
Abstract
Chemotherapy-induced peripheral neuropathy (CIPN) is an adverse effect caused by several classes of widely used anticancer therapeutics. Chemotherapy-induced peripheral neuropathy frequently leads to dose reduction or discontinuation of chemotherapy regimens, and CIPN symptoms can persist long after completion of chemotherapy and severely diminish the quality of life of patients. Differences in the clinical presentation of CIPN by widely diverse classifications of anticancer agents have spawned multiple mechanistic hypotheses that seek to explain the pathogenesis of CIPN. Despite its clinical relevance, common occurrence, and extensive investigation, the pathophysiology of CIPN remains unclear. Furthermore, there is no unequivocal gold standard for the prevention and treatment of CIPN. Herein, we review in vivo and in vitro models of CIPN with a focus on histopathological changes and morphological features aimed at understanding the pathophysiology of CIPN and identify gaps requiring deeper exploration. An elucidation of the underlying mechanisms of CIPN is imperative to identify potential targets and approaches for prevention and treatment.
Keywords
Introduction
Peripheral neuropathy is a common neurological disorder affecting peripheral nerve fibers including autonomic, motor, or sensory fibers. 1 Underlying etiologies of peripheral neuropathy are widely diverse and include genetic, infectious, metabolic, and toxic causes (Table 1). Peripheral neuropathy can be classified as a spectrum of diseases that are heterogenous and complex. 1 The peripheral autonomic, motor, and sensory neurons, their afferent and efferent axons, myelinating and nonmyelinating Schwann cells, and connective tissue components (blood and lymphatic vessels, endoneurium, epineurium, and perineurium) can all be differentially involved; however, an axonal lesion accompanied by Wallerian degeneration-like is a common feature that characterizes peripheral neuropathy, regardless of the underlying cause. 1,15,16
Examples of Agents and Conditions Associated With Peripheral Neuropathy.

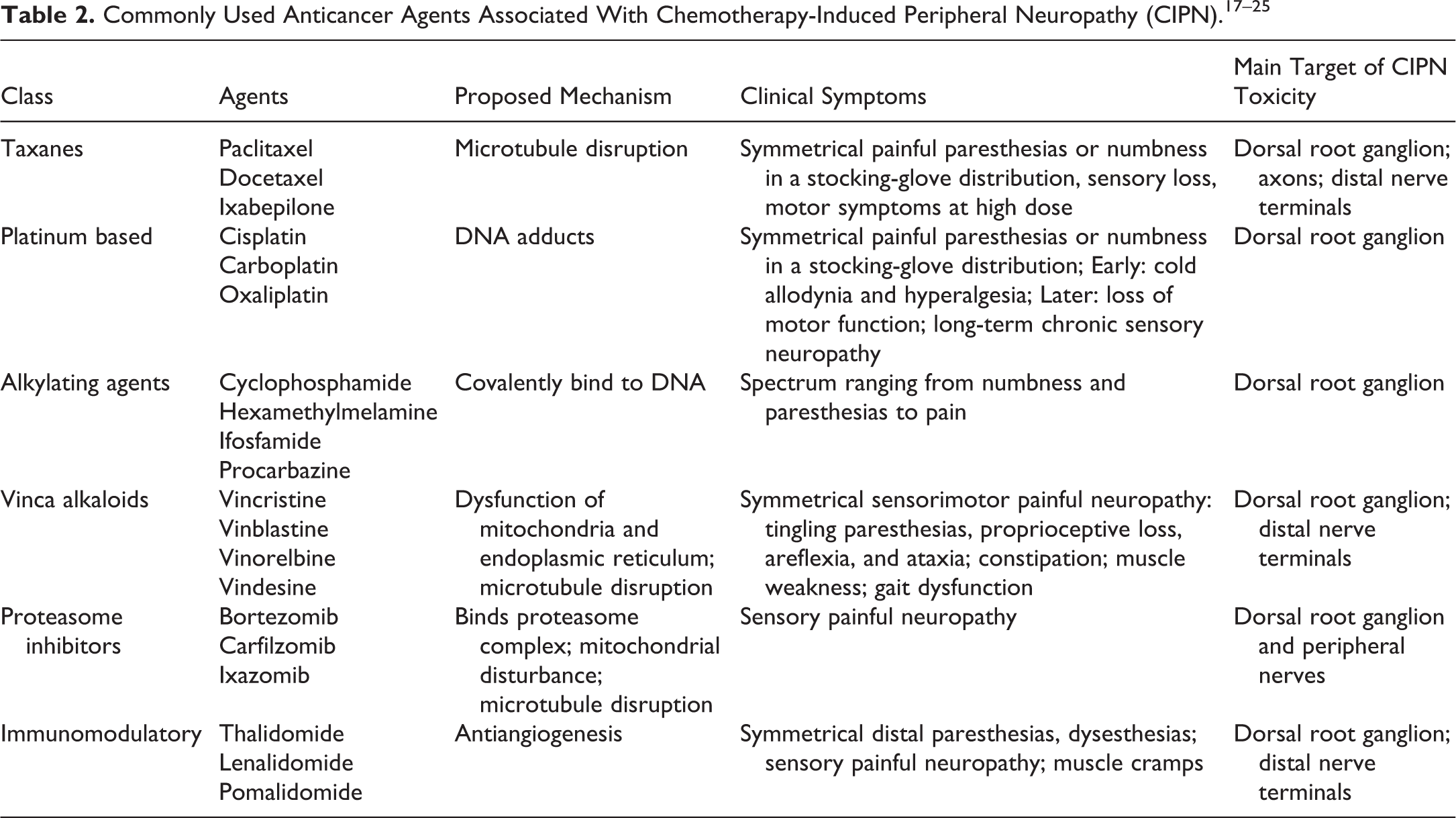
Chemotherapy-induced peripheral neuropathy (CIPN) is an adverse effect caused by a spectrum of classes of widely used anticancer therapeutics including platinum-based agents, microtubule disruptors (taxanes and vinca alkaloids), proteasome, and angiogenesis inhibitors (Table 2). 17 –26 Peripheral nervous system damage has also been reported with targeted cancer therapies; however, most reports are anecdotal and combination therapies with CIPN drugs are frequently used with target-specific therapeutics. 27 Chemotherapy-induced peripheral neuropathy is often dose dependent and progressive during and after treatment. 17 It frequently leads to dose reduction or discontinuation of chemotherapy regimens containing CIPN-causing agents. 23,28,29 Further, CIPN symptoms can persist long after completion of chemotherapy (“coasting”) and severely diminish the quality of life of patients. 18,30 As the number of long-term cancer survivors increases, the long-term effects of CIPN are becoming even more important and mechanistic explanations are needed to find effective treatments.
