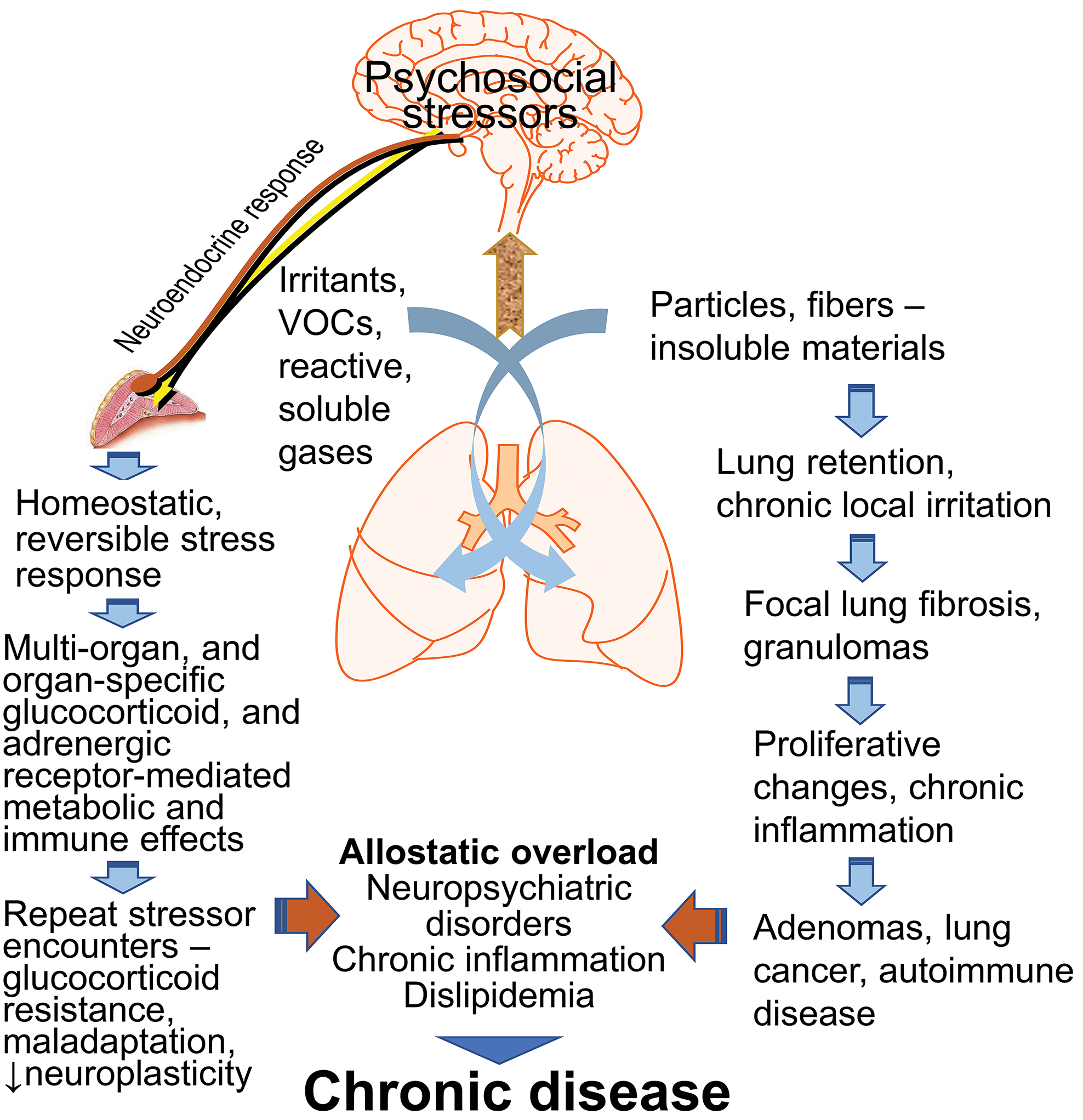
A proposed mechanistic paradigm of how air pollutants with other stressors contribute to individual variations in disease susceptibility. Acute exposure to all air pollutants may produce reversible homeostatic systemic metabolic and immune effects in a stressor-specific manner through neuroendocrine activation. Upon repeated encounter of stressors, however, there is likely a loss of adaptation/plasticity adding to allostatic load which can exacerbate disease. The pollutants that accumulate in the lung over time and produce low chronic irritation in lung epithelial cells additionally can induce local proliferative changes with the loss of adaptive stress response that can ultimately lead to active granuloma/fibrosis and/or lung cancer. This can have added contribution to the allostatic load and contribute to variation in susceptibility. VOCs = volatile organic chemicals.
