
Abstract
Immunohistochemistry (IHC) is a valuable tool in pathology. This review provides a brief description of the technical aspects of IHC and a detailed discussion on the variables that affect the results, interpretation, and reproducibility of IHC results. Lists of antibodies that have and have not worked in IHC on various mouse and rat tissues in our laboratory are provided as a guidance for selection of antibodies. An approach to IHC method optimization is presented. Finally, the critical information that should be included as a part of peer-reviewed manuscript is also discussed.
Immunohistochemistry (IHC) has remained an invaluable tool in the field of diagnostic, investigative, and toxicologic pathology. Since the first use of IHC in 1942, there have been substantial improvements to the methodology, which have led to widespread use of this technique. The current review will present some simplified basic principles of IHC, a detailed discussion of the variables that impact the results and reproducibility, a list of antibodies that have and have not worked on various rat and mouse tissues in our laboratory, sources of nonspecific staining and remedies, approaches to method optimization, and critical information that should be included in the published peer-reviewed papers. Information on the antibodies provided in this article is purely based on our experience of using them on rat and mice tissues in our laboratory conditions. A positive comment or a negative comment on a specific antibody does not necessarily mean that we endorse or disapprove that particular antibody. Also, information on the antibodies should be used as a guide to select an antibody rather than proof of specificity. Successful and unsuccessful use of antibody depends on the conditions, equipment, reagents used, and personnel in a particular laboratory.
Brief History of IHC
The use of antibodies to detect an antigen by IHC was first performed and reported in 1942. Coons et al. (1942) used a fluorescent tagged antibody to demonstrate pneumococcal antigen in tissue sections. Subsequently, Nakane and Pierce (1967) introduced the labeling of antibodies with enzymes such as alkaline phosphatase (AP) and horseradish peroxidase (HRP). In addition to the works of Nakane and Pierce, others started using HRP-conjugated antibodies (Davey and Busch 1970). In these early studies, immunohistochemical staining was performed on frozen sections and was later followed by the demonstration of proteins in paraffin-embedded tissue sections (Taylor and Burns 1974). The development of the hybridoma technique to produce monoclonal antibody advanced the use of IHC (Kohler and Milstein 1975). For this seminal discovery, Kohler, Milstein, and Jerne were awarded Nobel Prize in Medicine or Physiology in 1984 (Alkan 2004). At this point, many proteins could not be detected in formalin-fixed tissues, which led to the use of enzymes to enhance detection of antigens/proteins in tissue sections (Huang 1975). Although use of enzymes improved detection of some proteins, it was not helpful for many other proteins. An important breakthrough in the field of IHC came with the invention of antigen retrieval (AR; heat-induced epitope retrieval) method (Shi, Key, and Kalra 1991), which essentially revolutionized the field of IHC and led to IHC becoming an integral and indispensable tool in pathology.
Basics of IHC
Numerous publications exist on the technical aspects of IHC (Ramos-Vara 2005; Taylor, Shi, and Barr 2011; Elias 1990). Therefore, various methods available for IHC will only be discussed in brief. Antibodies are an integral component of IHC; monoclonal and polyclonal are the two types of antibodies used. Monoclonal antibodies are a homogeneous population of immunoglobulins directed against a single epitope (Greenfield 2012; Eisenbarth 1981). They are generated from a single B cell clone fused with a myeloma cell line (Kohler and Milstein 1975). This is referred to as a hybridoma. Monoclonal antibodies are either produced as a tissue culture supernatant or mouse ascites fluid. Most monoclonal antibodies are of mouse or rabbit origin. Polyclonal antibodies are heterogeneous population of immunoglobulins directed against multiple epitopes of a protein. They are generated by immunizing animals with the antigen of interest. Rabbit is the most commonly used species; however, goat, horse, and other animals are also used. Polyclonal antibodies are used either as serum or as affinity purified immunoglobulins. Monoclonal antibodies tend to produce less background staining compared to polyclonal antibodies (Taylor, Shi, and Barr 2011). However, some proteins are not detectable by monoclonal antibodies because of structural modifications induced by the fixation. In such instances, polyclonal antibodies are usually effective.
It is not possible to visualize the interaction of antigen–antibody in a tissue section without the help of a tag at the site of antigen–antibody interaction. Routinely, antibodies tagged to enzymes are used, and when an appropriate substrate is used, colored product develops at the site of antigen–antibody reaction. The two most commonly used enzyme tags are HRP and AP. The color of the reaction depends on the substrate used in the reaction. Some of the commonly used substrates for HRP include 3,3’ Diaminobenzidine, 3-Amino-9-Ethylcarbazole, and 4-Chloro-1-Naphthol, which produce brown, red, and blue color, respectively. Common substrates used with AP are fast red, fast blue, and new fuchsin which produce red, blue, and red color, respectively.
In IHC, irrespective of the type of antibody and substrate used, detection methods can be classified as direct (Figure 1A) or indirect (Figure 1B). In the direct method, the primary antibody is directly tagged with an enzyme to visualize the protein of interest. In the indirect method, primary antibody is unconjugated and is followed by a secondary antibody or tertiary reagents tagged with an enzyme.
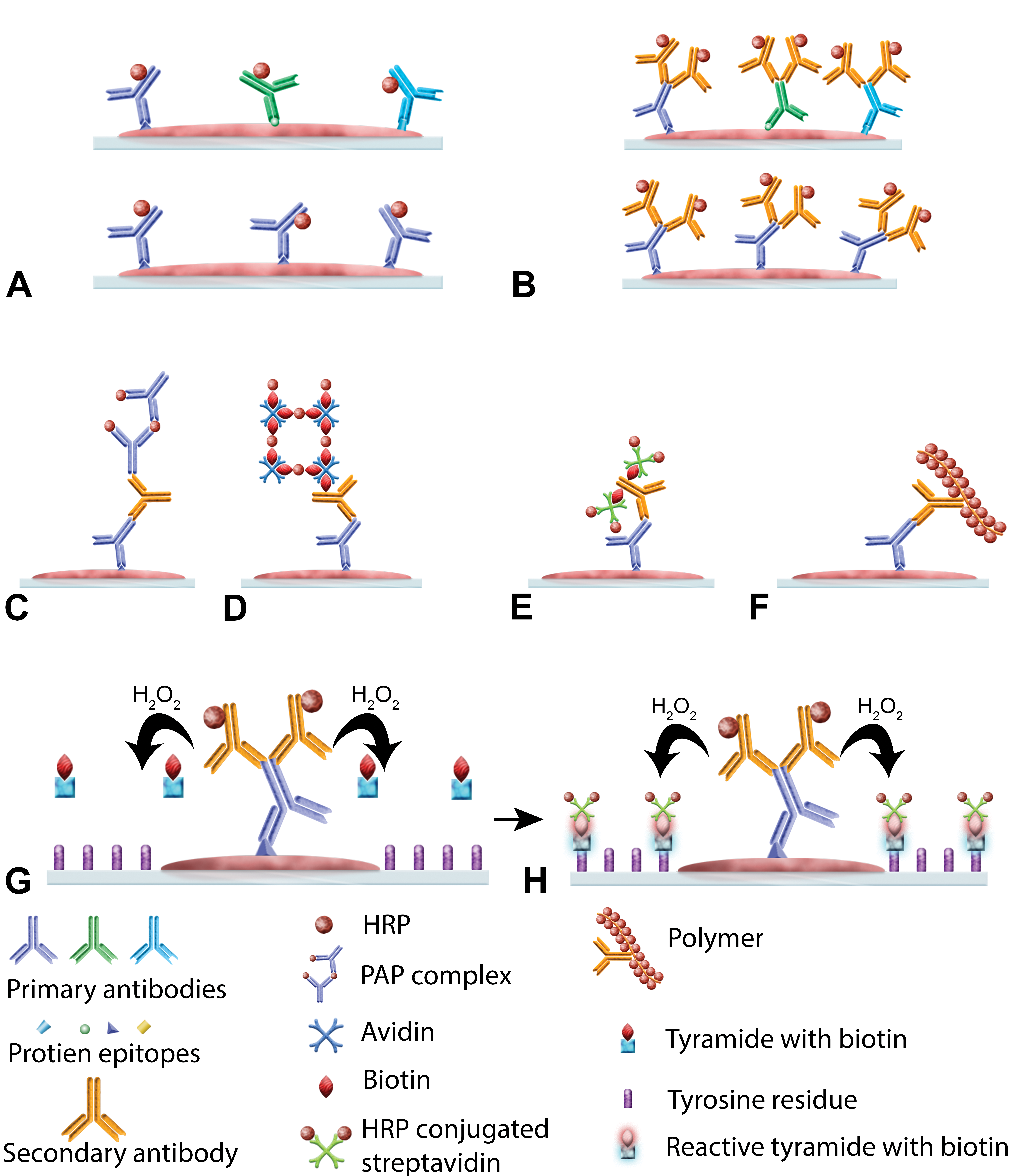
Basics of immunohistochemistry. (A) Illustration of the direct method of detecting a protein by immunohistochemistry where a polyclonal (upper) or monoclonal (lower) antibodies tagged to an enzyme such as horseradish peroxidase (HRP) are used. (B–F) Illustration of various indirect methods of detecting a protein by immunohistochemistry. A simple indirect method is shown in (B). In this method, primary antibody (polyclonal—top and monoclonal—bottom) is unconjugated and secondary antibody is conjugated to HRP or alkaline phosphatase. (C) Illustration of the peroxidase–antiperoxidase (PAP) method. First, unconjugated primary and secondary antibodies are added to the reaction, followed by addition of PAP complex. (D) Illustration of the avidin-biotin complex (ABC) method. After the addition of unconjugated primary antibody, biotinylated secondary antibody is added followed by addition of HRP-conjugated ABC. (E) Illustration of the labeled streptavidin-biotin method is shown. Unconjugated primary antibody, biotinylated secondary antibody, and HRP-conjugated streptavidin are added sequentially. (F) Polymer-based method is demonstrated. After adding unconjugated primary antibody, secondary antibody and enzyme conjugated polymer is added to the reaction. (G and H) Illustration of the catalyzed signal amplification method which is also known as tyramide signal amplification. After addition of unconjugated primary antibody and HRP-conjugated secondary antibody, biotinylated tyramide and hydrogen peroxide are added to the reaction. In the presence of hydrogen peroxide, HRP converts biotinylated tyramide to a reactive molecule which binds to the tyrosine residues present in the proximity of the protein of interest. This is followed by addition of HRP-conjugated streptavidin.