Peripheral neurotoxicity mechanisms of anticancer drugs are not fully understood but may result from interactions with DNA, mitochondria, ion channels, glutamate neurotransmission, and/or kinases, involving various targets in the peripheral nervous system such as dorsal root ganglia (DRG), sensory neurons, Schwann cells, and/or satellite glial cells. 31 –34 While the underlying mechanisms for CIPN are not known, current data identify a “dying back” axon degeneration of distal nerve endings as the major pathology in this disorder. 22 Mechanistic understanding of axonal degeneration will provide insights into pathways and molecular dynamics responsible for CIPN. The pathogenesis of CIPN and pathways shared with injury-induced Wallerian degeneration-like have been reviewed. 35 Development of effective mechanism-based therapies will need to rely on appropriate use of models that replicate essential features of peripheral neuropathy and use appropriate end point measurements that are relevant to the pathogenesis of the disease. 36 This review aims to focus on in vivo and in vitro model systems used to explore the underlying pathophysiology and mechanisms that lead to CIPN.
Clinical Assessment of CIPN
Differences in the clinical presentation of CIPN have spawned multiple mechanistic hypotheses to explain the pathogenesis of CIPN. Clinical symptoms of peripheral neuropathy are just as diverse as the causes and depend on the type of the nerve fiber affected: involvement of autonomic nerve fibers leads to autonomic dysfunction, which may manifest as orthostatic hypotension, cardiac dysthymia, abnormalities of sweating, incontinence, erectile dysfunction, or gastrointestinal symptoms, such as persistent nausea, vomiting, constipation, or diarrhea; motor nerve fiber involvement can lead to muscle weakness and wasting; sensory fiber involvement can manifest as ataxia, poor balance, and paresthesias, which can be extremely painful. 33,36
Clinically, CIPN is manifested predominately with the loss of sensory function 18,23,37 and presents in a “glove-and-stocking” distribution (Figure 1). 24,26 Diagnosis of peripheral neuropathy is often made based on patient history of symptoms, functional and electrophysiological features to corroborate involvement of specific nerve fiber types, and characteristic neuropathologic features derived from histopathologic methods. 1 However, sural or other whole nerve biopsies are rarely indicated in the evaluation of CIPN in patients. 22 Nerve conduction velocity testing is routinely used to assess CIPN in patients and animal models. 17,29,36,38 –41 The reduction in amplitude of sensory nerve action potentials and the “dying back” degeneration of nerve terminals of sensory neurons residing in DRG are both hallmarks of the neurophysiological and histopathological basis of CIPN. 18
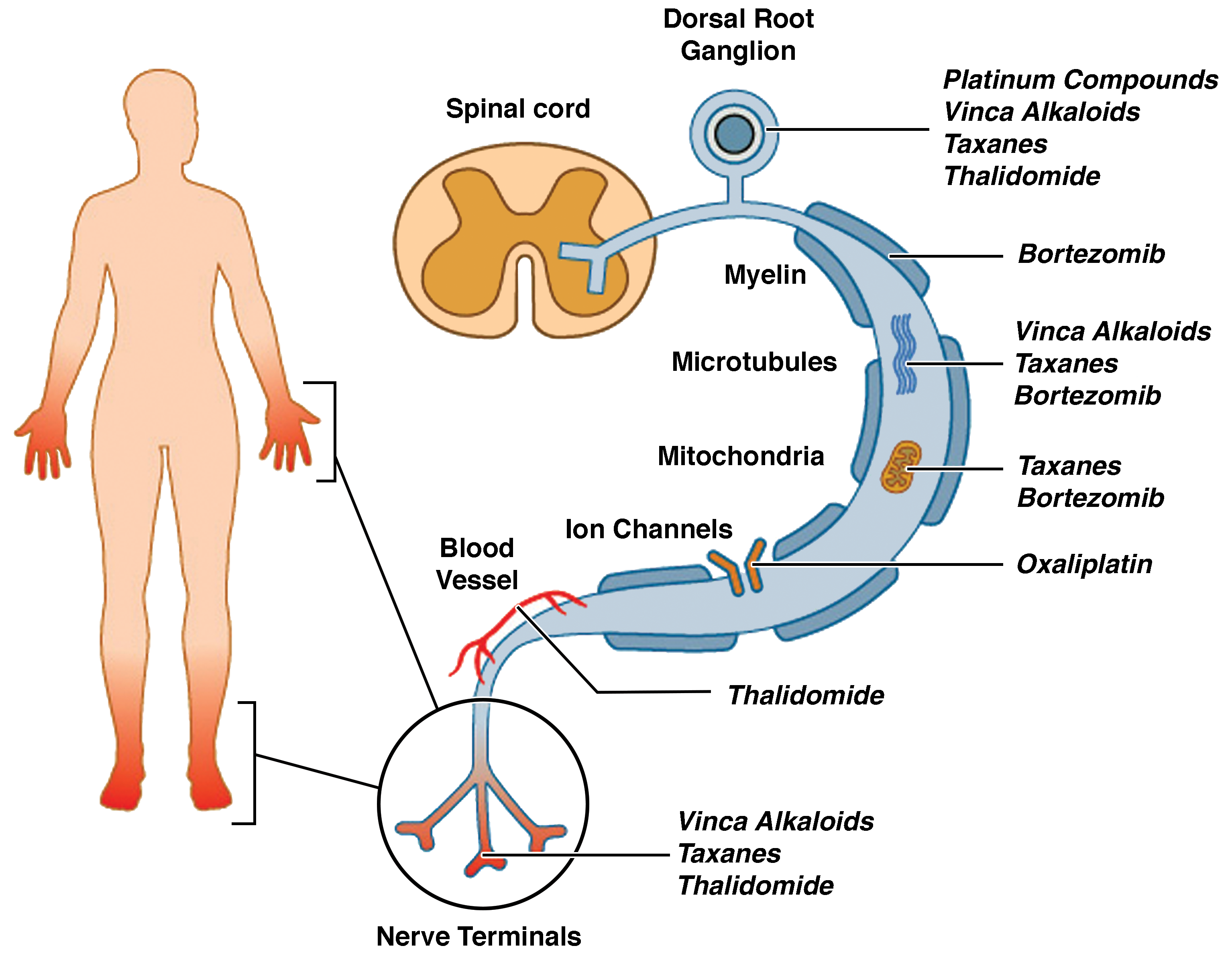
Depiction of the typical “glove-and-stocking” distribution of chemotherapy-induced peripheral neuropathy (CIPN) symptoms with putative targets for CIPN toxicity in the peripheral nervous system depicted from the dorsal root ganglion to axon and axonal components (myelin, microtubules, mitochondria, ion channels, and vascular network) and the distal nerve terminals. Reprinted from Park et al. 24 Reproduced with permission from John Wiley and Sons.
