
Abstract
The development of biomaterials, medical device components, finished medical products, and 3-D printed and regenerative medicine products is governed by a variety of international and country-specific standards and guidelines. Of greatest importance to planning, executing, and reporting biocompatibility, safety and efficacy studies for most biomaterials and medical components or products are the International Organization for Standardization guidelines, U.S. Pharmacopeial Convention, ASTM International, and Conformité Européenne (European Conformity) marking. The International Medical Device Regulators Forum publishes harmonized standards similar to the International Council for Harmonization. Good Laboratory Practices are applicable and guidance documents for the development of drugs and biologics can also be relevant to biomaterials, medical device components, and medical products and more recently to products produced by 3-D printing or additive manufacturing. Regenerative products may have medical device–based scaffolding and may be treated as biologics, reflecting the cell and tissue components. This compilation of international standards and guidelines provides toxicologic pathologists, toxicologists, bioengineers, and allied professionals with an overview of and source for important regulatory documents that may apply to the nonclinical development of their products.
Keywords
The action of a medical device is not dependent on chemical, pharmacological, immunological, or metabolic processes. Medical device materials and products may be composed of single or multiple biomaterials that require: biocompatibility testing of the biomaterials by in vivo implantation or other studies, safety testing (local and systemic toxicology of the finished device or clinical product), and efficacy testing of the functioning finished device or clinical product.
The necessity for establishing biocompatibility of individual materials and efficacy and safety testing of the finished medical device is distinct from the primary focus on safety, with limited to no efficacy testing, for drugs and biologics. For medical devices, all biocompatibility and efficacy testing occurs prior to any clinical testing. Being aware of and applying the proper testing standards to development and testing of the device can facilitate entry into the desired national or global market place. As standards organizations are the source of the majority of testing documents for medical devices, these documents are referred to as standards rather than guidelines/guidances that originate from governmental authorities. The International Organization for Standardization (ISO) authors the major regulatory standards, particularly the ISO 10993 series, for nonclinical biocompatibility and medical device testing. However, there are parallel national and regional standards that provide supplemental testing and considerations and independent international and national testing documents that may need to be integrated into safety and efficacy evaluation. This overview and compilation of international and national standards, along with potentially applicable guidance documents, is provided with Internet links and selected annotations. These annotated links should assist toxicologic pathologists, toxicologists, bioengineers, and allied professionals in identifying and using regulatory documents for nonclinical development of biomaterials, medical devices, and 3-D printed and regenerative products. This tabular compilation, along with a short suggested reading list, is not intended as a comprehensive manual but provides the reader with tabulated and annotated lists of source website information and regulatory documents for self-study. These standards and guidelines are current as of October 10, 2018. but the relevant organizations and their websites should always be consulted to confirm that the most recent standards are being applied.
Global Risk Classification
The ultimate goal for nonclinical testing is to provide a summary report that adequately documents that the biomaterials or finished medical device or products are biocompatible, safe, and efficacious. Risk classification terminology and regulatory controls needed for medical devices are based on use, level of surgical invasion or body contact, service duration, activity, and use of energy sources or technologies. Risk classification varies slightly between countries (Table 1), but Class II to IV and active implantable devices or Class B to D devices are the focus of nonclinical testing, with low-risk and noninvasive Class I or A medical devices generally not subject to nonclinical testing. Regenerative and 3-D printed devices are generally classified as custom devices. Appropriate categorization of a device is generally the first step in identifying the correct regulatory pathway and study needs.
Comparative Global Risk Classification of Medical Devices Exclusive of Special Rules that May Apply to Individual Devices in Different Countries.