The direct method is not used routinely for some important reasons: (1) it is not practical to conjugate every single primary antibody produced for IHC, (2) if primary antibody is unconjugated, it can be used for multiple applications as opposed to a conjugated antibody, and (3) the intensity of the signal produced by the direct method is much weaker than that produced using the indirect method as there will be less enzyme per epitope required for the color development at the site of antigen–antibody interaction. Therefore, the indirect method is the most commonly used method, and there are several variations of the indirect method.
Secondary antibody conjugated to HRP or AP
This is the simplest of the indirect methods. In this method, the primary antibody is unconjugated, and secondary antibody is conjugated to HRP or AP enzyme (Figure 1B).
Peroxidase–antiperoxidase (PAP) method and alkaline phosphatase–antialkaline phosphatase (APAAP) method
The PAP method, first reported in 1970 (Sternberger et al. 1970), used HRP as the enzyme of choice. A similar method using AP was reported in 1984 (Cordell et al. 1984). In these methods, instead of tagging the secondary antibody with an enzyme, untagged secondary antibody is added followed by a complex of HRP and anti-HRP or AP and anti-AP (Figure 1C). First, the antibody is generated against HRP or AP and allowed to form a soluble complex before using it to stain tissue sections. The anti-HRP antibody should be produced in the same species as the primary antibody. These complexes are commercially available; however, this method is not routinely used.
Avidin–biotin complex (ABC) method
The method was first reported in 1981 (Hsu, Raine, and Fanger 1981) and is one of the most commonly used IHC staining methods. The ABC method is more sensitive than the simple indirect or PAP/APAAP methods. Avidin, which is a protein found in egg white, has greater affinity for the protein biotin (Vitamin B7). Avidin has 4 binding sites for biotin, whereas biotin can bind to only one avidin molecule. Both proteins are mixed in a proportion such that avidin will have at least one binding site for biotin present on the secondary antibody. The primary antibody, biotin-conjugated secondary antibody, and ABC complex are applied to tissue sections in a sequential order (Figure 1D). ABC can be prepared using commercially available kits, 30 min before applying to tissue sections, or commercially available ready-to-use reagents can be used. Biotin used for the complex is conjugated to HRP, the enzyme, which results in color development when a substrate is added.
Labeled streptavidin–biotin (LSAB) method
In some instances, avidin can bind nonspecifically to tissue sections due to ionic and electrostatic interactions. Using the LSAB method (Bratthauer 1995) in such instances can resolve nonspecific binding of avidin. Streptavidin is a protein produced by Streptomyces avidinii, and it has an affinity for biotin, similar to avidin. Streptavidin has a neutral isoelectric point and hence does not tend to be involved in nonspecific electrostatic interactions. Also, unlike avidin, streptavidin does not bind lectins, thus preventing nonspecific staining resulting from avidin/lectin interaction. In this method, instead of ABC, streptavidin conjugated to either HRP or AP enzyme is used. The primary antibody, biotin-conjugated secondary antibody, and HRP-conjugated streptavidin are added in sequence to the tissue sections (Figure 1E).
Polymer-based method
The polymer-based technique has been the most recent development in the field of IHC (Sabattini et al. 1998). In the biotin-based methods such as ABC and LSAB, nonspecific binding of biotin or endogenous biotin can be an issue. The polymer-based method is devoid of such issues, as biotin is not involved in detection. In this method, following incubation with a primary antibody, tissue sections are incubated with a polymer on which the secondary antibody and large numbers of enzyme molecules are conjugated (Figure 1F). The polymer provides larger amount of enzyme per epitope to increase the sensitivity. In addition, the time required to complete the staining procedure is shorter. The size of the polymer, resulting in steric hindrance, is one of the disadvantages of this system. Therefore, all protein epitopes may not be accessible by this method. However, more recently polymers have been modified to reduce the effect of steric hindrance (Buchwalow et al. 2013).
Catalyzed signal amplification (CSA) method
This method is not routinely used in IHC but might be useful when the antigen of interest is present in very low quantity (Bobrow et al. 1992). The CSA method is also known as tyramide signal amplification method since tyramide is used as a substrate. In this method, after incubating the tissue sections with primary antibody and HRP-conjugated secondary antibody, biotinylated tyramide and hydrogen peroxide are added. The HRP enzyme on the secondary antibody converts tyramide to a reactive intermediate. The reactive tyramide intermediate binds to the tyrosine residues on the proteins in the immediate vicinity of the antigen–antibody complex. After this step, tissue sections are incubated with HRP-conjugated streptavidin to visualize the reaction (Figure 1G and H).
Variables That Impact the Result and Reproducibility in IHC
Although IHC seems to be a simple tool, there are numerous variables contributing to intra- and interlaboratory reproducibility issues (Baker 2015; Marx 2013). Each laboratory producing its own antibody is not a practical solution in a fast-paced research environment, in which investigators are under constant pressure to publish quality data in the shortest possible time. This has led investigators to rely on commercially available antibodies. Since there is a huge market for antibodies, numerous companies have surfaced to offer a variety of antibodies for different applications (Baker 2015; Delpire 2015). Whether it is a monoclonal antibody produced in culture or a polyclonal antibody produced in animals, variability in response to an antigen is intrinsic to the immune system. This can be problematic without the proper quality control, and unfortunately, many companies selling the antibodies may lack stringent internal quality control measures (Marx 2013; Baker 2015). The onus is then on investigators to address all of the variables during method development and interpretation to ensure that the results are reproducible (Bordeaux et al. 2010). Fixation, AR, detection method, choice of antibody, lot number, and nonspecific staining in certain cell types are some of the important variables that need serious consideration.
Fixative
The most important variable that can affect immunohistochemical staining is fixation. There is no single fixative that is optimal for all antigens. In our experience and based on the literature, no fixative matches 10% neutral-buffered formalin (NBF) in terms of providing superior morphology and IHC staining (Taylor and Levenson 2006; Ramos-Vara 2005; Paavilainen et al. 2010). One should be aware that too little fixation as well as prolonged fixation with NBF can create problems for IHC (Ramos-Vara 2005). It has been shown that at least 24 to 48 hr are required to achieve maximal formaldehyde binding (Helander 1999; Fox et al. 1985). Samples should be less than 5 mm thick and fixed for no less than 24 to 48 hr (Hayat 2002). If these fixation procedures are consistently practiced, it is possible to achieve reliable and reproducible IHC results using most of the antibodies. There will always be exceptions, and the fixation time may have to be tweaked for some specific proteins.
Another commonly used fixative is 4% paraformaldehyde (PFA). It is similar to 10% NBF in terms of formaldehyde content. However, unlike 10% NBF, it does not contain 1.5% methanol as a stabilizer. A disadvantage of PFA is that it has to be freshly prepared and over time polymerizes in solution, thereby reducing the fixation rate. Furthermore, there is no data to show that PFA is superior to 10% NBF. Other fixatives such as ethanol, Davidson’s fixative, and Bouin’s fixative are variably used. Many antibodies do not produce optimal IHC staining when these fixatives are used (Howat and Wilson 2014). While optimizing IHC for a new protein, the ideal scenario would be to fix one piece of tissue in 10% NBF and use another piece of tissue to prepare a frozen tissue block, in case the antibody of interest is not compatible with formalin fixed tissue.
Antigen Retrieval
Partial or complete masking of antigenic/epitope immunoreactivity as a result of fixation is a common problem in IHC. Since 10% NBF is the most commonly used fixative, and in the foreseeable future, it will not be replaced by any other fixative, AR is an important variable that can influence the outcome, interpretation, and reproducibility in IHC. In most cases, IHC is not successful without AR. AR refers to any technique that reverses masking of an antigen/epitope to restore immunoreactivity. There are two commonly used AR methods: (1) enzymatic and (2) heat-induced. In some cases, the combination of heat-induced and enzymatic retrieval is useful. The mechanism involved in the enzyme-based method is considered to be cleavage of proteins by enzymes (Leong, Cooper, and Leong 2010). This in itself may negatively affect detection of certain antigens. Proteinase-K, pronase, pepsin, and trypsin are some of the enzymes used in the enzymatic method.
Heat-induced antigen retrieval (HIAR) is the most commonly used method; however, the mechanism of HIAR is not completely understood (Krenacs et al. 2010). HIAR involves heating the tissue sections in a buffer. This can be achieved by the use of a pressure cooker, microwave, steamer, or water bath. In our experience, the pressure cooker method has been the most effective in producing consistent and reproducible results as pressure and temperature can be well controlled. Others have also found this to be true (Pileri et al. 1997). In certain instances, the pressure cooker HIAR is not without some problems. Tissue sections detaching from the slide may occur especially with collagen rich tissues such as skin (Figure 2) or bone. With these tissues, steamer, microwave, or water bath HIAR may be preferable.