Histopathologic evaluation of nerve biopsies is clearly the definitive tool for diagnosing and detecting pathologic changes in peripheral nerves. Since CIPN shows more pronounced distal involvement, the distally located, conveniently accessible, and almost purely sensory sural nerve is the optimal choice for nerve biopsy 1 ; however, nerve biopsies are rarely indicated in patients with CIPN. 22 The role of skin biopsy in patients with CIPN is an emerging, minimally invasive tool for assessment of epidermal unmyelinated sensory fibers and small myelinated fibers of the dermis. 1,42 A consistent pathologic finding in patients with CIPN is intraepidermal nerve fiber (IENF) loss in hands and feet. 28 The IENF consists of unmyelinated axons from small-diameter sensory neurons. 43 Using immunohistochemistry and image analysis tools, IENF density can be measured to assess small fiber neuropathy in patients with CIPN as well as in animal and in vitro models of CIPN. 28,44,45 A “dying back” axon degeneration is the hallmark of CIPN in patients and rodent models that is assessed by histopathological examination of peripheral nerve biopsies or skin biopsy and measurement of IENF density. 38
Sensitive, noninvasive functional techniques to specifically assess small fiber neuropathy include nerve excitability threshold tracking in patients with CIPN 46 and Doppler flowmetry to determine axon reflex flare area in patients with peripheral neuropathy of several different etiologies. 47 A limitation of these techniques is that structural loss of innervation does not necessarily correlate with the presence of painful neuropathy symptoms in patients. 48
Pathophysiology of CIPN
Over the past 2 decades, a compelling body of evidence points to 4 main trends that have emerged from preclinical research on CIPN: neurotoxic anticancer drugs affect the peripheral sensory nerve by (1) directly targeting the mitochondria and producing oxidative stress, (2) functionally impairing ion channels, (3) triggering immunological mechanisms through activation of satellite glial cells, and/or (4) disruption of microtubules. 21,25 It is important to note that these various neurotoxic events are not necessarily related to the anticancer mechanisms of action for these agents and may account for the lack of effective treatment. It is hypothesized that a polytherapy which targets multiple mechanisms will most likely be the means to achieve neuroprotection. 21
Different chemotherapies affect distinct components of the peripheral nervous system, from the level of the sensory cell bodies in the DRG to the distal axon (Figure 1). 24 Dorsal root ganglions are a prominent target as they are less protected by the blood–nerve barrier and more vulnerable to neurotoxic damage, 49 potentially explaining the predominance of sensory involvement in patients with CIPN. Platinum compounds form DNA adducts that accumulate in DRGs 50,51 and lead to cell death in sensory neurons. 52,53 Vinca alkaloids, taxanes, and thalidomide have also been associated with DRG damage. 54 –57
Disruption of microtubules is another common mechanism of neurotoxicity. 19 Microtubules are central to axonal transport of proteins from the cell body into and down the length of the axon. 21 Taxanes bind to β-tubulin components of microtubule assemblies, producing overpolymerization and interference with normal microtubule dynamics, which has been linked to disruption of axonal transport. 58,59 Taxanes have also been shown to induce increased microtubule bundling in axons, which leads to alterations in peripheral nerve mechanical properties when treated in vitro or in vivo. 60,61 Vinca alkaloids bind tubulin and inhibit microtubule dynamics, leading to interference with the mitotic spindle. 57 Bortezomib, a proteasome inhibitor, also affects tubulin polymerization independent of its anticancer mechanism. 62 Bortezomib CIPN may be caused by an indirect increase in stabilization of microtubules due to increased expression of microtubule-associated proteins. 63