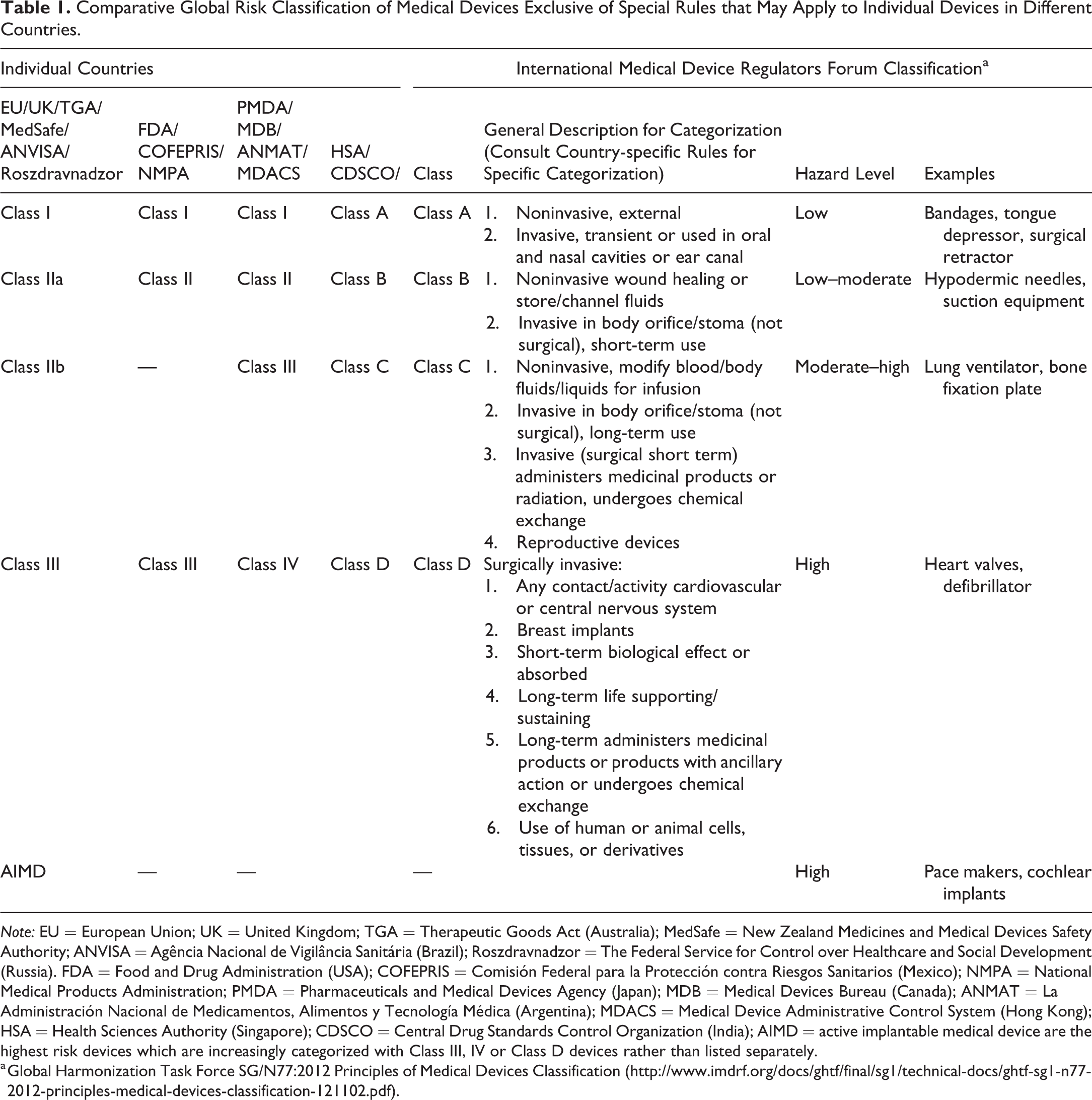
Note: EU = European Union; UK = United Kingdom; TGA = Therapeutic Goods Act (Australia); MedSafe = New Zealand Medicines and Medical Devices Safety Authority; ANVISA = Agência Nacional de Vigilância Sanitária (Brazil); Roszdravnadzor = The Federal Service for Control over Healthcare and Social Development (Russia). FDA = Food and Drug Administration (USA); COFEPRIS = Comisión Federal para la Protección contra Riesgos Sanitarios (Mexico); NMPA = National Medical Products Administration; PMDA = Pharmaceuticals and Medical Devices Agency (Japan); MDB = Medical Devices Bureau (Canada); ANMAT = La Administración Nacional de Medicamentos, Alimentos y Tecnología Médica (Argentina); MDACS = Medical Device Administrative Control System (Hong Kong); HSA = Health Sciences Authority (Singapore); CDSCO = Central Drug Standards Control Organization (India); AIMD = active implantable medical device are the highest risk devices which are increasingly categorized with Class III, IV or Class D devices rather than listed separately.
a Global Harmonization Task Force SG/N77:2012 Principles of Medical Devices Classification (http://www.imdrf.org/docs/ghtf/final/sg1/technical-docs/ghtf-sg1-n77-2012-principles-medical-devices-classification-121102.pdf).
Access to Medical Device Written Standards
Independent nongovernmental bodies are the source of many written testing standards for medical device-related products accepted by governmental agencies. These standards are usually from nonprofit scientific testing organizations, which use sales to pay for development, production, and regular review of standards and testing documents. The ISO, national pharmacopeias, and national standard organizations written standards are most important to study design and biological testing of biomaterials and medical devices. Toxicologic pathologists and toxicologists involved in safety and efficacy testing of biomaterials and medical devices need access to these standards, even though standards must be purchased and replaced if the document is updated. Standards such as the ISO 10993 series are reviewed every 5 years but may be confirmed or amended rather than substantively changed, and for toxicologic pathologists, the focus will be only on several selected standards so there is limited cost (e.g., 138 CHF [approximately US$145], as of June 1, 2018, for ISO 10993-6:2016) to maintain these important references. ISO webpages for individual standards provide a preview button that will show the table of contents to assist in determining the application of the standard. Similarly, ASTM International webpages for individual standards summarize the significance, use, scope, and cross-referenced ASTM documents. Other paid access documents are frequently less transparent. With the issuance of new standards, there is no obligation in the United States for a biomaterial or device to meet the new standards. However, in the European Union (EU) and other European Economic Area (EEA) countries using the Conformité Européenne ( ; European Conformity) standard and marking, changes in a product or changes in a directive or legislation may require a device to meet new standards for continued registration (https://www.nist.gov/standardsgov/compliance-faqs-ce-marking).
