
Abstract
Diabetes mellitus (types 1 and 2) is the leading cause of glomerular disease and end-stage renal disease in most developed countries, with estimates that one-third of people living with diabetes will develop diabetic kidney disease (DKD). The current standard of care medications slow but do not arrest progression of kidney disease, and therefore, therapy for DKD is a highly unmet medical need for patients. To discover and test novel and durable new therapies, it is necessary to develop animal models of human DKD, which authentically recapitulate the human disease state and provide translatable efficacy to human patients. Here, we review selected mouse models of human DKD, which demonstrate many of the features of type 2 human DKD.
Keywords
Diabetic Kidney Disease (DKD)
In the United States, renal failure is the ninth leading cause of mortality with approximately 30 million Americans, 15% of the adult population, living with chronic kidney disease (CKD). Nearly half of these cases are undiagnosed. Diabetes mellitus (types 1 and 2) is the leading cause of glomerular disease and end-stage renal disease (ESRD) in the United States, with estimates that one-third of people living with diabetes will develop CKD (Centers for Disease Control and Prevention 2018). Globally, the incidence of CKD is rapidly increasing and was the 12th leading cause of death with 1.2 million deaths attributed to the condition in 2015 (GBD 2015 Mortality and Causes of Death Collaborators 2016). DKD has dramatically increased by a staggering 39.5% worldwide from 2005 to 2015 (GBD 2015 Mortality and Causes of Death Collaborators 2016). In patients with diabetes, symptomatic renal disease generally ensues 15 to 25 years postdiagnosis with diabetes, though early histologic findings such as glomerular basement membrane thickening may be seen as early as 2 to 3 years postdiagnosis with type 1 diabetes (Mac-Moune Lai et al. 2004). The standard of care for DKD involves two classes of drugs, angiotensin converting enzyme inhibitors and angiotensin II receptor blockers. Both classes slow but do not arrest progression to ESRD, thus DKD represents a highly unmet medical need for novel and more durable therapies.
The second major cause of renal failure is hypertension, which is a frequent comorbidity in individuals with diabetes of either type 1 or 2. Hypertension increases glomerular pressure causing additional glomerular injury and contributes to the severity of microvascular disease in the hyperglycemic state. Other comorbidities include dyslipidemia of diabetes characterized by increased very low-density lipoproteins and plasma triglycerides, decreased high-density lipoproteins, and normal or increased low-density lipoproteins leading to acceleration of atherosclerosis (Mooradian 2009). Diabetic dyslipidemia further exacerbates macrovascular disease causing an increased risk of congestive heart failure, myocardial infarction, coronary artery disease, and stroke. The interplay between hyperglycemia and hypertension in renal injury is complex and multifactorial with interwoven pathways further aggravating the injury. However, the multifaceted nature also presents opportunities for therapeutic intervention.
Early alterations in DKD include glomerular hyperfiltration, glomerular and tubule epithelial hypertrophy, and microalbuminuria. With disease progression, glomerular basement membranes thicken, the mesangium and interstitium accumulate extracellular matrix, and there is an increase in urinary loss of albumin. In advanced disease, worsening glomerulosclerosis leads to progressive loss of renal function. Histologically, features of DKD include glomerular basement membrane thickening, mesangial matrix expansion, and arteriolar hyalinosis. The classic histologic finding in DKD is Kimmelstiel–Wilson nodules characterized by periodic acid–Schiff positive, nodular glomerular sclerosis often with a distinctive acellular central region surrounded by palisading cells (Reidy et al. 2014).
The Animal Models of Diabetic Complication Consortium (AMDCC) was formed in 2001 to identify the features of animal models that reproduce diabetic complications, including DKD, similar to the syndromes identified in people with diabetes. The Consortium developed criteria for animal models of DKD that include (Diabetic Complications Consortium Protocol): Progressive renal insufficiency in the setting of hyperglycemia (>50% decline in glomerular filtration rate [GFR]) Albuminuria (>10- fold increase compared with age-, gender-, and strain-matched controls) Histologic changes Basement membrane thickening (electron microscopy) Advanced mesangial matrix expansion with or without mesangiolysis and nodular mesangial sclerosis Interstitial fibrosis Arteriolar hyalinosis
It is recognized that many of the earlier diabetic mouse models did not present histologically with features of human DKD or meet the goals set forth by the AMDCC. A few key features that frequently did not occur in these models include the development of nodular glomerulosclerosis, arteriolar hyalinosis, and interstitial inflammation. Comorbidities, hypertension in particular, are often absent. Other considerations include the slow nature of DKD development in patients with diabetes, which usually does not become symptomatic until 15 to 25 years postdiagnosis with diabetes. Mouse models develop an accelerated disease state in comparison, and hyperglycemia in these models is persistent for only weeks to months versus decades. As previously mentioned, the multifactorial nature including genetic components may not exist in the models. Table 1 has representative mouse models of both type 1 diabetes mellitus and type 2 diabetes mellitus (T2DM). For the purposes of this synopsis, only the T2DM models will be discussed further.