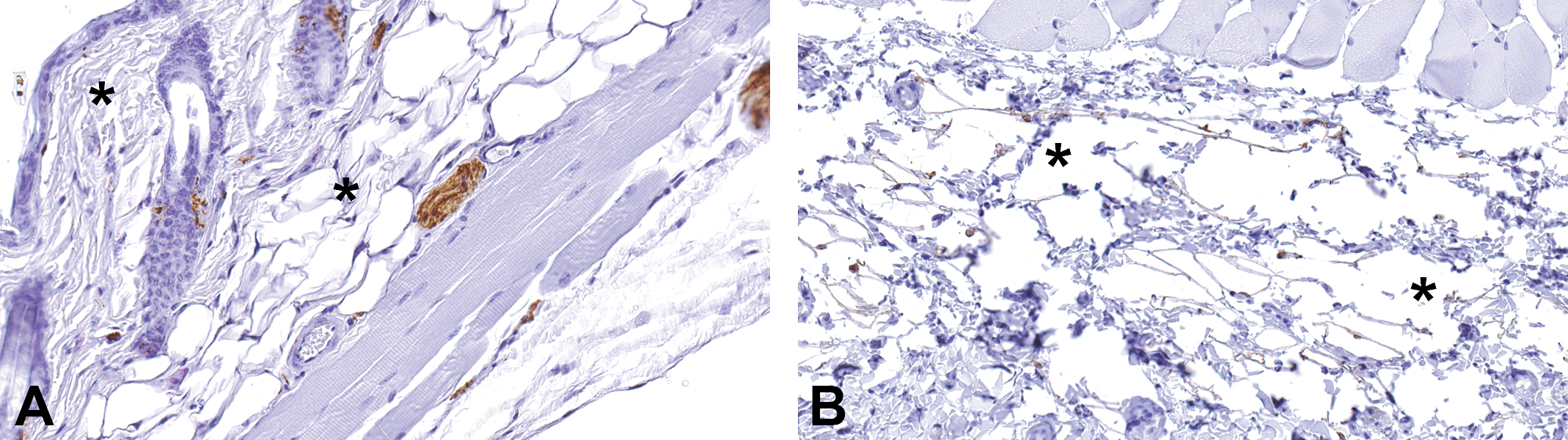
Illustration showing tissue lifting in collagen rich tissues such as skin. This is one of the disadvantages of using pressure cooker for heat-induced antigen retrieval in immunohistochemistry (IHC) of collagen rich tissues. (A) IHC for S-100 protein performed without antigen retrieval. Notice the lack of any artifactual changes in the collagen and adipose tissues (asterisks). (B) IHC for S-100 protein performed using pressure cooker and citrate buffer for heat-induced antigen retrieval. Notice the excessive lifting of collagen (asterisks) which interferes with staining and interpretation.
The effectiveness of HIAR seems to depend on the pH of the buffer used. Routinely, citrate buffer (pH 6.0) and Ethylenediaminetetraacetic acid (EDTA) buffer (pH 8.5) are used in our laboratory. In our experience, many antibodies seem to work best with citrate buffer at pH 6.0 on rat and mouse tissues. In human literature, citrate, Tris-HCL, and EDTA buffers are the commonly used buffers (Krenacs et al. 2010). However, the results vary among the published literature on human tissues (Yamashita 2007; Pileri et al. 1997; Shi et al. 1995; Taylor, Shi, and Barr 2011).
A single buffer that works the best for all antigens has not been identified, and the results of IHC staining may vary depending on the AR method and type of buffer used. Thus, it is recommended to test or validate various combinations of HIAR and buffers at 2 or 3 different pHs during the optimization. This is more critical while evaluating new proteins for which much information is not available in the literature. In such circumstances, it is equally important to support the IHC findings by other methods such as Western blot, in situ hybridization, polymerase chain reaction, and so on. To highlight the variability with AR, some examples are discussed below.
Peroxisomal membrane protein 70 on rat liver
Some proteins do not require AR. In such cases, choose the method that provides the most intense staining (Figure 3). This will allow the user to increase the specificity (by increasing the dilution) and to use lesser amount of primary antibody.
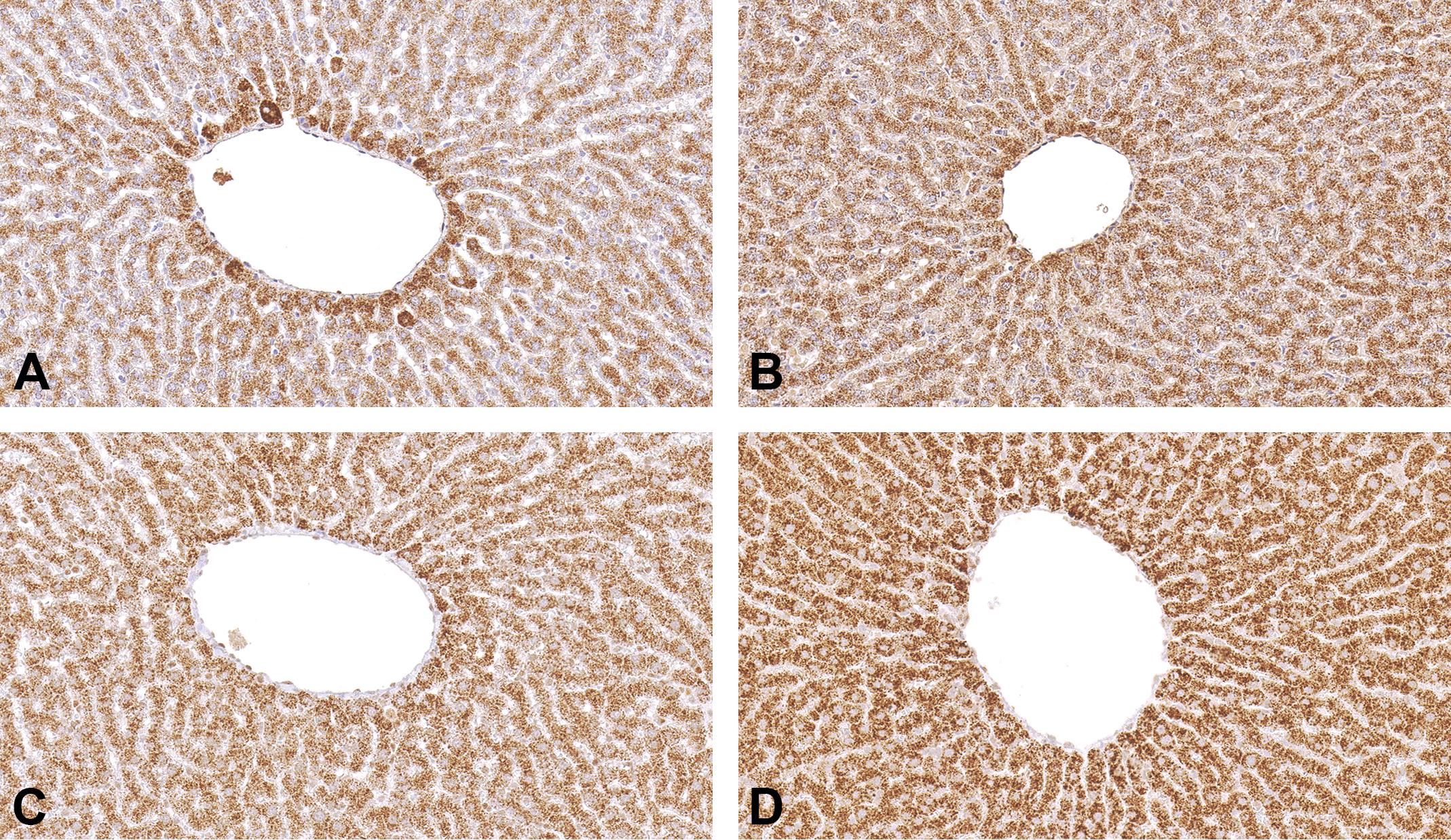
Immunohistochemistry for peroxisomal membrane protein 70 on rat liver demonstrates that certain proteins do not require antigen retrieval (AR). (A) No AR. (B–D) Pepsin, pressure cooker/citrate buffer, and pressure cooker/EDTA buffer were used for AR, respectively. Presence of immunostaining in (A) indicates that AR is not required. However, using the method which produces specific, higher intensity staining (in this case D) will allow the user to increase the specificity by increasing the antibody dilution.
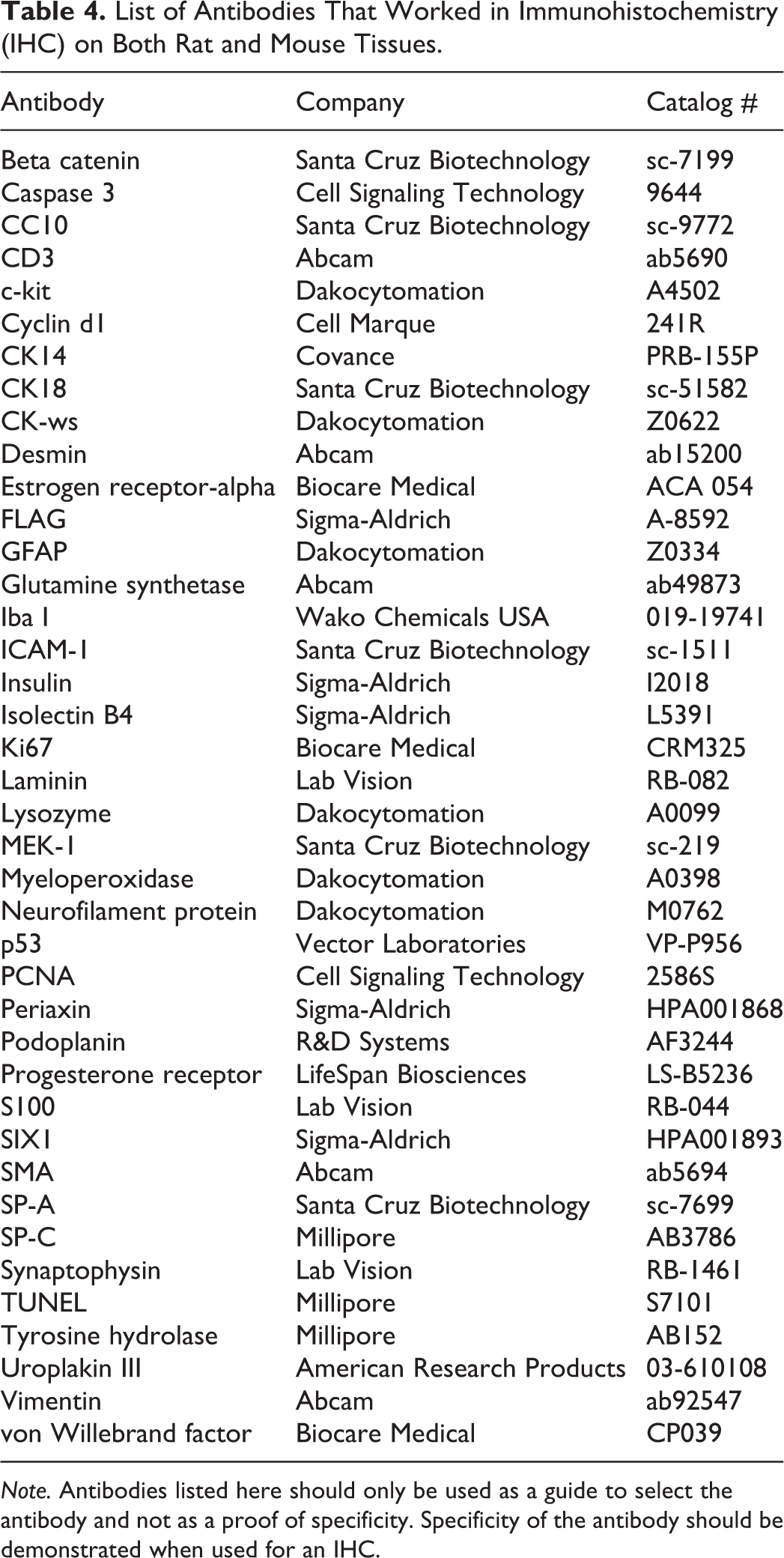
Paired box protein 5 on rat spleen
In formalin fixed paraffin-embedded tissues, most proteins are not detectable without the use of AR (Figure 4A and B). If a protein is not detectable without AR, tissue must be tested with at least one of the AR methods before deciding the presence or absence of a protein in any tissue.