; European Conformity) standard and marking, changes in a product or changes in a directive or legislation may require a device to meet new standards for continued registration (https://www.nist.gov/standardsgov/compliance-faqs-ce-marking).
International Standards Organizations
Table 2 summarizes website sources for country-specific, international, and regional standards. These should be consulted for planning, executing, and reporting biocompatibility, safety, and efficacy testing in the listed countries. In general, most biomaterial and device testing will be based on the standards from the ISO (ISO-10993 series and others), ASTM International, and the International Medical Device Regulators Forum (IMDRF). Countries with and without dedicated laws and medical device regulatory authorities may have supplemental national or regional standards or trade policies that need to be met. Israel provides an example of a country with regulations that require registration and approval by the Medical Device Division of the Israeli Ministry of Health (AMAR), but prior approval in Australia, Canada, Europe ( mark), Iceland, Japan, or United States (PMA or 510(k)) forms the basis for and simplifies the approval process (http://www.loc.gov/law/help/medical-devices/index.php). When working in new geographic regions, in countries with no specific legislation for medical devices or when introducing novel products, a consulting firm with expertise in the regional and international regulations may be of great value and prevent missed regulations or costly mistakes in study design and study-to-market strategy. The regulatory landscape is in flux as more countries are beginning to adopt medical device–specific regulations (South Africa), harmonizing with regional or international standards (ISO and IMDRF in Asia), and introducing their own medical device authority and regulations (India in 2017). Other international and regional standard organizations, such as the International Electrotechnical Commission and International Telecommunication Union, European Committee for Standardization, and PanAmerican Standards Commission, often regulate material and electrical component testing but not the safety and efficacy testing of medical devices.
Global Regulatory Agencies and Their Major Regulatory Documents.

Note: EU = European Union; UK = United Kingdom; ISO = International Organization for Standardization; IMDRF = International Medical Device Regulators Forum; OECD = Organization for Economic Co-operation and Development; GLP = Good Laboratory Practices; CFR = Code of Federal Regulations; USP-NF = U.S. Pharmacopeia and National Formulary; CSA Group = Canadian Standards Association Group; JSA = Japanese Standards Association; ABNT = Brazilian Association of Technical Standards; AHWP = Asian Harmonization Working Party; APEC = Asia-Pacific Economic Cooperation; GOST = Gosudarstvennyy standart.
ISO
The current ISO standards, technical subjects (TS), and technical reports (TR) for biological and clinical evaluation of medical devices applicable to nonclinical testing are listed in Table 3. The ISO 10993 series are most applicable to nonclinical animal studies and many of these documents, particularly ISO 10993-1, contain algorithms to guide study needs and help structure development plans. ISO 10993-6:2016 is of paramount importance to toxicologic pathologists for evaluating implanted biomaterials. ISO 10993-11:2017 for systemic testing and 10993-3:2014 for carcinogenicity are also valuable resources. It should be noted that ISO 10993-6:2016 was recently updated with important changes including:
International Organization for Standardization (ISO) Documentsa Applicable to Biological Testing of Biomaterials and Medical Devices (Paid Access at https://www.iso.org).