Representative Mouse Models of Both Type 1 Diabetes Mellitus (T1DM) and Type 2 Diabetes Mellitus (T2DM).
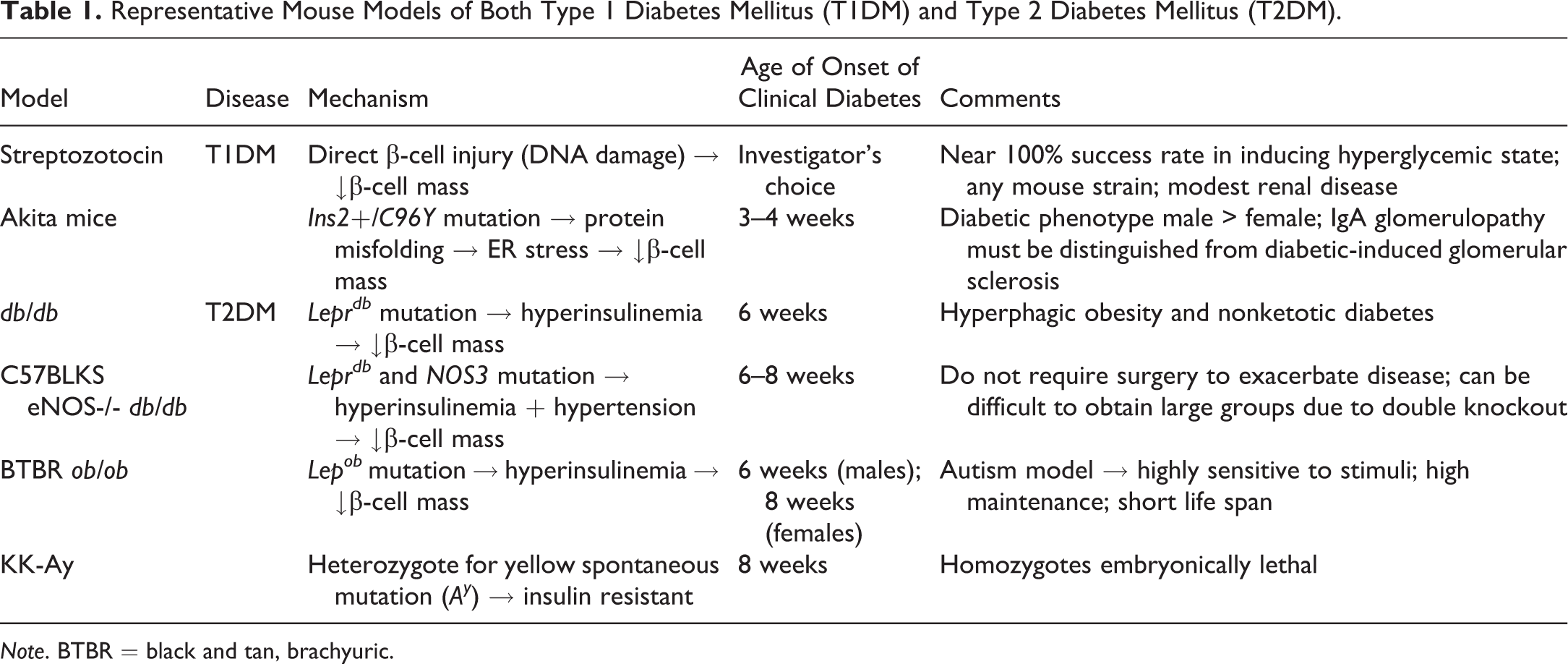
Note. BTBR = black and tan, brachyuric.