Bortezomib is also thought to cause peripheral neuropathy through mitochondrial toxicity and endoplasmic reticulum stress in Schwann cells, leading to pathological adaptive responses including demyelination and macrophage recruitment. 64 Finally, accumulation of ubiquitin-conjugated proteins leading to inhibition of transcription, transport, and cytoplasmic translation of messenger RNAs (mRNAs) is yet another potential mechanism of bortezomib. 54
Damage to the mitochondria and impairment of mitochondrial function may also play a vital role in the onset and development of CIPN. 65 Paclitaxel administration has been reported to cause prominent abnormalities in axonal mitochondria, 66 whereas bortezomib has also been reported to affect endoplasmic reticulum and mitochondrial integrity, particularly in Schwann cells. 64,67 In addition to energy deficiency, chemotherapies may damage the peripheral vasculature as exemplified by thalidomide, which reduces peripheral nerve blood supply via antiangiogenic effects that lead to axonal degeneration. 68
The role of neuroinflammation in CIPN pathology is becoming increasingly evident. 69 Penetration of the blood–nerve barrier and accumulation of chemotherapy agents in the DRG may cause neuroinflammation through activation of immune-like cells, such as satellite glial cells, 70 followed by secretion of mediators that enhance neuronal excitability and generate pain hypersensitivity. 71
Additional targets of neurotoxicity include direct axonal toxicity at the distal terminals, which may induce neurotoxicity and Wallerian degeneration-like following treatment with paclitaxel, 72 vincristine, 73 and thalidomide. 74 Oxaliplatin may directly alter axonal voltage-gated sodium (Na+) ion channel function, inducing an acute neurotoxicity manifested by peripheral nerve hyperexcitability. 75 –77 In some patients, bortezomib may induce primary myelin sheath degeneration. 67 However, despite potential diverse mechanisms underlying the development of CIPN, common degenerative pathways may be triggered when the normal processes and energy delivery mechanisms of the peripheral nervous system become disrupted.
Although it is well known that anticancer drugs may act on various subcellular targets of peripheral sensory nerves and that mechanisms of CIPN may be shared by several chemotherapeutic agents independent of their antitumor properties, there remains no consensus on the molecular mechanisms culminating in CIPN. The development of in vivo and in vitro models has provided valuable tools for studying the pathogenesis of CIPN and intervention strategies. 28,29,40,78,79
Animal Models
The first animal model of CIPN was described in 1992 using cisplatin in rats. 80 Since then, animal models have been used to reproduce neurotoxic effects of nearly all of the conventional chemotherapeutic agents and investigate the pathophysiology of CIPN using clinically relevant doses (Table 3), 23,28,29,38,40,84,85 with only the exception of thalidomide. 36,86 The outcome measure most commonly reported is evoked limb withdrawal to mechanical monofilaments, and the greatest increase in pain-related behavior is associated with vincristine. 86 Other outcomes measured include assessment of locomotor function, memory, reward, and attention. 86 In animal studies modeling CIPN, mice and rats show similar clinical symptoms and histopathologic features with no significant heterogeneity between them (Table 3). 86 Notably, histomorphologic changes observed in animal models of CIPN are consistent with those revealed in patients with CIPN. 29
Comparative Pathologic Features of Select Chemotherapeutic Agents That Cause CIPN in Humans and Rodent Models.