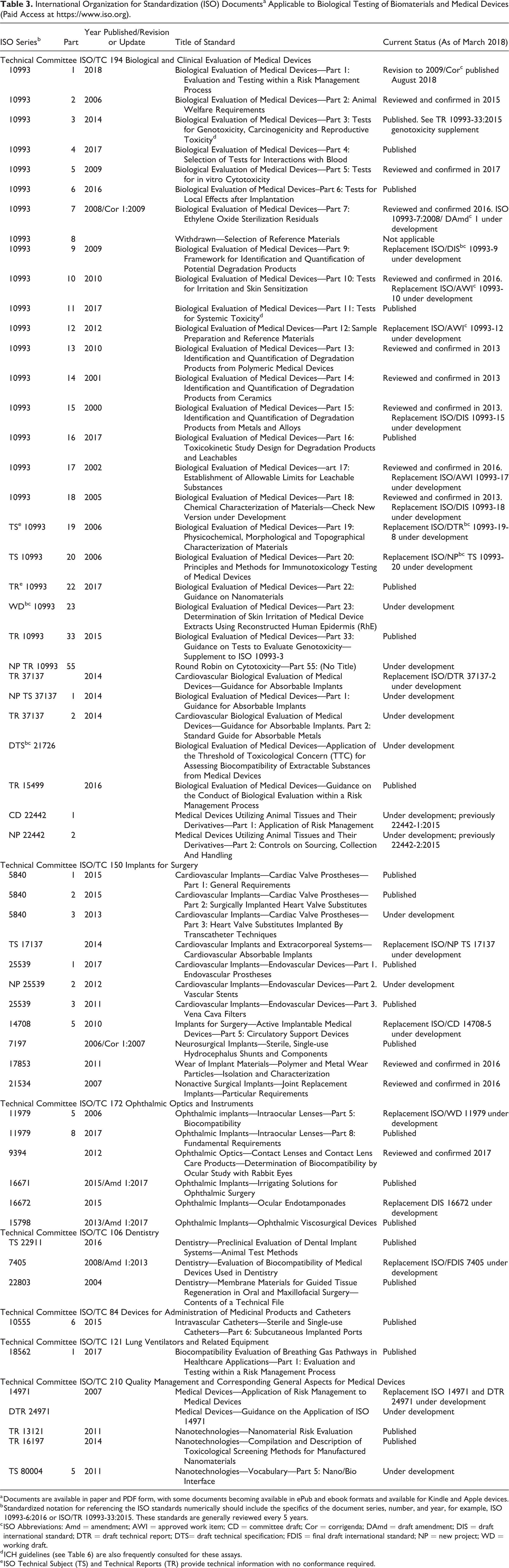
a Documents are available in paper and PDF form, with some documents becoming available in ePub and ebook formats and available for Kindle and Apple devices.
b Standardized notation for referencing the ISO standards numerically should include the specifics of the document series, number, and year, for example, ISO 10993-6:2016 or ISO/TR 10993-33:2015. These standards are generally reviewed every 5 years.
c ISO Abbreviations: Amd = amendment; AWI = approved work item; CD = committee draft; Cor = corrigenda; DAmd = draft amendment; DIS = draft international standard; DTR = draft technical report; DTS= draft technical specification; FDIS = final draft international standard; NP = new project; WD = working draft.
d ICH guidelines (see Table 6) are also frequently consulted for these assays.
e ISO Technical Subject (TS) and Technical Reports (TR) provide technical information with no conformance required.
a suggestion to collect and evaluate lymph nodes draining the implantation site, a need to evaluate tissue remodeling out to the point of tissue normalcy when appropriate, a notation that additional observations beyond semiquantitative scoring may be needed to characterize implant reactions, changes to interpretive terminology for implant scores (reaction scores rather than irritancy scores), inclusion of an example of semiquantitative scoring template for neural tissues in Annex E, and an example scoring reference for poly-
Other ISO 10993 standards are of interest for general safety and efficacy testing design and conduct, local tolerability (dermal and vascular), specialized testing (immunotoxicology and nanomaterials), in vitro testing, and degradation and impurities (extractables and leachables). There are also multiple standards, TS, and TR documents from other ISO Technical Committees that apply to the biocompatibility and testing of specialized biomaterials and organ-specific devices (surgical implants, ophthalmic, and dentistry). The ISO standards also cross-reference other standards organizations (ASTM International and U.S. Pharmacopeia [USP]) and governmental guidance documents, including the International Council for Harmonization (ICH).
ASTM International
Formerly called the American Society for Testing and Materials, ASTM International standards (guides, test methods, and practices) with the most relevance to biological testing are from committee F04 and its subcommittees (Table 4). The ASTM International standards often predate and are referenced by ISO standards and provide the technical framework for long-term host and implant interactions of extracts, implants, and systemic interactions. More recently, standards for tissue-engineered medical products have been under development.
ASTM International Standards Applicable to Biological Testing of Biomaterials and Medical Devices (Paid Access at https://www.astm.org/standard/index.html).