Clinical Features
Key clinical features of selected T2DM models are listed in Table 2, including the magnitude (or absence) of hyperglycemia, microalbuminuria, azotemia, and hypertension. The db/m lean mouse, the heterozygote, nondiabetic littermates of the db/db are used as baseline. Hyperglycemia is most profound in the db/db derivatives except the C57BLKS eNOS-/- db/db, which is hyperglycemic but not as marked in comparison. Microalbuminuria is pronounced in both the db/db uNx ReninAAV and the C57BLKS eNOS-/- db/db. Azotemia is not substantial in any of the models but mild in the db/db ReninAAV, db/db uNx ReninAAV, and C57BLKS eNOS-/- db/db. Hypertension was greatest in db/db ReninAAV, db/db uNx ReninAAV, and C57BLKS eNOS-/- db/db, while the BTBR ob/ob are reported as hypotensive (Hudkins et al. 2010).
Clinical Features of Selected Type 2 Diabetes Mellitus Mouse Models.
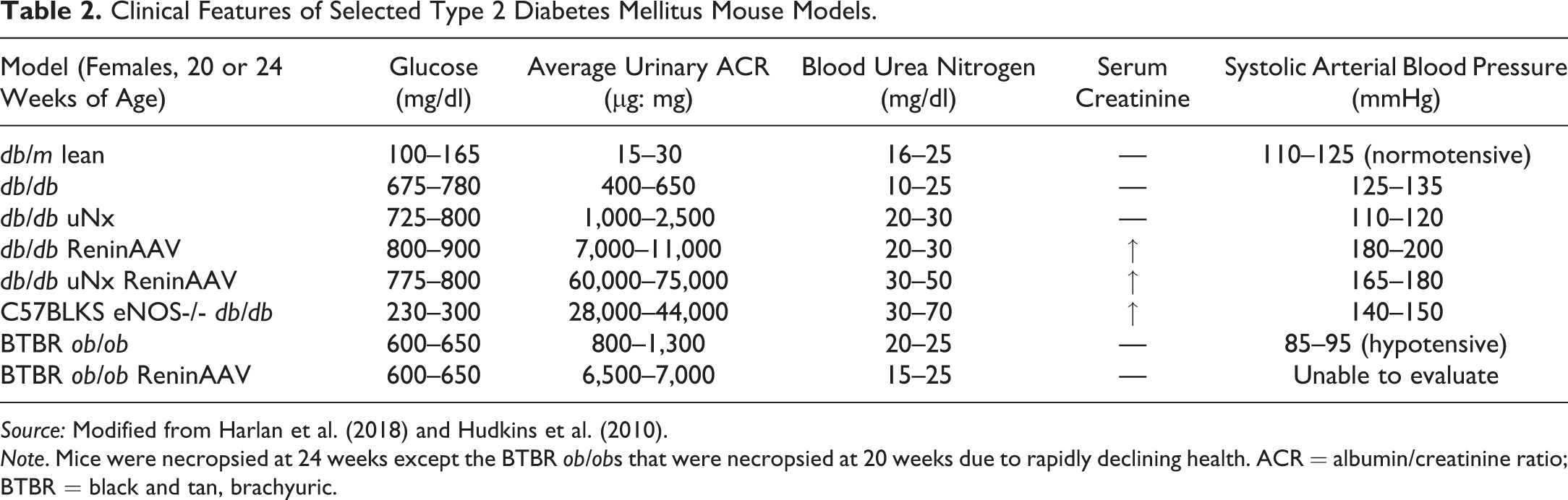
Source: Modified from Harlan et al. (2018) and Hudkins et al. (2010).
Note. Mice were necropsied at 24 weeks except the BTBR ob/obs that were necropsied at 20 weeks due to rapidly declining health. ACR = albumin/creatinine ratio; BTBR = black and tan, brachyuric.
db/db Mouse
The db/db mouse was developed at Jackson Laboratory over 50 years ago as a hyperphagic and obese mouse leading to a progressive diabetic state as a result of an autosomal recessive point mutation of the leptin receptor (Leprdb ). These animals have high circulating levels of leptin and insulin due to the defective leptin signaling. The db/m lean mouse, the heterozygote, nondiabetic littermate of the db/db can be used as a control comparison. While this model features many DKD elements and meets many of the AMDCC criteria including renal hypertrophy, glomerular tuft enlargement, progressive increase in mesangial matrix, and albuminuria, it lacks arteriolar hyalinosis, nodular glomerulosclerosis, tubulointerstitial fibrosis, and tubular atrophy (Sharma, McCue, and Dunn 2003). Glomerular basement membrane thickening takes 18 to 20 months to develop (Sharma, McCue, and Dunn 2003). An additional consideration when using this mouse in efficacy studies is the extended time required to establish nephropathy. As a T2DM model, these mice lack common comorbidities in the human condition, mainly hypertension. To accelerate the progression of renal disease, a uninephrectomy (uNx) is performed at four weeks of age to increase the burden on the remaining kidney. This procedure results in a profound increase in mesangial matrix and albuminuria compared with age-matched intact db/db mice. Recognizing the critical role chronic hypertension plays in the progression of DKD, persistent hypertension can be induced via adeno-associated virus delivery of renin (ReninAAV) at 12 weeks of age to produce the db/db uNx ReninAAV (Harlan et al. 2018). In comparison to the db/db uNx, the nephropathy is much more severe with rapid progression (Figure 1).