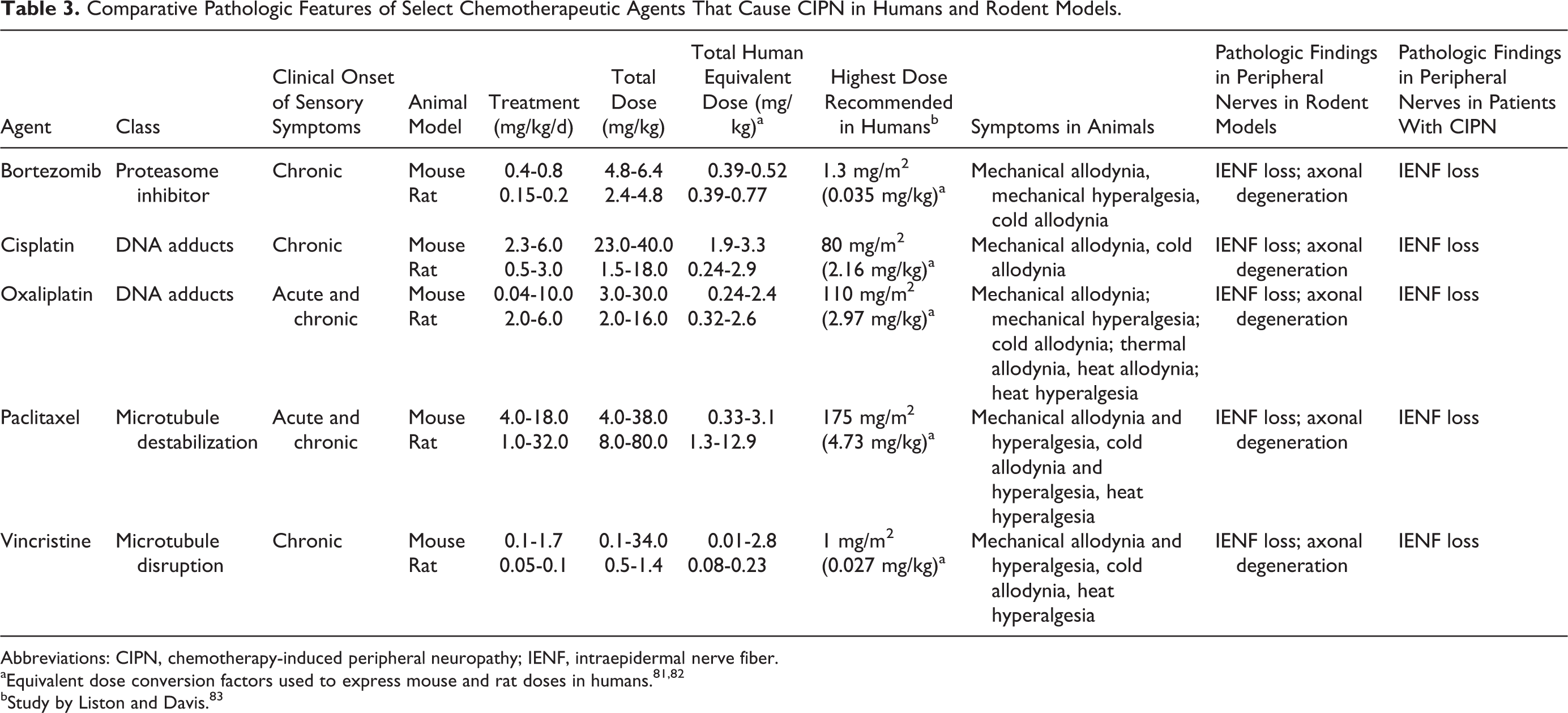
Abbreviations: CIPN, chemotherapy-induced peripheral neuropathy; IENF, intraepidermal nerve fiber.
bStudy by Liston and Davis. 83
Animal models of CIPN generally model the acute phase of CIPN, 86 whereas chronic (“coasting”) CIPN is frequently reported clinically. 87 –89 The most frequent behaviors reported in animal models of CIPN are estimates of gain in sensory function with hypersensitivity in paw withdrawal evoked by mechanical stimuli being the assay most often employed. 86 The most commonly reported pain-related behavior in animals is also reflex withdrawal responses. 90 This starkly contrasts with chronic clinical CIPN, where the predominant clinical sensory phenotype in patients with CIPN is sensory loss. 91 Thus, there is a dichotomous sensory profile reported from animal studies and what is observed in chronic patients with CIPN, potentially compromising the clinical relevance of these models for chronic CIPN; however, they may have more relevance to acute CIPN. 86
Very little information has been reported regarding CIPN in animals after chronic drug treatment. Electrophysiology and histopathology were examined after chronic chemotherapy treatment in mice given cisplatin, paclitaxel, epothilone B, or bortezomib for 4 weeks. 92 All drugs caused a significant reduction in sensory/motor nerve conduction velocities. Functional toxicity was confirmed by histopathological examination at both the light and electron microscopic level as axonal degeneration. 92
Höke and Ray provide a thorough review of animal models used to study CIPN. 40 Many of these animal studies demonstrate similar neuropathological changes seen in human CIPN and exhibit typical electrophysiological abnormalities expected in peripheral neuropathy, such as reduced nerve action potentials, and in some cases reduced conduction velocities. 36 However, there are unsettling differences in outcomes measured, including pathological, electrophysiological, and behavioral abnormalities observed in different laboratories. The source of such discrepancies may include genetic background of animals used, mode of administration (intravenous vs intraperitoneal), dose and duration of drug administration, and extent and detail of outcome measurements used in the evaluation of peripheral neuropathy. 36
Three tubulin-targeting anticancer drugs known to cause CIPN were compared at the maximum tolerated dose (MTD) for 2 weeks in mice. 93 Paclitaxel and ixabepilone produced significant reductions in caudal nerve conduction velocity, caudal amplitude, and digital nerve amplitude, whereas eribulin mesylate produced no significant deleterious effects on the same parameters. 93 All 3 agents showed axonal degeneration in DRGs and sciatic nerves with ixabepilone causing the most severe lesions, followed by paclitaxel and eribulin mesylate inducing mild changes (Figure 2). Ixabepilone, at its MTD (3 mg/kg), caused severe and frequent morphologic alterations, both in neuronal and in glial compartments of DRGs. Neurons exhibited dark cytoplasmic inclusions, often localized to the perinuclear area. Clear vacuolations and cytoplasmic swelling were evident in satellite glial cells, which were indicative of severely injured and degenerating cells. Additionally, adverse effects were evident at lower doses of ixabepilone. Cytoplasmic dark inclusions and degenerating neurons, as well as vacuoles of satellite glial cells, were evident in the 0.75 × MTD and MTD ixabepilone groups. Moreover, rare episodes of clear cytoplasmic vacuolation of neurons were also observed.