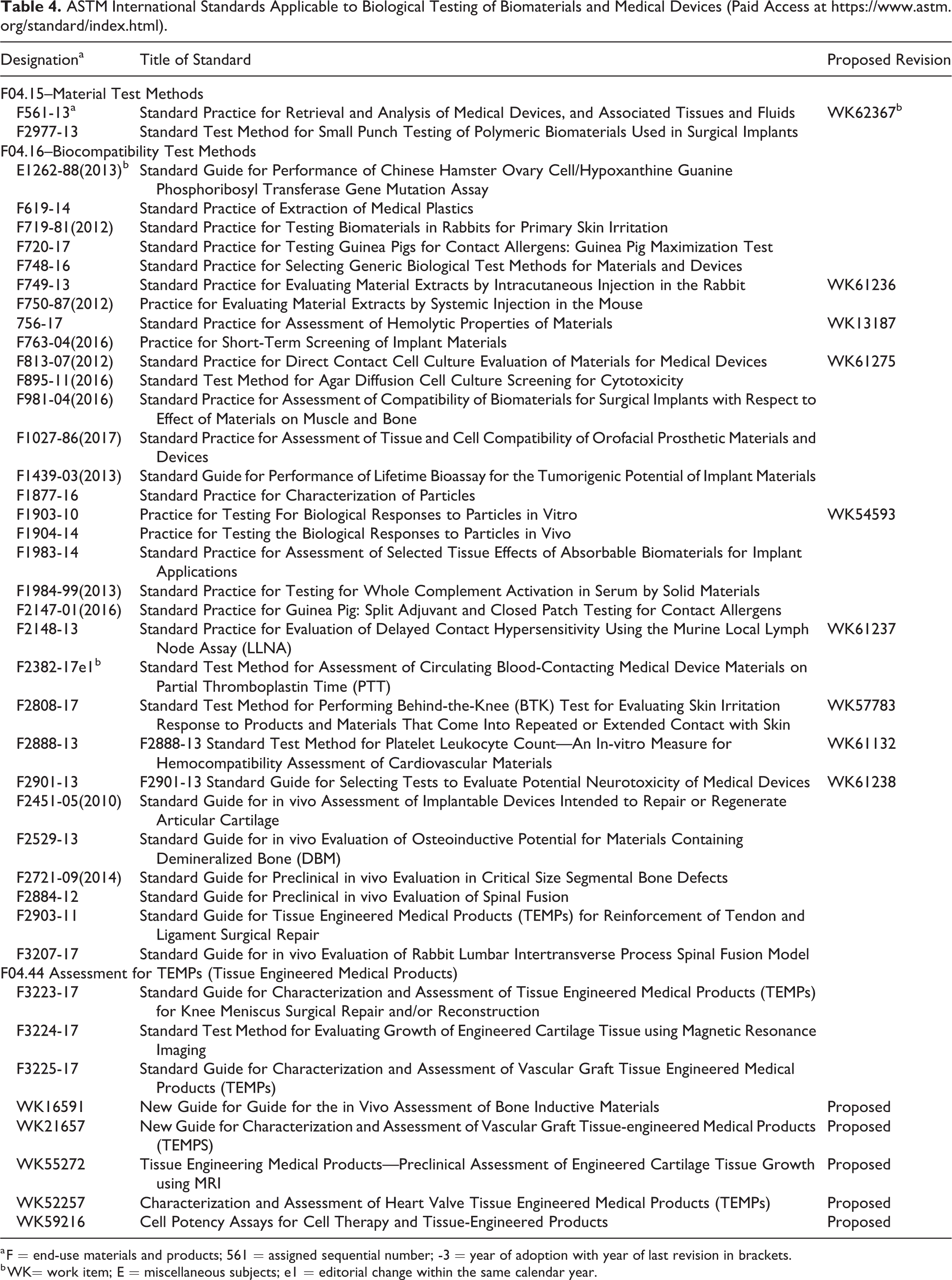
a F = end-use materials and products; 561 = assigned sequential number; -3 = year of adoption with year of last revision in brackets.
b WK= work item; E = miscellaneous subjects; e1 = editorial change within the same calendar year.
IMDRF
The IMDRF is charged with producing and publishing harmonization standards for raw materials, biomaterials, and medical device testing (Table 5), and these represent the accepted standards or supplement ISO standards for member countries. The IMDRF was preceded by the Global Harmonization Task Force (GHTF), and GHTF standards remain active until they are updated by the IMDRF. The IMDRF assists countries without a defined body of laws or limited laws to enact regulations and they provide support for regional harmonization. In the absence of a dedicated national regulatory medical device division, regulation of biomaterials and devices may be controlled by the country drug division or rely on importation of devices approved regionally or internationally. Regardless, device standards in minimally regulated countries often default to the ISO, ASTM International, and IMDRF standards, with and without modification by national or regional standards.
International Medical Device Regulators Forum (IMDRF) and Global Harmonization Task Force (GHTF)a Documents Applicable to Biological Testing of Biomaterials and Medical Devices.