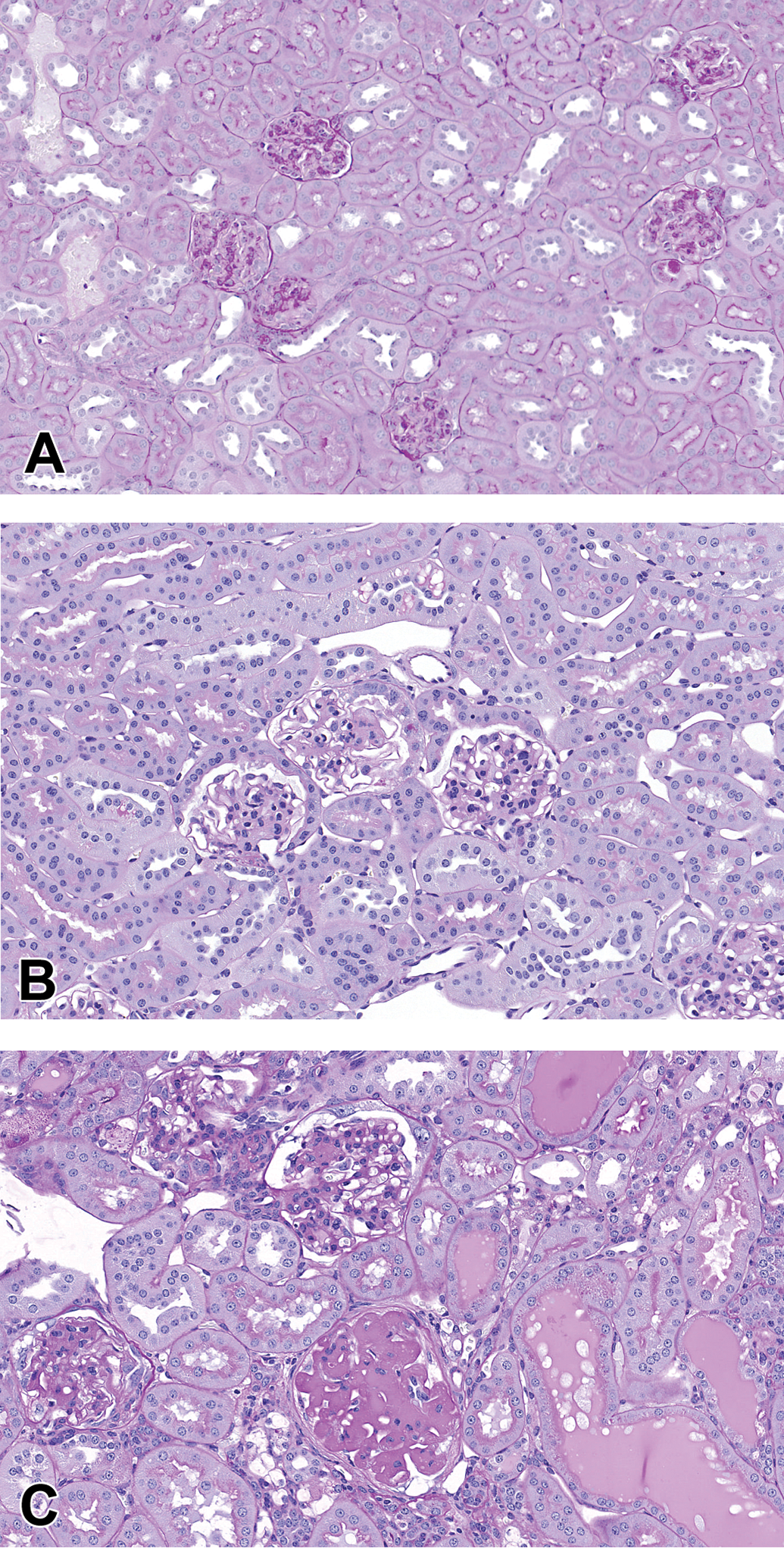
Comparison of db/m lean (A) with the db/db uNx (B), which have minimal mesangial matrix expansion, while the db/db uNx ReninAAV (C) shows extensive mesangial matrix expansion, nodular sclerosis, and tubular casts (PAS). Both the db/db uNx and db/db uNx ReninAAV have glomerular tuft hypertrophy compared with the db/m lean. Mice underwent uninephrectomy at 4 weeks of age (db/db uNx and db/db uNx ReninAAV) and ReninAAV (db/db uNx ReninAAV) induction at 12 weeks of age. Mice were necropsied at 24 weeks of age. PAS = periodic acid-Schiff stain; ReninAAV = adeno-associated virus delivery of renin; uNx = uninephrectomy.