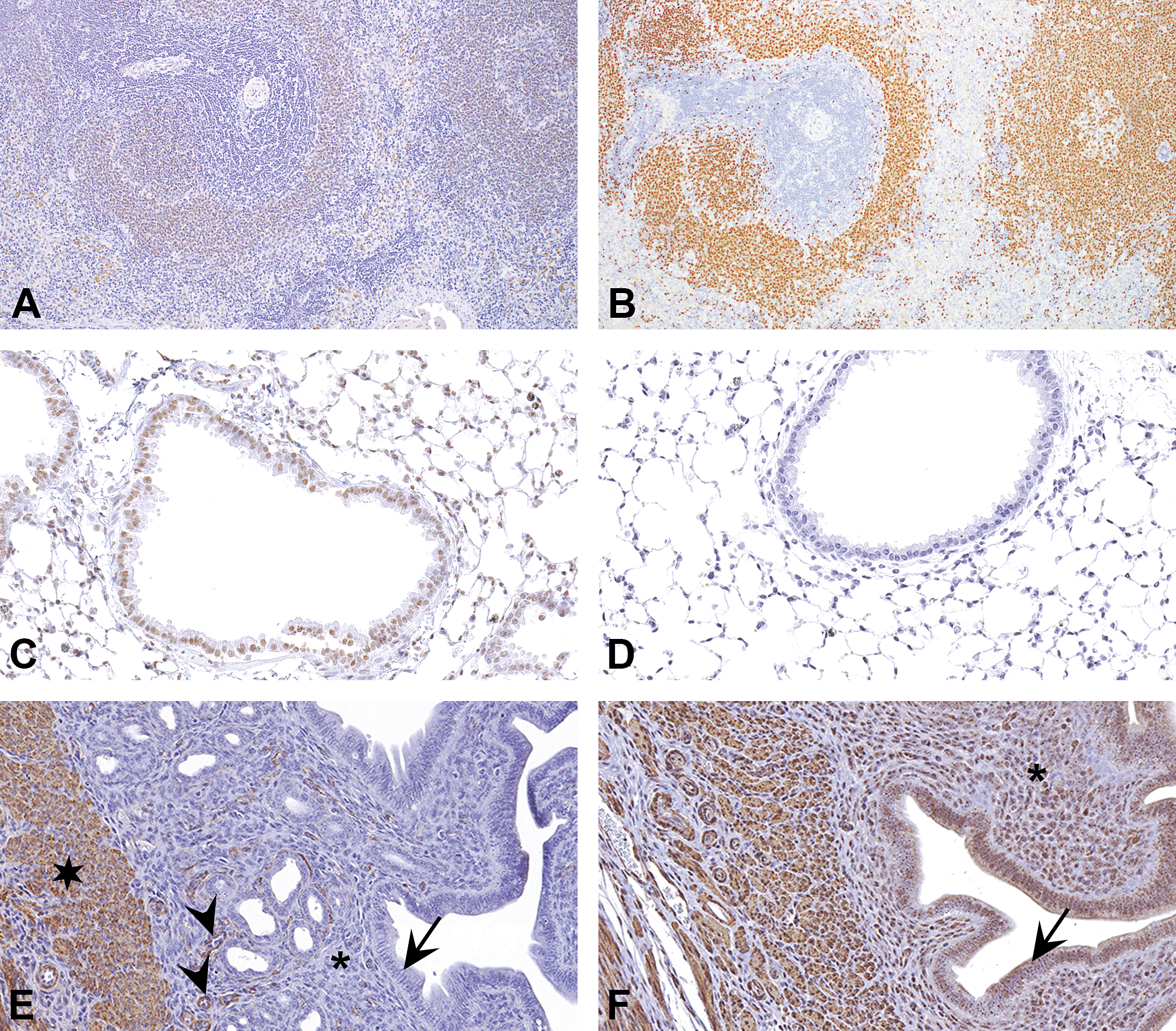
Influence of antigen retrieval (AR) on the results of immunohistochemistry (IHC). (A) and (B) show IHC using paired box protein 5 antibody, a B-cell marker, on rat spleen without and with AR, respectively. The staining is very weak when AR is not preformed (A) compared to the staining after AR (B). Use of an incorrect AR method can result in misinterpretation. (C) and (D) demonstrate TUNEL method in mouse lung sections in which pressure cooker/citrate buffer and proteinase K were used for AR, respectively. Use of pressure cooker in TUNEL method induces nonspecific nuclear staining in all the cells as seen in (C). Based on the protein biology and histomorphology, this should be interpreted as nonspecific staining. When proteinase K is used for AR, no nonspecific staining is observed in the same lung tissue (D). Use of AR can induce nonspecific staining for certain proteins. Specific staining for smooth muscle actin in the myometrium (star) and blood vessels (arrowheads) of mouse uterus is present in (E). Note that the epithelium (arrow) and stromal cells (asterisk) are not staining. Use of AR, when not required, can induce nonspecific staining as seen in (F). Here, in addition to the staining in myometrium and blood vessels, nonspecific staining is present in epithelium (arrow) and stromal cells (asterisk).
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining on a mouse lung
Not all AR methods are equal. To make a valid interpretation, it is important to have an understanding on the biology of a protein and the histomorphology associated with the protein of interest. Figure 4C and D illustrates staining of lung tissue with the TUNEL method to identify apoptotic cells. Using citrate buffer and pressure cooker, HIAR induced staining in the nuclei of all the cells in the section (Figure 4C). Although the nuclear location is accurate for this protein, none of the stained cells have the histomorphology of apoptotic cells. Therefore, the observed staining is nonspecific. When another section from the same tissue was stained using proteinase K for AR, none of the cell nuclei stained (Figure 4D). This agrees with the absence of apoptotic cells in the tissue section. This example highlights the importance of validating different AR methods and critical importance of knowledge on protein biology during optimizing the AR method for IHC.
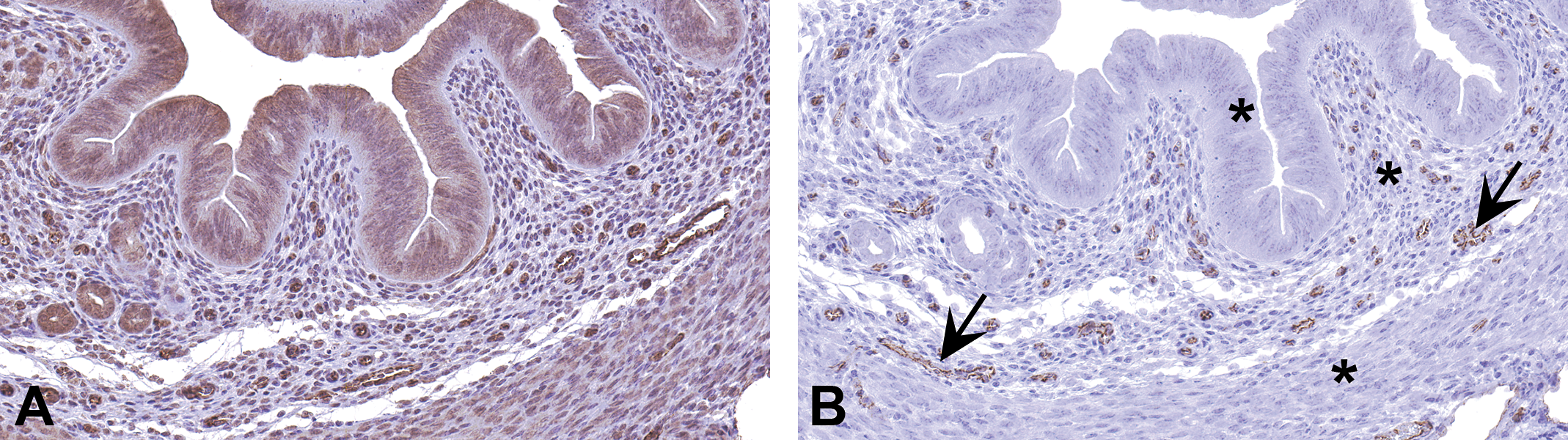
Smooth muscle actin staining in uterus
This protein does not require AR. However, if AR is performed, it induces nonspecific staining in the tissue. This is evident in Figure 4E and F. Without AR, specific staining for Smooth Muscle Actin (SMA) is present (Figure 4E). When AR is performed, nonspecific staining is present in the epithelium and stromal cells (Figure 4F). This information is essential and should be obtained during optimization. It is useful in instances where dual or multiple stainings are performed to detect two or more antigens within one tissue section, and the other protein(s) of interest require AR. In such situations, having information on the effect of AR is essential in deciding which stains can be combined.
Detection method
More recently, polymer-based systems have been promoted as a better detection system for IHC as it significantly reduces or completely eliminates endogenous avidin/biotin background staining. This may be true for many antibodies and one such example is shown in Figure 5A and B. However, it is important to remember that polymer is not always the best choice for all proteins. At least, one nonpolymer-based detection method (such as the ABC method) and one polymer-based detection method should be tried during the optimization of an antibody. Figure 5C and D shows a section of intestine stained for Ki67 using the ABC method and a polymer. The ABC method produces crisp and intense staining compared to staining with the polymer. In addition, not all cells stain with the polymer. Determining the optimal detection method will significantly reduce the chances of misinterpreting the IHC results.