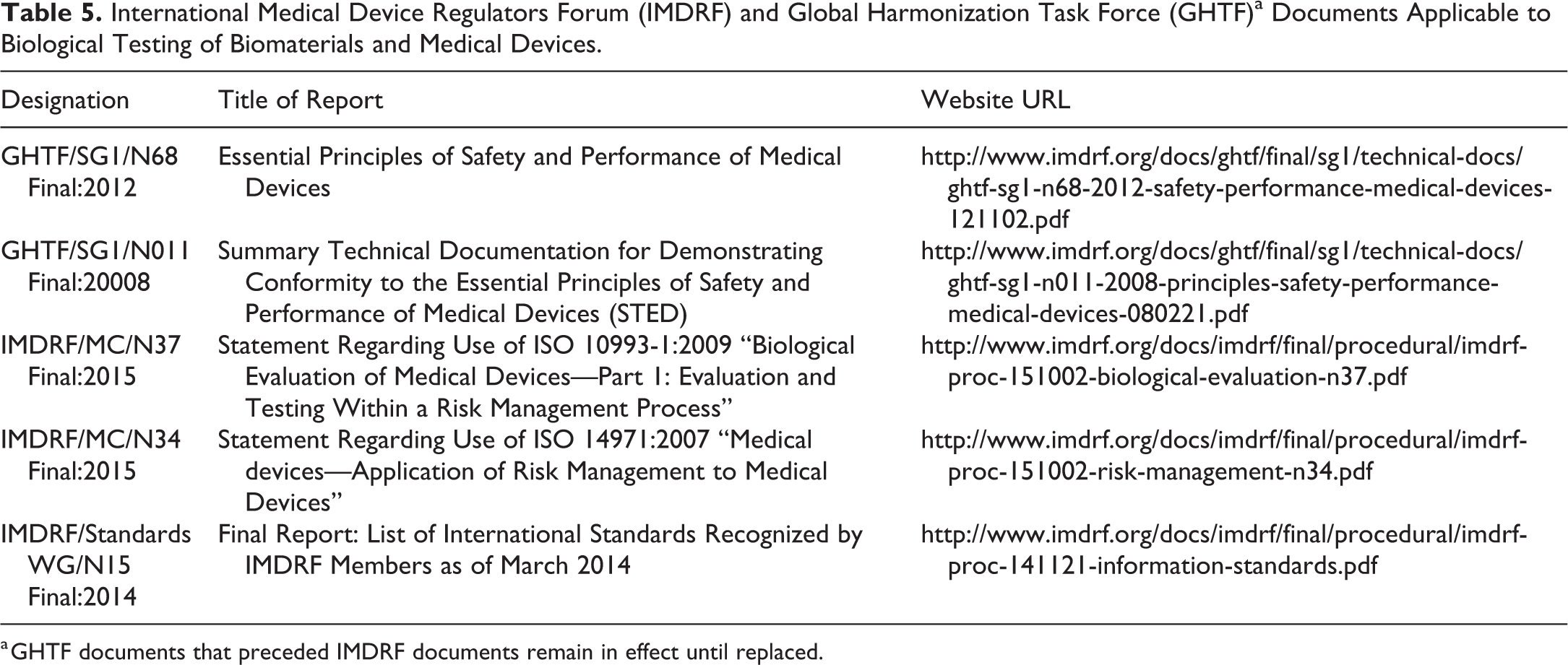
a GHTF documents that preceded IMDRF documents remain in effect until replaced.
World Health Organization (WHO)
The WHO has published a global regulatory framework (http://www.who.int/medical_devices/publications/global_model_regulatory_framework_meddev/en/) that recommends guiding principles, harmonized definitions and specifies the attributes of effective and efficient regulations for member states to ensure quality and safety of medical devices. The WHO recommendations rely on international harmonization documents developed by the GHTF and its successor, the IMDRF. The WHO also advocates for regional harmonization programs and provides support to counties without current regulations for medical devices.
Regional Standards Organizations
 (European Conformity) Marking
(European Conformity) Marking
Product markings are a specialized form of product certification that often applies to geographic regions. There are many differences between Europe and the United States, as well as between other countries, in reviewing and approving new medical devices. In particular, the Food and Drug Administration (FDA) takes a centralized approach in contradistinction to decentralization in the EEA. New medical devices are often first approved in the EEA through a specialized certification marking, before undertaking the separate U.S. FDA approval process. The marking in EU member countries and states identifies medical devices that meet safety, health, and environmental requirements of the EU directives and are tested by an accredited third party (Notified Body). The mark should not be confused with the similar but compressed and unregistered China Export logo that does not identify testing to standards. Product markings in North America such as Underwriters Laboratory and Federal Communications Commission or CSA Group (formerly Canadian Standards Association) may apply to health-care and biomaterial components but are generally not relevant to biological safety and efficacy testing of medical devices.
Asian, African, and Latin American Regional Harmonization
The Asian Harmonization Working Party and Asia-Pacific Economic Cooperation are working with the IMDRF to harmonize medical device regulations in Abu Dhabi, Brunei Darussalam, Cambodia, Chile, China, Taiwan, Hong Kong, India, Indonesia, Jordan, Saudi Arabia, Korea, Laos, Malaysia, Myanmar, Pakistan, Philippines, Singapore, South Africa, Thailand, and Vietnam. The Pan African Harmonization Working Party, with representatives from the East African Community, Nigeria, and South Africa, is also using international support to begin regional harmonization of medical device regulations. Latin America has made little effort toward harmonization which has resulted in a patchwork of enforced and harmonized regulations.
National Standards
A number of countries have developed internal standards for regulation and importation of a variety of materials and products. The majority of these standards govern physiochemical properties of materials and electrical components, but some countries do have standards directly applicable to biological testing (Table 2). Of particular importance are the ASTM International standards referenced in the ISO and IMDRF standards and the national pharmacopeias, particularly the U.S. Pharmacopeia.