Figures 1 and 2 demonstrate the range of histologic changes produced in the db/db uNx ReninAAV including extensive mesangial matrix expansion, nodular glomerulosclerosis, interstitial fibrosis and inflammation, and proteinaceous casts. Arteriolar hyalinosis (not pictured) also occurs (Harlan et al. 2018). Mesangiolysis with or without microaneurysms is not a prominent feature of this model, since it represents late-stage DKD.
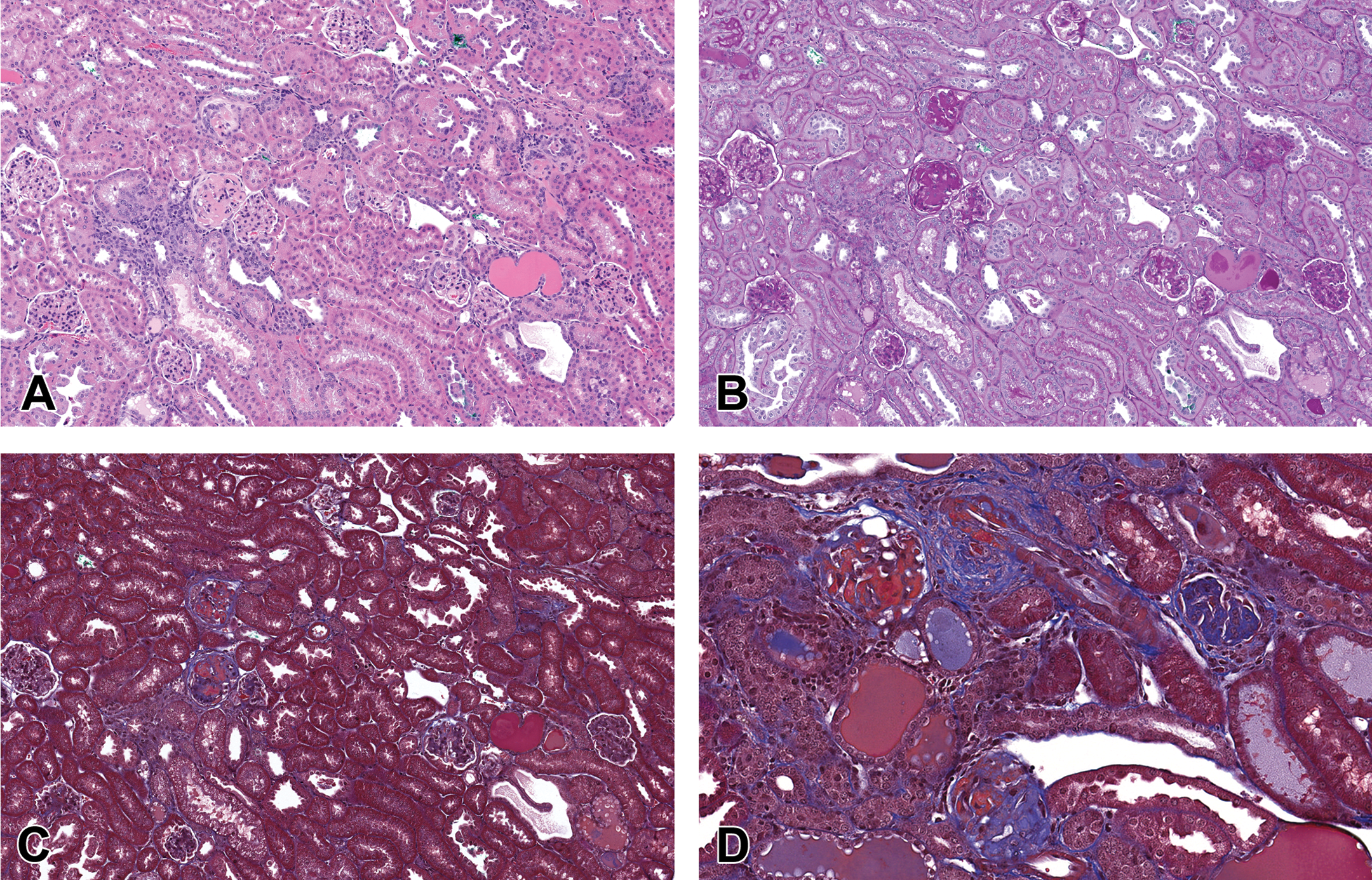
(A, B, C) Db/db uNx ReninAAV mice display multiple features of DKD including interstitial fibrosis and inflammation, tubular dilatation with proteinaceous casts, and mesangial matrix expansion. (D) Higher magnification of the Masson’s trichrome stain illustrates nodular glomerulosclerosis, interstitial and periglomerular fibrosis (hematoxylin and eosin, PAS, and Masson’s trichrome stain). DKD = diabetic kidney disease; PAS = periodic acid-Schiff stain; ReninAAV = adeno-associated virus delivery of renin.
C57BLKS eNOS-/- db/db
Endothelial nitric oxide synthase (eNOS) encoded by the NOS3 gene is the major NOS enzyme in renal vasculature. eNOS is upregulated early in DKD, then decreases with prolonged disease. The double knockout mouse coupled with leptin receptor mutation leads to rapid progression of DKD (Zhao et al. 2006). This model has