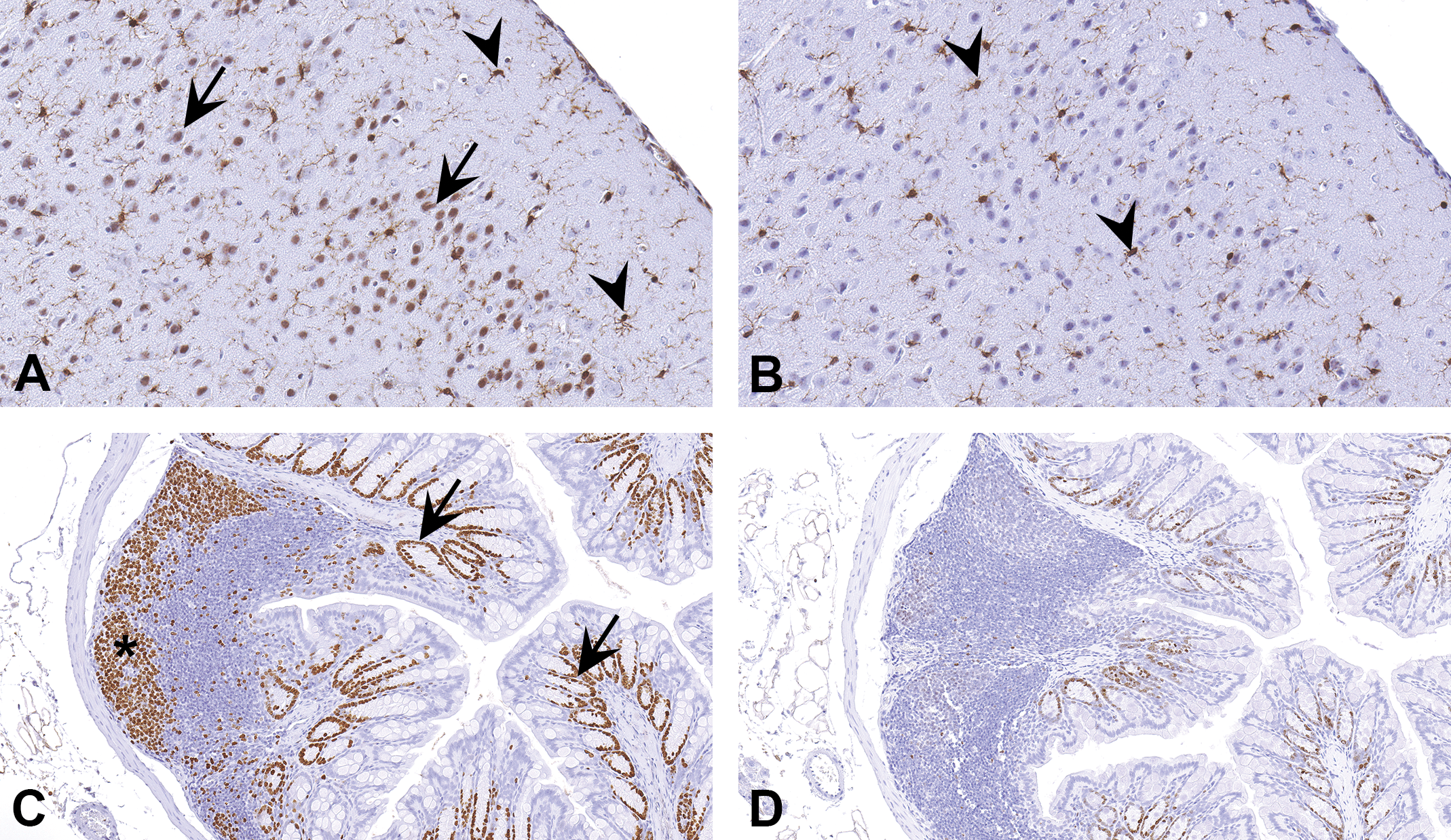
Choice of detection method can influence the immunohistochemistry (IHC) results. In some instances, using a polymer-based detection method is better than other methods such as avidin-biotin complex (ABC) method to avoid nonspecific staining. (A) Mouse brain stained with ionized calcium-binding adapter molecule 1 antibody using ABC method. Note nonspecific staining in the nuclei of neurons (arrows) along with specific staining of microglial cells (arrowheads). When a polymer-based method is used (B), only specific staining in the microglial cells (arrowheads) is present. However, polymer-based method is not the best for all proteins. In (C), IHC for Ki67, a marker of proliferating cells, by ABC method demonstrates intense staining in crypt epithelium (arrow) and lymphocytes (asterisk). However, only small proportion of cells are stained in (D), when the section is stained using a polymer-based method. This comparison allows the user to decide that the method used in (C) is optimal for Ki67 staining. This example highlights the need to determine optimal detection system at the method optimization stage.
Choice of antibody
It is a very common practice to use commercially available antibodies for IHC. The purchase of antibody is made on the basis of either published literature or a vendor’s recommendation(s). However, it is critical to ensure that the antibody is performing in the prescribed manner by using the appropriate controls and by understanding the biology of the protein. It is especially important in the investigative pathology setting where a pathologist is not part of the evaluation. The importance of this variable is highlighted using some examples described below.
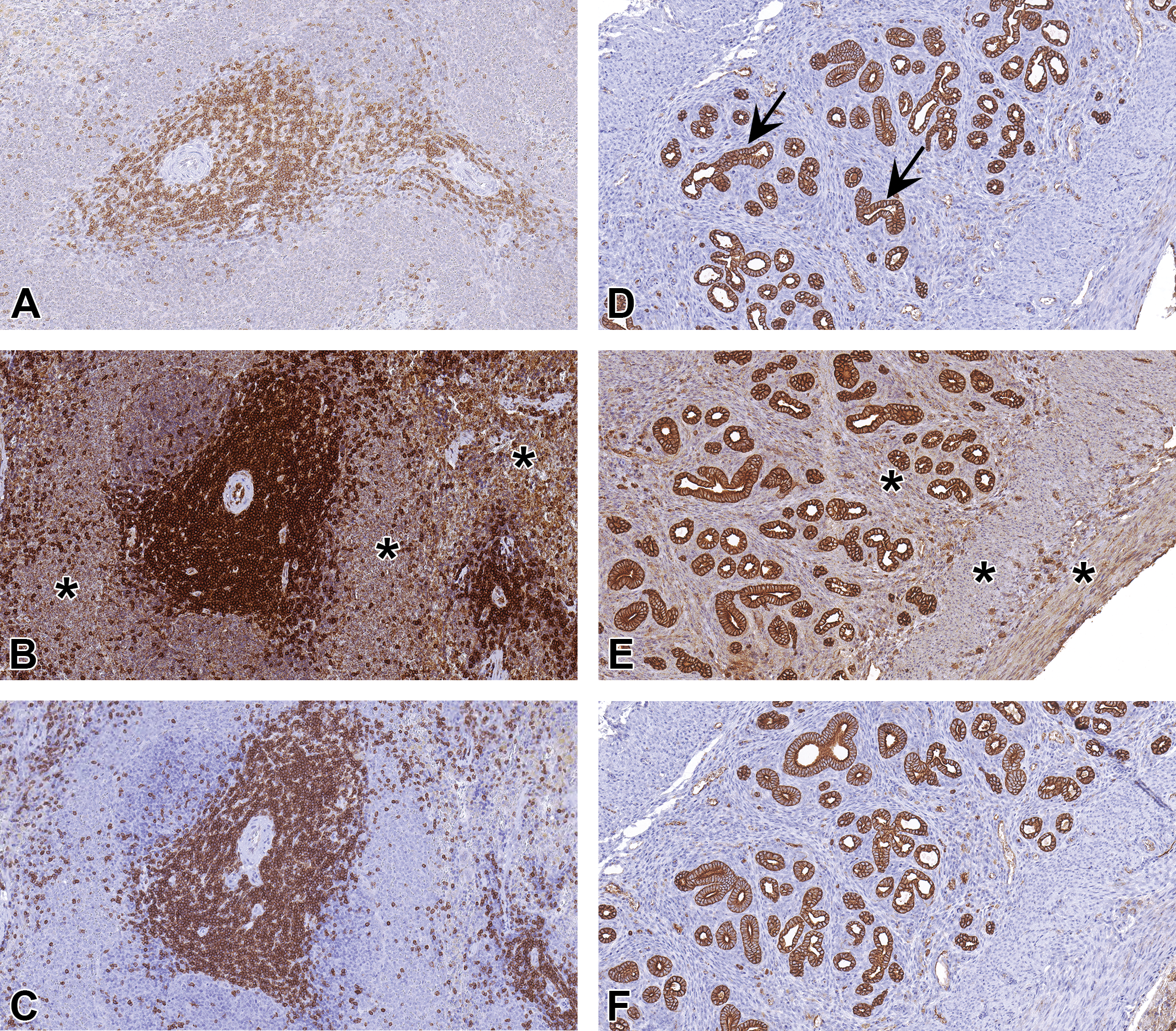
Mucin-2 (Muc-2)
Muc-2 is a protein secreted by the goblet cells in the intestine (Gendler and Spicer 1995). In Figure 6A, staining for Muc-2 (Abcam; catalog # ab11197) appears real as the negative control stained using nonspecific immunoglobulins has no staining (Figure 6B). However, if observed closely, the goblet cells that should stain specifically with the Muc-2 antibody are not staining. Therefore, the staining should be interpreted as nonspecific. Figure 6C demonstrates another section of intestine stained with a separate Muc-2 antibody (Novus Biologicals, catalog # NBP1-31231). Specific staining is present in the goblet cells and the epithelial cells are not stained. In this example, the first antibody that produced nonspecific staining was claimed by the company to specifically mark mouse MUC2. Now, based on our and probably others’ feedback, the company has removed mouse from the list of “species reactivity” (http://www.abcam.com/muc2-antibody-9961-ab11197.html; accessed on December 28, 2017).