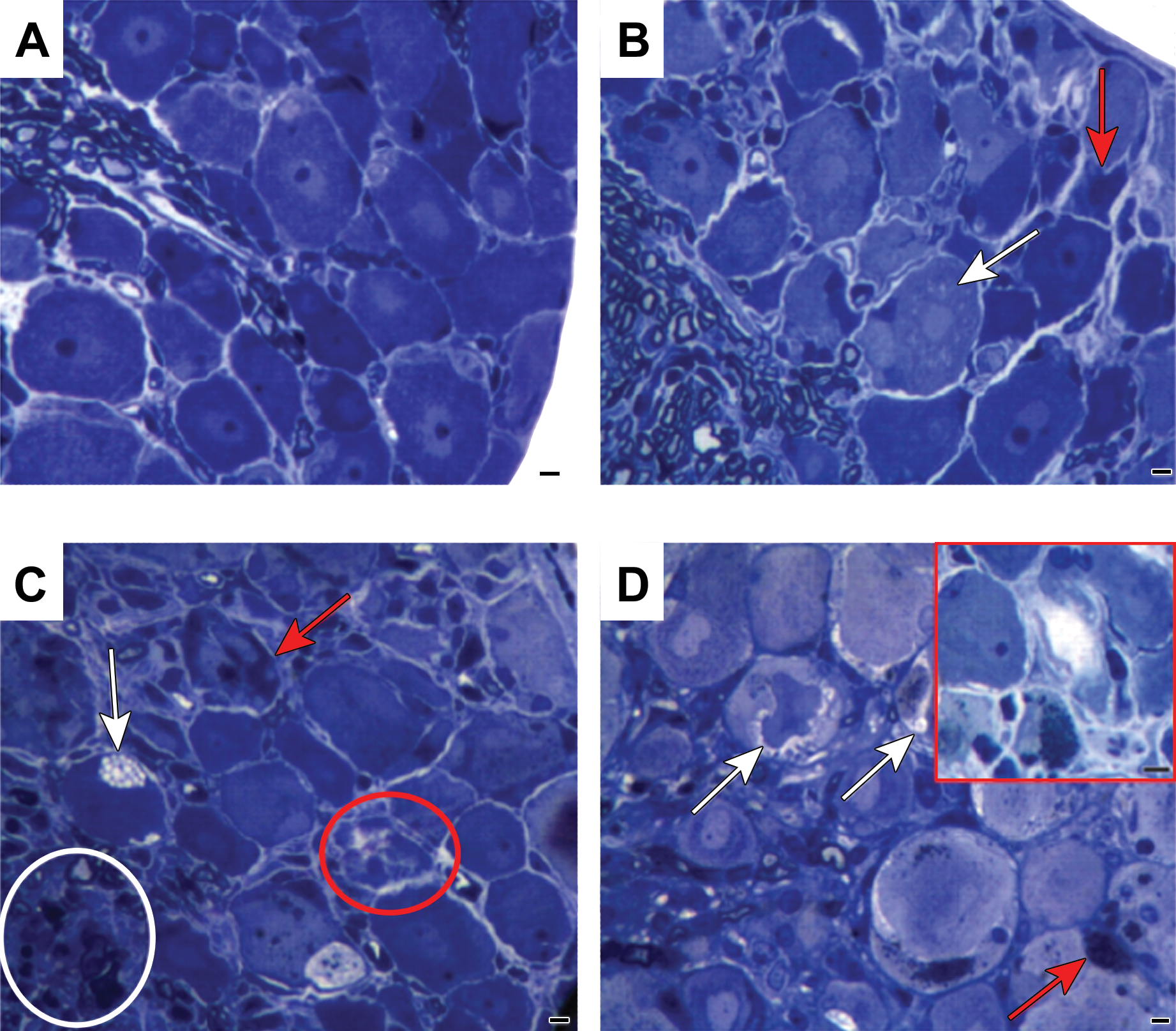
Effect of maximal tolerated dose (MTD) of eribulin mesylate, paclitaxel, and ixabepilone on dorsal root ganglion (DRG) morphology. Dorsal root ganglion morphology at the light microscopic level showed changes after each chemotherapy (at its MTD). Ixabepilone (D) caused most severe and frequent morphologic changes both in neuronal and glial compartments. Severely injured, degenerating neurons are outlined (red box). Dark cytoplasmic inclusions were evident and were often localized in the perinuclear area. Vacuolations (white arrows) and swelling phenomena (red arrowheads) were evident in the cytoplasm of satellite cells (D). Dorsal root ganglions from paclitaxel-treated mice (C) also displayed degenerating nerve cells (outlined by red circle) with dark cytoplasm (red arrows) and clear vacuolations in cytoplasm of satellite cells (white arrow). Alterations in the proximal axons of DRG were also observed (white circle). Dorsal root ganglions from eribulin mesylate-treated mice (B) showed mild pathologic changes evidenced by some cytoplasmic vacuolation (white arrow) and degenerating nerve cells (red arrow), as compared to vehicle-treated mice (A). Scale bar, 20 μm. Reprinted from Wozniak et al. 93 Reproduced with permission from American Association for Cancer Research.