the features of the db/db with moderate, persistent hypertension. This model has the advantage over the db/db uNx ReninAAV model in that it does not require uNx to enhance renal disease; however, it is difficult to obtain sufficient numbers of mice because this is a double genetic knockout. Histologically, the model provides a dynamic range of renal injury including mesangial matrix expansion, nodular glomerulosclerosis, proteinaceous casts, interstitial inflammation and fibrosis, and arteriolar hyalinosis. Mesangiolysis and microaneurysms are observed (Figure 3).
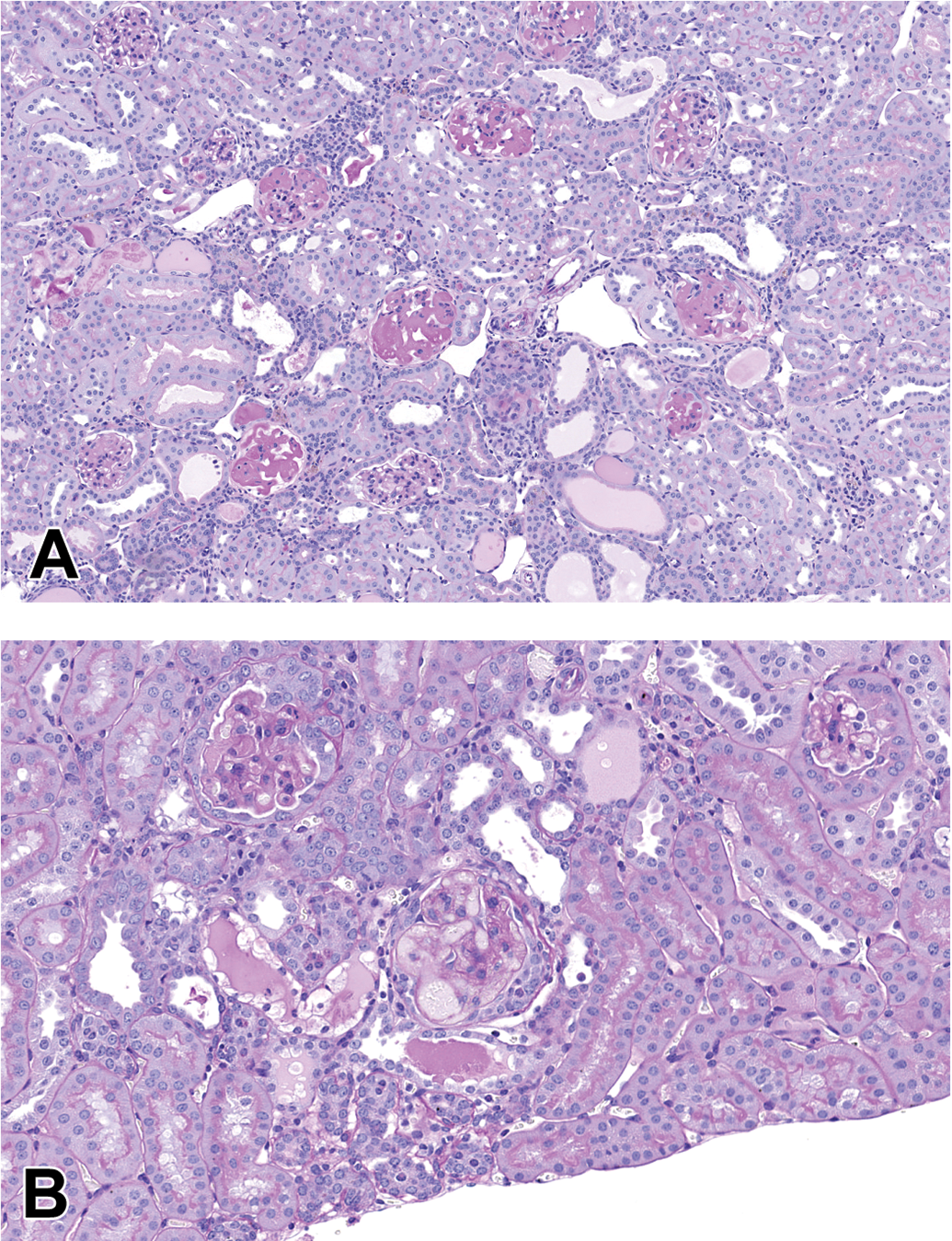
Glomeruli in the C57BLKS eNOS-/- db/db necropsied at 24 weeks of age. (A) Multiple glomeruli have mesangial matrix expansion with nodular sclerosis. Protein casts are within dilated tubules (PAS). (B) The mesangium has patchy eosinophilia characteristic of mesangiolysis leading to the formation of microaneurysms (PAS). eNOS = endothelial nitric oxide synthase; PAS = periodic acid-Schiff stain.
BTBR ob/ob
The black and tan, brachyuric (BTBR; meaning “short-tailed”) mouse is an autism model displaying behaviors consistent with autism spectrum disorder such as diminished social and communication abilities, repetitive behaviors, and high sensitivity to stimuli (Meyza et al. 2013). The BTBR ob/ob is also homozygous Lepob resulting in a deficiency in leptin production but with intact leptin signaling. The mice develop severe obesity, moderate hyperglycemia with glucose intolerance, and hyperinsulinemia with insulin resistance. One of the challenges working with these mice is their high sensitivity to stimuli making handling and end point recordings difficult for some procedures such as tail cuff blood pressure measurements. Blood pressure has been reported by other investigators, and these mice are hypotensive rather than hypertensive compared to the BTBR wild-type or heterozygous controls (Hudkins et al. 2010). Like db/db mice, ReninAAV can be introduced to increase blood pressure and augment the renal disease (Harlan et al. 2018). Histologically, glomerular changes in the BTBR ob/ob ReninAAV are less severe than either db/db uNx ReninAAV or C57BLKS eNOS-/- db/db. Mesangiolysis with or without microaneurysm occurs with some frequency (Hudkins et al. 2010). Nodular glomerulosclerosis and mild interstitial fibrosis are also features. As compared to the other two models, protein casts, tubular basophilia, and interstitial inflammation are not prominent or not observed. Within studies conducted at Eli Lilly and Company, arteriolar hyalinosis and progressive renal insufficiency did not occur (Figure 4).