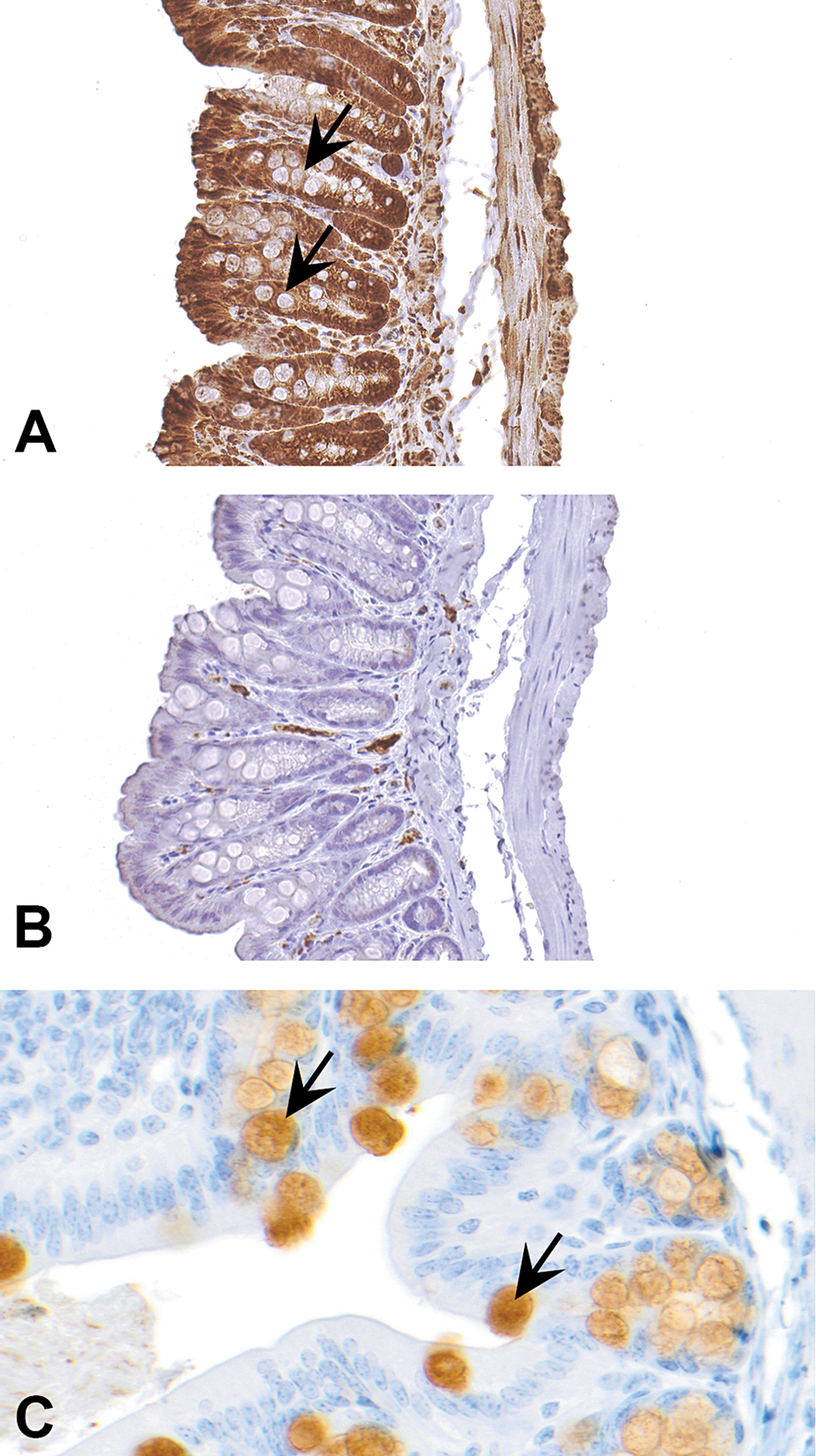
(A)–(C) demonstrate how choice of an antibody can influence the outcome of immunohistochemistry. In (A), an antibody claimed by the company (Abcam; catalog # ab11197) to detect mucin 2 (MUC2) in mouse produced nonspecific staining and did not recognize goblet cells which contain MUC2 (arrows). Although the negative control stained with an isotype matched nonspecific immunoglobulin produced no staining (B), staining in (A) can’t be interpreted as positive as the staining is present everywhere except goblet cells. Section of intestine in (C), stained by another MUC2 antibody (Novus Biologicals, catalog # NBP1-31231) shows specific staining in goblet cells (arrows).
SRY Box 9 (Sox-9)
These examples illustrate the importance of understanding protein biology even when the antibody is chosen based on the literature. SOX-9 protein is a transcription factor that is essential in testicular development. Its nuclear expression in Sertoli cells can be used to identify Sertoli cells (Barrionuevo et al. 2016). In this case, sections of mouse testis were stained using a Sox-9 antibody (Santa Cruz Biotechnology; catalog # sc-20095). The section stained with Sox-9 antibody had staining in the seminiferous tubules (Figure 7A), and the negative control section stained with a nonspecific immunoglobulin had no staining (Figure 7B). To the untrained eye, the staining in Figure 7A appears to be in the nuclei of some cells. However, close examination shows that the staining in most of the cells is actually not in the nucleus, but in the perinuclear area. Further, it is not staining the Sertoli cells but germ cells. The distribution and morphology of Sertoli cells are highlighted in Figure 7C by another Sertoli cell marker, GATA binding protein 4 (Abcam, catalog # ab84593). If the knowledge of Sertoli cell morphology and distribution is applied, staining in Figure 7A can be interpreted as nonspecific staining. However, the antibody has been used in the literature and shown to specifically stain Sertoli cells (Wang et al. 2007). The antibody is also marketed for use on paraffin-embedded tissue sections. In these type of situations, it is wise to obtain a different antibody or use a different marker based on the research question.
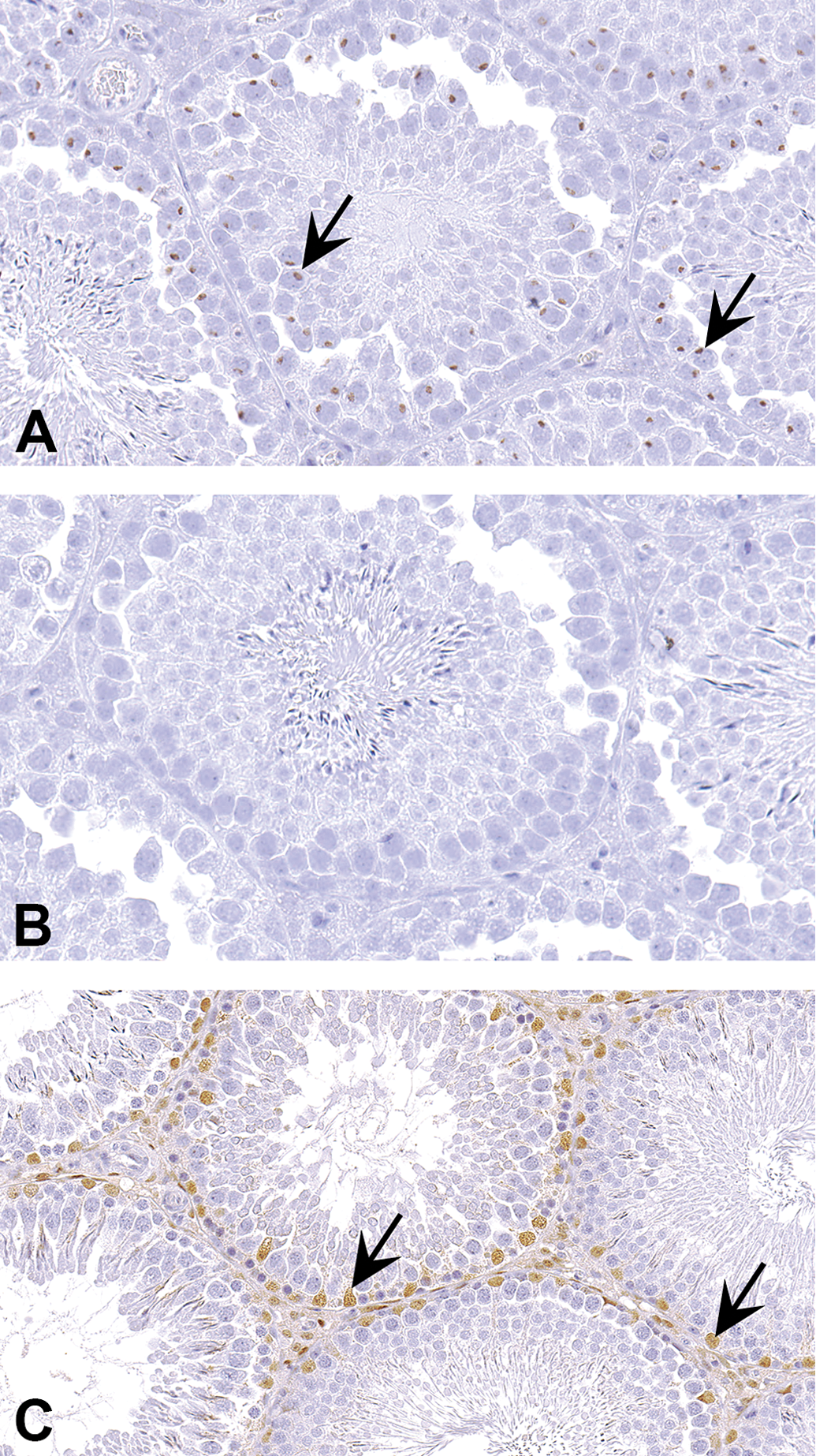
In addition to proper choice of an antibody, comprehensive knowledge on the protein of interest is important to avoid misinterpretations. (A) is a section of testis stained with an antibody claimed to be specific for SRY Box 9 protein (SOX-9; Santa Cruz Biotechnology, catalog # sc-20095), a Sertoli cell marker. Based on the negative control section stained with an isotype matched nonspecific immunoglobulin (B), staining in (A; arrows) can be interpreted as specific. However, applying the knowledge of Sertoli cell distribution and morphology (arrows) shown in (C) using another Sertoli cell marker, GATA binding protein 4 (GATA4; Abcam, catalog # ab84593), staining in (A) doesn’t seem to be in Sertoli cells. Further, SOX-9 is a transcription factor and is known to be localized to nucleus. However, in (A), the staining appears to be perinuclear. Considering all these together, staining in (A) must be interpreted as nonspecific.