A dose-dependent effect of paclitaxel on sciatic nerve morphology was observed at the MTD (30 mg/kg). 93 Severe pathologic changes consistent with axonal degeneration affected both large- and small-diameter fibers (Figure 2). Severity of axonopathy was milder than that seen with ixabepilone, but still clearly evident in groups treated with 0.75 × MTD (22.5 mg/kg) or 0.5 × MTD (15 mg/kg) paclitaxel. Paclitaxel at 0.5 × MTD (15 mg/kg) also caused formation of dark inclusions in the cytoplasm. In addition, DRGs from the 0.75 × MTD (22.5 mg/kg) group had clear vacuolations in the cytoplasm of neurons as well as in satellite glial cells. A proportion of neurons in the DRGs were degenerating and their cytoplasm appeared much darker than normal neurons at the MTD of paclitaxel (30 mg/kg). Dose-dependent axonal degeneration was present in proximal axons at 0.5 × MTD and greater.
Treatment with eribulin mesylate showed a dose-dependent effect on sciatic nerve morphology at 0.5 × MTD (0.875 mg/kg) to the MTD (1.75 mg/kg). 93 At these doses, eribulin mesylate induced mild-to-moderate pathologic changes consistent with axonal degeneration that affected both large- and small-diameter fibers (Figure 2). Clear cytoplasmic vacuolation of DRG neurons was evident between 0.75 × MTD and the MTD of eribulin mesylate doses, whereas dark inclusions were only rarely observed at the MTD.
Bortezomib-induced sensory neuropathy in rats revealed axonal degeneration in sciatic nerves after 8 weeks of treatment (0.15 or 0.20 mg/kg 3 times/week) and at the end of a 4-week follow-up period (0.20 mg/kg only). 94 Complete recovery of morphologic changes was observed in rats treated with 0.15 mg/kg, whereas only partial recovery was observed in the 0.20 mg/kg treated group. Sensory neuropathy in mice treated with 0.40 mg/kg bortezomib 3 times per week for 4 weeks showed the same axonal degeneration in sciatic nerves as in rats and a significant reduction in nerve fiber density. 39
The anticancer effects of tubulin-targeting agents generally attributed to their ability to bind microtubules, interfere with mitotic spindle formation, and ultimately block mitosis, resulting in cell death. 95 However, somatic neurons do not divide; therefore, neurotoxic effects of tubulin-targeting agents appear to be independent of their anticancer activity. An understanding of the mechanisms behind the neurotoxic effects of tubulin-targeting and other CIPN-causing agents is far from complete and further studies of the peripheral neuropathy of tubulin-targeting chemotherapies are warranted.
In Vitro Models
Primary DRG explants or dissociated cell cultures obtained from rodents have been used as in vitro models to recapitulate the pathophysiological feature of a predominantly sensory neuropathy and axonal degeneration in DRG neurons and to elucidate underlying mechanisms of CIPN. 28,35 Image analysis of neurite outgrowth is the end point most commonly employed to quantify morphological alterations caused by CIPN-inducing agents. 28,35,44,79,96 –100 Using adult DRG neurons isolated from 3- to 6-month-old male Wistar rats, Malgrange et al observed significant inhibition of neurite outgrowth by cisplatin, vinblastine, taxol, and vincristine at a clinically relevant concentration not altering the overt cell viability. 101 Time- and dose-dependent inhibition of neurite outgrowth was also observed in DRG explants exposed to cisplatin and/or taxol. 102,103 Toxic effects of vincristine and taxol that preferentially target neurites, especially distal axons, and contribute to the commonly featured pathological pattern of “dying back” in clinical CIPN were reproduced by a compartmentalized microfluidic culture platform that enables culture of DRG neuronal cell bodies (somas) and their axons (neurites) in separate chambers, hence, allowing local treatment of drugs to identify the primary site of action. 73,104
Measurement of electrical activity from compartmentalized somas and neurites with a multielectrode assay substrate demonstrated a progressive loss of electrophysiological function in neurites but not somas in cultured neurons from 15-day-old rat embryos exposed to vincristine, 105 substantiating the finding of reduced conduction velocity that has been observed frequently in clinical and animal models of CIPN. Ultrastructural analysis of taxol-treated DRG organotypic cultures or explants revealed an unusual aggregation of microtubules in neurons and the presence of microtubule-endoplasmic reticulum arrays and necrotic features in the neuronal somas and neurites, 103,106 whereas apoptotic cell death was noted in rat DRG neurons and PC12 cells exposed to cisplatin. 107,108 Mitochondrial impairment caused by taxol and cisplatin in DRG neurons was evident with loss of mitochondrial membrane potential; induction of mitochondrial DNA damage, cristae lysis, and autophagic vacuoles 108 –110 ; and abnormalities in mitochondrial bioenergetics, glycolysis, or mRNA transport in axons in DRG neurons isolated from rodents that developed taxol-induced painful neuropathy. 111,112
Exposure of rat DRG explants or dissociated neurons to bortezomib at clinically relevant doses caused time- and dose-dependent inhibition of neurite outgrowth without overt cytotoxicity, but was accompanied by somatic aggregation of β-tubulin and disruption in axonal trafficking of mitochondria. 113 In cultured rat DRG neurons, augmentation of the stimulated release of neuropeptide calcitonin gene-related peptide was observed with taxol treatment, which may play a role in taxol-altered neuronal sensitivity. 114 Inhibition of neurite outgrowth by cisplatin and bortezomib varied in DRG neurons of different rodent strains, indicating genetic background may contribute to variability in patient susceptibility to CIPN. 115