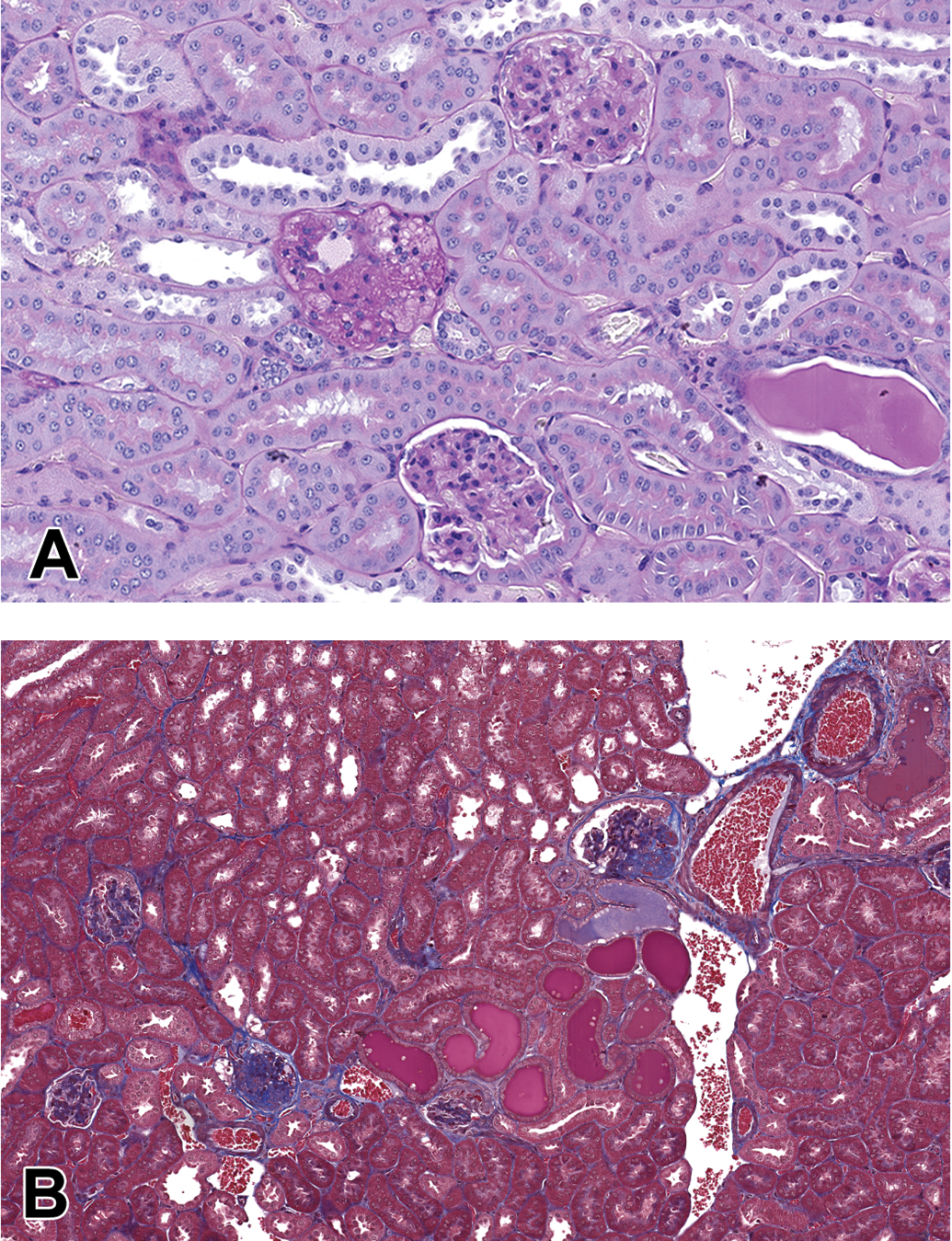
Glomeruli in the BTBR ob/ob ReninAAV necropsied at 20 weeks of age. (A,B) Mesangial matrix expansion, nodular glomerulosclerosis, protein casts are present, as well as mesangiolysis (A, PAS) with microaneurysms and interstitial fibrosis (B, MTS). BTBR = black and tan, brachyuric; PAS = periodic acid-Schiff stain; ReninAAV = adeno-associated virus delivery of renin.
Concluding Remarks
In conclusion, mouse DKD models recapitulate many of the features of human DKD, are reproducible, consistent and dynamic for demonstration of robust therapeutic response for drug development. Many newer models meet most or all of the AMDCC criteria (Table 3). The addition of ReninAAV offers reliable hypertension similar to human T2DM DKD.
Comparison of the Three Models with the AMDCC Criteria for Animal Models of Diabetic Kidney Disease.

Note. AMDCC = Animal Models of Diabetic Complication Consortium; BTBR = black and tan, brachyuric; Met = met AMDCC criteria.
Footnotes
Author Contributions
Authors contributed to conception or design (KH, SH, ZQ, JH); data acquisition, analysis, or interpretation (KH, SH, ZQ, JH); drafting the manuscript (KH); and critically revising the manuscript (KH, SH, ZQ, JH). All authors gave final approval and agreed to be accountable for all aspects of work in ensuring that questions relating to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Declaration of Conflicting Interests
The author(s) declared no potential, real, or perceived conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.