American National Standards Institute (ANSI)
The ANSI provides a neutral venue for other organizations to develop voluntary national consensus standards. Additionally, they work with international standards groups (ISO and IEC) and oversee a sales library of various standards that includes bundling of topic-specific medical device standards from national and international sources.
USP—National Formulary (NF)
The USP is a nongovernmental independent and nonprofit scientific organization that played an early role in biomaterial and medical device testing, along with other national pharmacopoeias, in providing guidance documents. The basis for the ISO 10993-6:2016 implantation studies is the USP test which evaluates short-term exposure to implanted materials. The USP compendium combined with the NF (USP-NF) monographs provide written standards for individual materials and excipients. As with other standards published by nongovernmental agencies, written standards from this source must be purchased (www.uspnf.com). The USP also sells physical reference materials such as defined polymers as implantable control material to compare to test biomaterials and for quality control.
Governmental and International Guidelines
Freely available governmental guidance documents including Good Laboratory Practices (GLP) and international guidance documents for the development of drugs, biologics, gene and cellular products, and nanomaterials can also be relevant to testing biomaterials, medical device components, and finished medical products (Table 6).
International Guidelines Applicable to Biological Testing of Biomaterials and Medical Devices.

GLP
The FDA has a stated preference for safety testing of medical devices under GLP and has issued specific guidelines and considerations for medical device testing. Of particular interest is the FDA Use of International Standard ISO 10993-1, “Biological evaluation of medical devices Part 1: Evaluation and testing within a risk management process” guideline issued in 2016. This document supersedes the Blue Book Memorandum #G95-1 (1995) and details the FDA usage of the ISO 10993-1 guideline. Although not the primary standard for biocompatibility testing, GLP specifications (21 CFR) provide a framework for scientific conduct of all pivotal nonclinical studies including medical devices testing. Individual EU member states do not exclude the use of GLP and although the OECD principles of GLP generally only apply to chemical and chemical products, jurisdictional flexibility allows for the inclusion of medical device testing under these principles (Table 6).
ICH
The ICH guidelines (www.ich.org) for pharmaceuticals and biologics do not specifically address biocompatibility and efficacy of biomaterials and medical devices. However, the testing principles in selected ICH Safety, Quality, and Multidisciplinary Guidelines (Table 6) provide useful information and these testing guidelines can supplement or selectively supplant (e.g., genotoxicity) ISO standards (Table 3).
Pediatric Medical Device Testing
Although nonclinical testing is generally required in juvenile animals for pharmaceuticals intended for pediatric patients, medical device safety and efficacy evaluation in young patients has been more informal and nonclinical juvenile toxicity testing is seldom conducted. The FDA provides three guidelines that apply to pediatric indications, and they will allow clinical data to be leveraged as evidence of safety and efficacy for pediatric devices (Table 6).
Nanomaterials
The use of nanomaterials in medical devices and combination products is beginning to be addressed by the FDA (Table 6) and ISO TRs and specifications (Table 3). Particles in the nano range are also of interest due to the potential release of small particles from biodegradable materials and during corrosive and wear activity on permanent implanted devices such as metal release from joint replacements. These situations may also involve clinical testing and retrieval of implanted devices under ASTM International standards (Table 4).
3-D Bioprinting and Regenerative Medicine Products
Newer technologies are being used to produce discrete devices or devices as part of combination products and production of prosthetics and tissue replacements including regeneration of tissues using polymer scaffolds. Despite advances in regenerative possibilities and increasing use of bioprinting, few specific guidelines or standards exist, as these products are often custom devices in most countries. In the United States, cell and tissue products, combination products, or tissue engineering may be minimally regulated or regulated as drugs, biologics, or medical devices based on application of criteria in recent final and draft guidances (Table 7).
International Guidelines and Standards Applicable to 3D Bioprinting (Additive Manufacturing) and Regenerative Medicine Products.