An increasing body of evidence indicates that supporting, non-neuronal cells in DRG-derived cultures are also involved in the pathogenesis of CIPN. 24,116 Our laboratory established a multiparametric morphology-centered rat DRG culture model allowing assessment of toxic effects of CIPN drugs on both sensory neuronal and non-neuronal (mainly Schwann cells) cell populations simultaneously. 44 In this model, CIPN-inducing agents bortezomib, cisplatin, eribulin, taxol, and vincristine induced a dose-dependent loss of neurite process area without overt cytotoxic effects on cell bodies, recapitulating the feature of “dying back” axonopathy observed in clinical or animal models of CIPN. 44 Compound-specific effects, such as neurite fragmentation by cisplatin or bortezomib and enlarged neuronal cell bodies by paclitaxel, were also observed, supporting the usefulness of this multicellular culture model to identify risk and examine mechanisms of CIPN. 44
Recent advances in stem cell technology to differentiate human embryonic stem cells (hESCs) or human induced pluripotent stem cells (hiPSCs) into DRG-like, peripheral sensory neurons have provided novel human-based, clinically relevant, in vitro models to study peripheral sensory neuronal development and injury. 96,98 –100,117 –120
The application of hiPSC-derived DRG-like sensory neurons to assess CIPN-inducing agents has been evaluated by several laboratories using commercially available cells; results support the use of hiPSC DRG-like cells for toxicity testing of compounds with CIPN liability. 79,98,100
Prevention, Restoration, and Other Intervention Strategies
It is beyond the scope of this article to discuss the plethora of agents and strategies that have been tested to prevent or treat CIPN in patients and animal models of CIPN. The reader is referred to several review articles that cover the topic. 37,121 –124 Suffice it to say that to date, there is no unequivocal gold standard for the prevention and/or treatment of CIPN. 37,124
Unmet Needs
There is a need for reliable, standardized, and validated clinical assessment end points or diagnostic tools to better identify the presence and severity of CIPN. 84,125 The clinical utility of such tools is critical when employed as end points in clinical trials designed to measure outcomes of prevention, mitigation, or treatment strategies for CIPN. 125
Factors to explore that likely contribute to the risk of CIPN include cumulative dose, therapy duration, synergistic neuropathy caused by previously given chemotherapies and concomitant chemotherapies, and the role of preexistent neuropathy. 19,126 Further complicating the epidemiology of CIPN is the existence of immediate, short-term “acute pain syndrome” with some chemotherapeutics, which may be severe enough to lead to reduced dosing or termination of life-saving treatment, whereas other chemotherapeutics induce pain after several treatment cycles. 23,126 A better understanding of the epidemiology of CIPN, combined with understanding of the anticancer mechanism of the chemotherapeutic, should help to clarify the mechanisms underlying CIPN and possibly define intervention strategies. 85
Although somatosensory symptoms of CIPN can generally be characterized as “neuropathic pain,” given that the symptoms share similarities with other painful peripheral neuropathies, such as diabetic neuropathy, with symmetrical tingling and “burning pain” that starts distally in the limbs, and with a stocking-and-glove distribution, 34 there are differential symptomologies evoked by different classes of chemotherapeutic agents. It is likely that each class or individual drug will have unique neurotoxicity mechanisms associated with them. Although a single therapy to treat CIPN is desirable, it is more realistic that several different therapeutics are required. Clearly, there is a need to understand the mechanisms of CIPN to develop effective therapeutic strategies. 85,126,127
Animal models of CIPN generally model the acute phase of CIPN, 86 whereas chronic (“coasting”) CIPN is frequently reported clinically. 87 –89 The mismatch between timing of clinical and nonclinical findings illustrates the need for closer scrutiny of animal models of CIPN to bridge the gap between nonclinical and clinical studies. 85
Typically, most animal models of CIPN involve administration of a chemotherapeutic agent in the absence of a tumor burden. 122 Although tumor-bearing rodent models of CIPN are more clinically relevant, the practical and ethical issues are not to be underestimated. 122 In situations where chemotherapy is administered after surgical removal of the tumor, modeling CIPN by chemotherapy administration alone is a valid approach. 122 The use of intermittent dosing schedules to generate CIPN models and consideration of the same dosing schedules across different laboratories would further understanding of causal mechanisms of CIPN and enhance reproducibility. 122
Conclusions
Despite the myriad etiologies and symptoms, and difficulty in diagnosing CIPN, histopathology is the same but is seldom used to characterize peripheral neuropathy. Axonal degeneration is the common histomorphologic change observed during CIPN and is the consistent pathological process in most drug-induced neuropathies. Although experimental studies have contributed to the understanding of the pathogenesis of certain drug-induced peripheral neuropathies, the basic mechanisms remain poorly understood. It is through mechanistic insights that new avenues for development of therapies to prevent or treat CIPN will be unveiled.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under Contract No. HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government. This research was supported [in part] by the Developmental Therapeutics Program in the Division of Cancer Treatment and Diagnosis of the National Cancer Institute.