Peroxisome proliferator-activated receptor-alpha (PPARα)
Figure 8 demonstrates staining of rat liver using a PPARα antibody (Abcam; catalog #ab8934). When AR is performed using pepsin, intense staining is present in the centrilobular hepatocytes (Figure 8A). Since the negative control is clean (Figure 8B), it can be presumed that the staining is real and interpreted as PPARα is highly expressed in centrilobular hepatocytes. However, when the same tissue is subjected to staining after AR using EDTA buffer (pH 8.5) and pressure cooker, staining is only present in bile ducts and not in hepatocytes (Figure 8C). In this scenario, the results can be interpreted as PPARα is expressed only in bile ducts and not in hepatocytes. From the earlier publications, it is known that PPARα is a nuclear receptor expressed in various tissues including liver, and the protein is localized to nucleus (Braissant et al. 1996; Desvergne and Wahli 1999). In light of these reports, both interpretations (Figure 8A and C) are incorrect as nuclear staining is not present in the hepatocytes. If the earlier publications regarding nuclear localization are to be believed, this antibody is not specific and can’t be used with confidence. If this type of antibody is used to describe localization of a novel protein, the type of AR can easily influence the location and cell type in which the protein’s localization gets reported. In addition to the choice of antibody, this example highlights the importance of testing various AR methods and the necessity of other tools to support the IHC results.
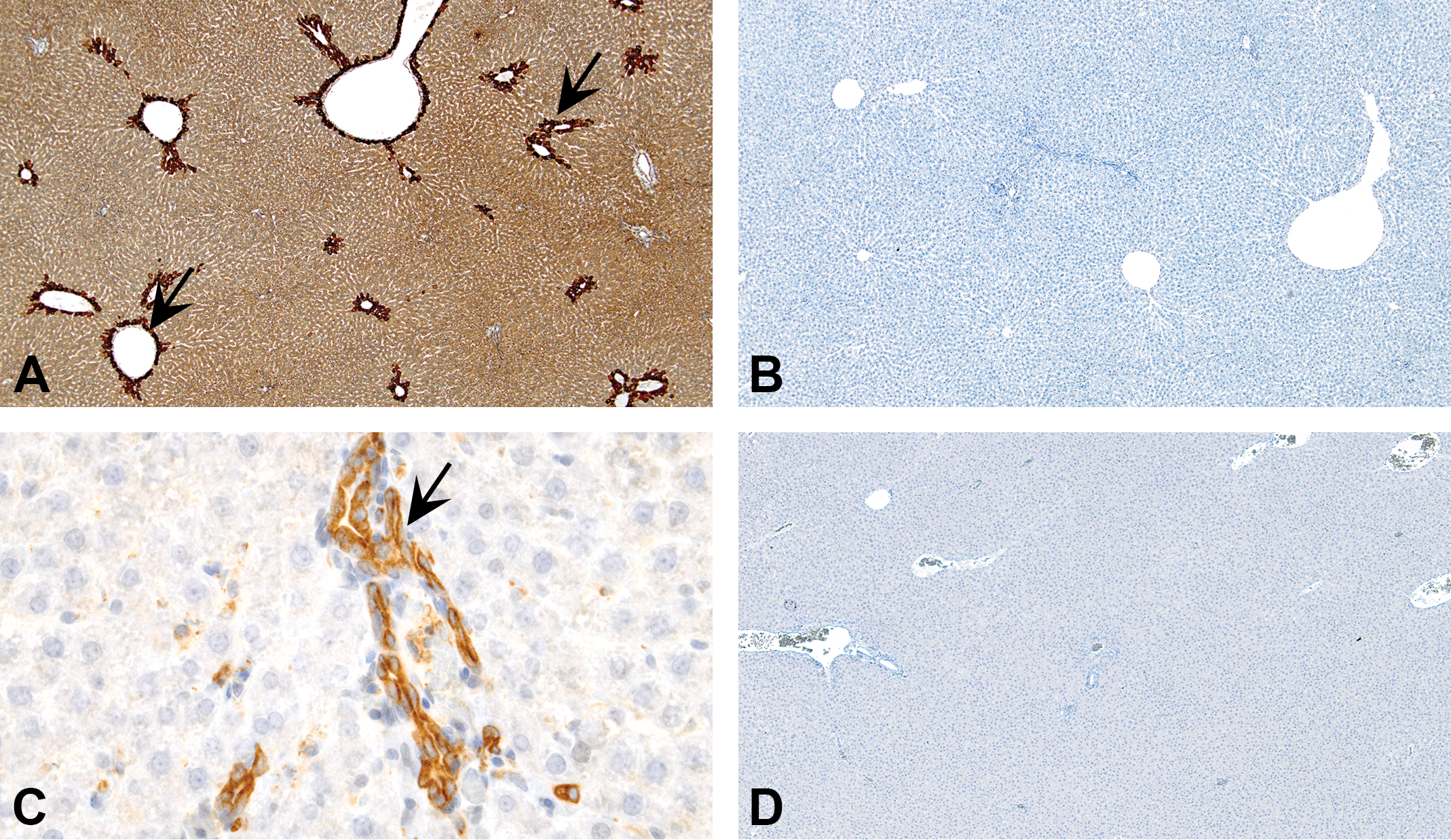
Importance of choice of antibody, antigen retrieval (AR) method and importance of protein biology to avoid misinterpretation in immunohistochemistry are highlighted in (A)–(D). Peroxisome proliferator-activated receptor-alpha (PPARα) is a nuclear receptor expressed in various tissues including liver, and the protein is localized to nucleus. (A) When a section of rat liver is stained with PPARα antibody (Abcam; catalog #ab8934) using pepsin for AR, intense cytoplasmic staining can be observed in centrilobular hepatocytes (arrows). The corresponding negative control section shown in (B) has no staining. However, when the liver section is stained with the same antibody but with a different AR (pressure cooker and EDTA buffer), the staining pattern changes. Now the staining is present only in bile duct cytoplasm (C; arrow) and not in hepatocytes. The corresponding negative control shown in (D) has no staining. The results vary based on the type of AR used. Further, staining observed in both (A) and (C), although appear specific, is not true because the localization seen here is cytoplasmic and not nuclear as reported for PPARα.
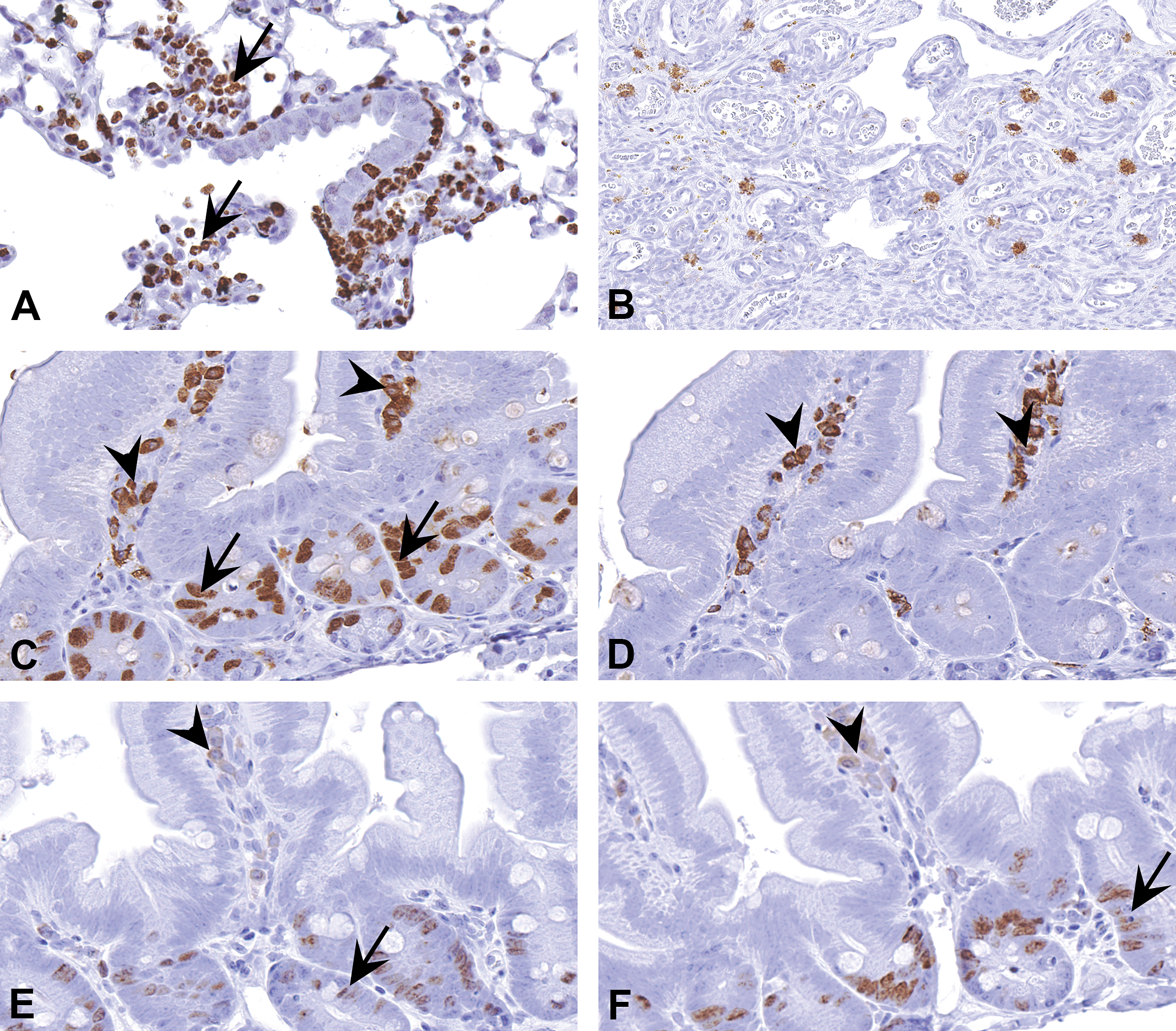
Octamer-binding transcription factor 4 (OCT4)
Pluripotent stem cells and cancer stem cells express OCT4 in the nucleus (Zeineddine et al. 2014). Since OCT4 is known to be expressed in human seminomas (Looijenga et al. 2003), rat seminoma was used as a positive control to develop a method for rat tissues. As shown in Figure 9A, there was no staining, except for some weak nonspecific staining in the surrounding inflammatory cells, in the seminoma. However, some nuclear staining was present in germ cells in the seminiferous tubules (Figure 9B). Since much is not known about the biology of this protein in rats, the staining was assumed to be specific. Using this method, a uterine tumor was stained with OCT4 antibody. Based on the negative control (Figure 9D), all the tumor cells appeared to be positively stained (Figure 9C). However, when other areas of the tissue section were examined, there was staining in the normal endometrium as well as the myometrium (Figure 9E and F). This finding is unlikely as these normal cells are well-differentiated cells, and OCT4 is associated with stemness. This clearly demonstrates that relying entirely on one type of negative control and ignoring the other components in the tissue as a whole can lead to misinterpretation of the staining results. The most appropriate interpretation in this scenario is that the antibody (Santa Cruz Biotechnology, catalog # sc-5279) is probably not appropriate for rat tissues.