Future Perspective
The epidemiological studies have provided associations between air pollution and biological as well as physiological indicators of chronic pulmonary and cardiovascular diseases, 57,58 metabolic diseases (including obesity), 66,67,146 neuropsychiatric disorders, and even low birth weight outcomes, 147 suggesting a mechanistic link. Given the evidence that the air pollutant effects are mediated through changes in endocrine activities, that the impairment of neuroendocrine function is linked to virtually all chronic dieases, and that the neurobehavioral disorders and chronic peripheral diseases coexist, necessitates more focused studies along neuroendocrine axes using systems approach. Epidemiological associations, such as those showing increases in blood glucose 148 and increases in pulmonary inflammation in children who are receiving bronchodilators, 149 can be linked to air pollution–induced activation of neuroendocrine response and release of stress hormones in the circulation. 12,119,146 Thus, the role of neuroendocrine system in air pollution and chronic disease susceptibility is mechanistically plausible; however, clear understanding of stressor-specific outcomes and interactive role of neural regulation in susceptibility variations remains an area needing further research. Although adrenergic and glucocorticoid receptor systems have been widely manipulated for currently used therapeutic interventions to treat chronic diseases, 150,151 further understanding of the role of central neuroendocrine mechanisms in air pollution health effects offers opportunities to identify new mechanistic pathways for therapeutic interventions. Other well-recognized interventions that could improve health of susceptible individuals include access to healthy living conditions, healthy diets, and reduced psychosocial stressors that are linked to health disparities.
Impaired neuroendocrine regulation of homeostasis can contribute to host susceptibility in the presence of other stressors, such as chronic diseases, obesity, psychosocial pressures, poor diets, genetic predisposition, and air pollution. Epidemiological association studies have begun to incorporate the allostatic load concept in air pollution studies. 152,153 The concepts of chemical and nonchemical stressors, external and internal exposomes, and one health have already emerged. 154 Given the role of neuroendocrine system in processing all perceived stress signals centrally and producing stressor-specific metabolic and immune responses through coordination of all organ systems, one can presume that its malfunction can lead to abnormal stress response and increase in host susceptibility. Assessing the biological plausibility for interactive influence of each of these risk factors and identifying the biological indicators or group of indicators of stressor-induced susceptibilities with or without air pollutants will contribute to mechanistic understanding of the overall health outcomes. 145,155,156 As modeling approaches have been applied to assess the risk of cumulative stressors on overall well-being, health, and longevity, more emphasis is needed to understand molecular mechanisms with systems concepts in mind. New insights have also emerged in the biology of developmental processes, and therefore, the research on understanding how early life stresses may predispose one to adulthood diseases has grown 157 (not covered in this article). The incorporation of the neuroendocrine homeostatic mechanisms and dynamicity of response with consideration of all life stressors, host physiology, and genetics, with or without incorporating air pollutant exposure, will be useful in understanding the mechanisms of susceptibility variations.
Footnotes
Authors’ Note
The research described in this article has been reviewed by the National Health and Environmental Effects Research Laboratory, US Environmental Protection Agency, and approved for publication. Approval does not signify that the contents necessarily reflect the views and policies of the Agency nor does the mention of trade names of commercial products constitute endorsement or recommendation for use.
Acknowledgments
The authors thank Drs Michael Madden, Colette Miller, and Ian Gilmour of the US EPA for their critical review of the manuscript. Most of the previously published work presented in the second half of the paper is performed at the National Health and Environmental Effects Laboratory of the US Environmental Protection Agency, Research Triangle Park, North Carolina, by my 3 excellent trainees, Drs Andres Henriquez, Samantha Snow, and Desinia Miller.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The preparation of this review paper was supported by the US Environmental Protection Agency’s Intramural Research Program.