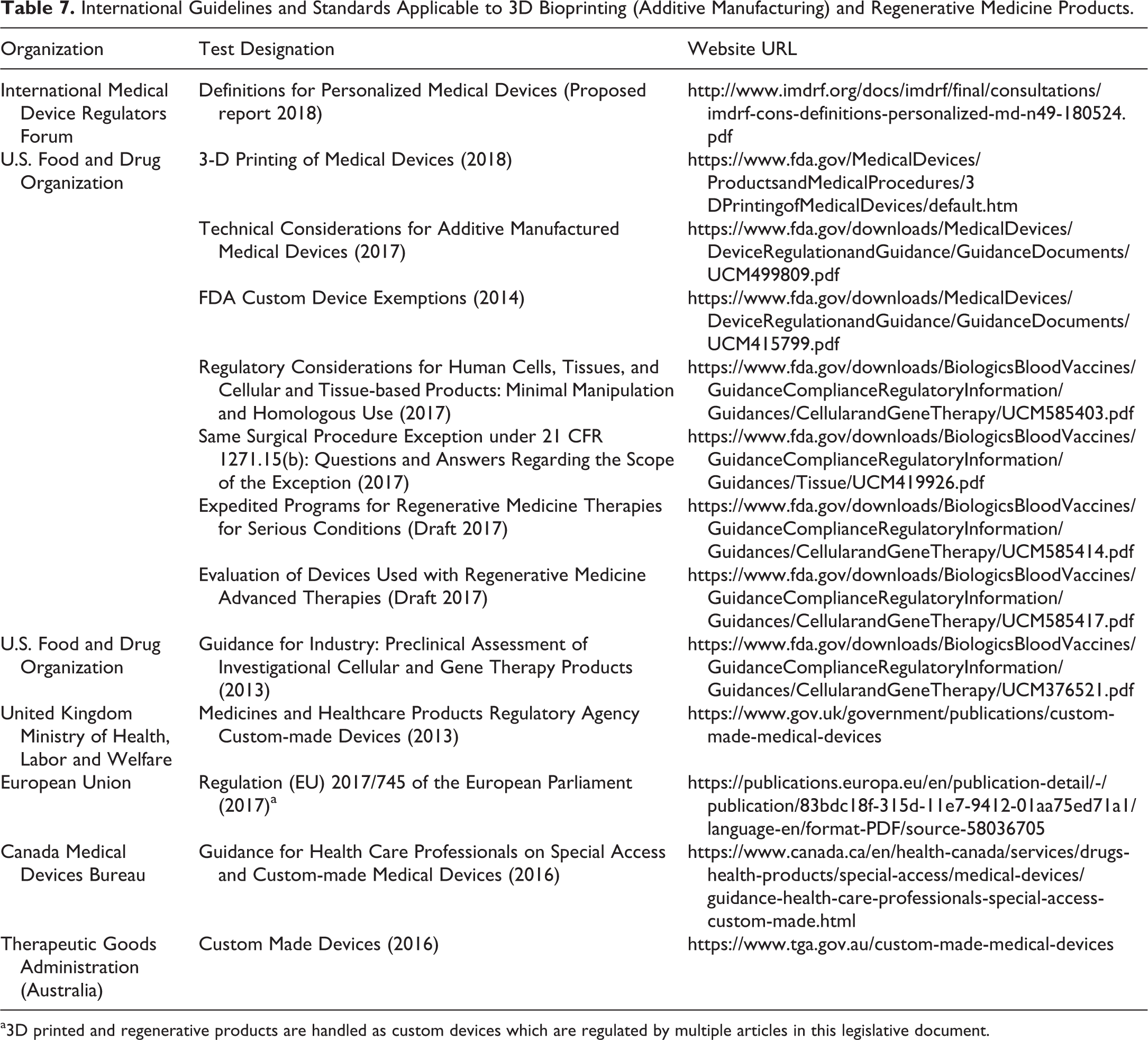
a3D printed and regenerative products are handled as custom devices which are regulated by multiple articles in this legislative document.
3-D Bioprinting
Three-dimensional bioprinting (also known as additive manufacturing) can be used to produce complex replacement tissues and body parts, prosthetics, appliances, and even create miniature organs for in vitro drug testing. Specific regulations to govern medical devices produced by 3-D printing are limited, and for the foreseeable future, the UK, EU, Canada, and Australia are treating these as custom-made devices (Table 7). In contrast, the FDA has begun to consider the broad category of 3-D printing and has produced a draft document for technical considerations for additive manufactured medical devices. This guidance recommends biocompatibility testing of the finished device, according to the FDA Use of International Standard ISO 10993-1 guidance document (Table 6). The ASTM has also begun to produce standards for tissue-engineered medical products (Table 4).
Regenerative Medicine Products
Regenerated tissues, such as bladder, blood vessels, esophagus, and ear, generally use the patient’s own cells to avoid rejection upon implantation. Although similar to tissue or combination products, regenerative products are becoming more complicated. Advancements in bioengineering, concurrent application of 3-D printing of cells on scaffolds, immuno-engineering, and “smart” biomaterials that participate in the formation of functional tissue also complicate classification of these products (Table 7).
In the UK and EU, development of regenerative products continues to be covered under guidelines for biologics and cell therapy with input from the standards that govern custom medical devices, if required. In the United States, the FDA has started to develop a broader framework for oversight of regenerative medicine with 4 guidance documents, of which 2 focus on safety and efficacy (Table 7). The ASTM (Table 4) standards on tissue engineering should be consulted in this category.
Footnotes
Author Contributions
All authors (JS and KF) contributed to conception or design; data acquisition, analysis, or interpretation; drafting the manuscript; and critically revising the manuscript. All authors gave final approval and agreed to be accountable for all aspects of work in ensuring that questions relating to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Declaration of Conflicting Interests
The author(s) declared no potential, real, or perceived conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.