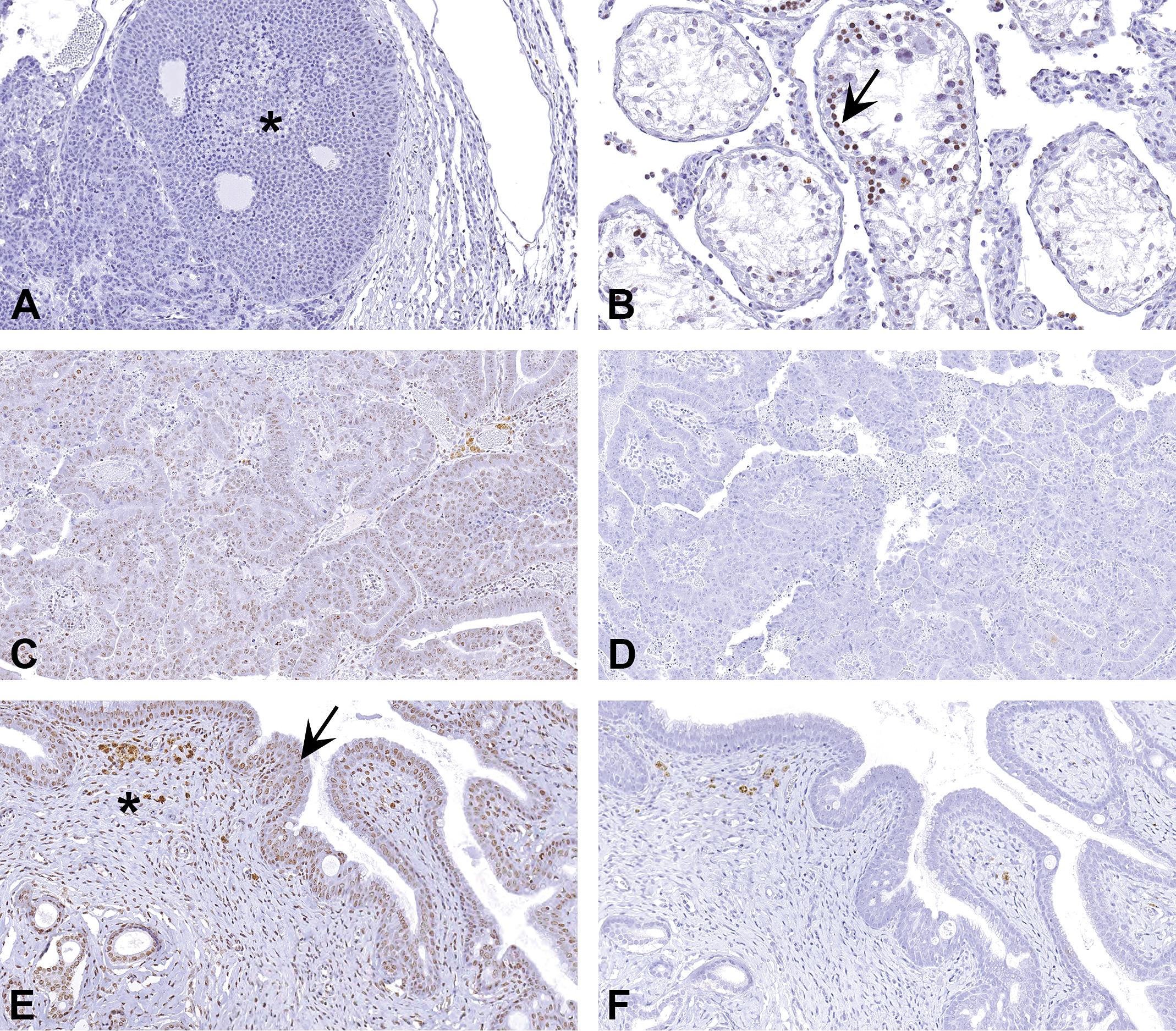
(A)–(F) demonstrate how tissue other than the region of interest can help in determining the specificity of an antibody. (A) Section of rat testis stained with octamer-binding transcription factor 4 (OCT4) antibody (Santa Cruz Biotechnology, catalog # sc-5279) showing no staining in a seminoma. (B) However, some cells in the surrounding seminiferous tubules (arrow) show nuclear staining. (C) Section of a uterine tumor stained with OCT4. The neoplastic cells appear to be positive based on the absence of any staining in the negative control (D). However, when the surrounding normal tissue (E) and corresponding area in the negative control (F) are evaluated, staining appears to be also present in the normal endometrial epithelium (arrow) and stroma (asterisk). Since these cells are terminally differentiated, and OCT4 expression is associated with stemness, this antibody can be interpreted to be not appropriate for rat tissues.
Like the above examples, many more examples can be shown to highlight how the vendor’s information on the antibodies, publications where the data are entirely based on IHC, and not supported by additional tools, information from just one type of negative control can’t be relied upon completely. In Table 1, we listed several antibodies that have been advertised to work in rats and/or mice but have failed to provide satisfactory results in our laboratory. In Tables 2 to 4, we have also provided a list of antibodies that have been successfully used on mouse and rat tissues in our laboratory.
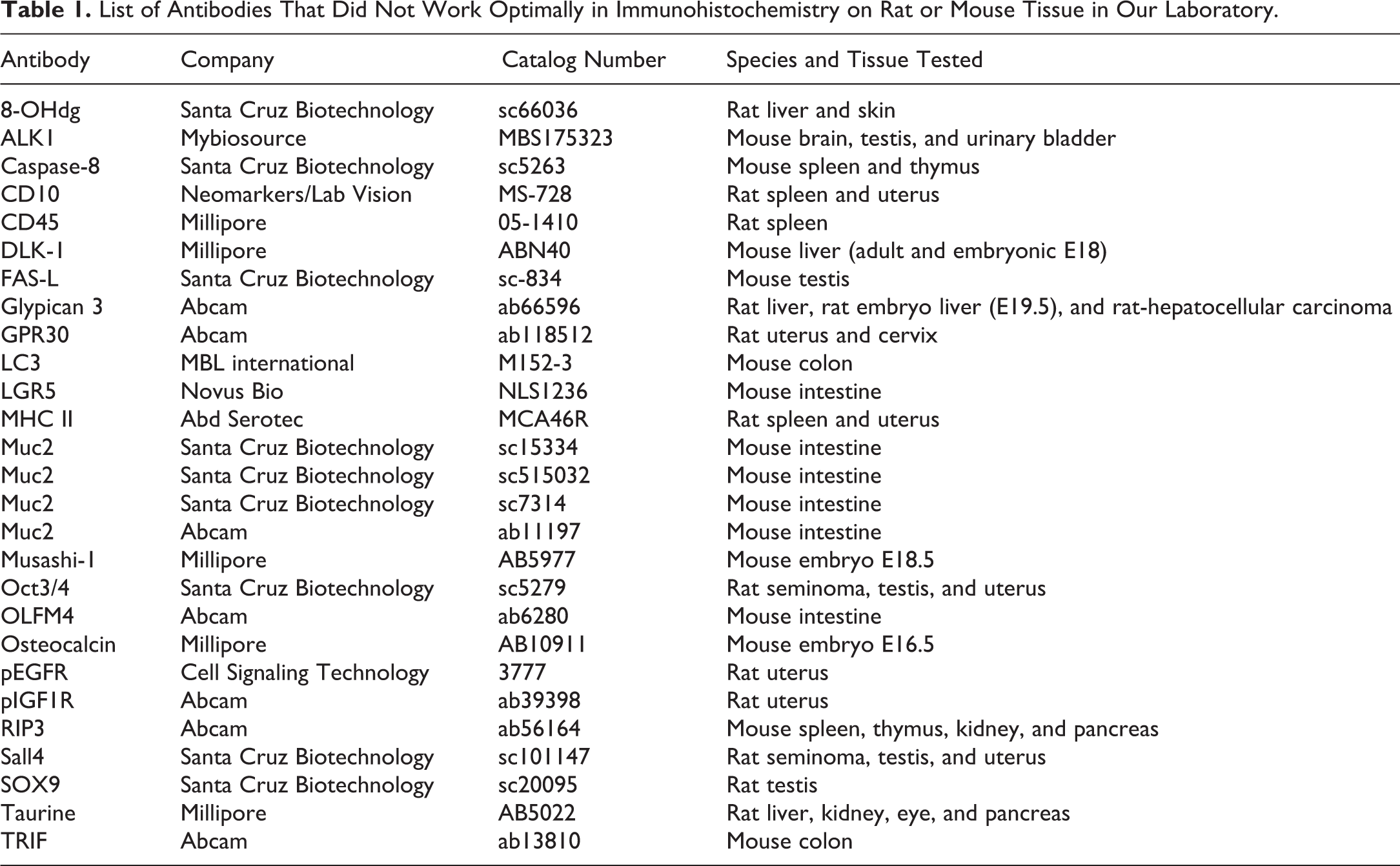
List of Antibodies That Did Not Work Optimally in Immunohistochemistry on Rat or Mouse Tissue in Our Laboratory.
List of Antibodies Used in Immunohistochemistry (IHC) on Mouse Tissues.
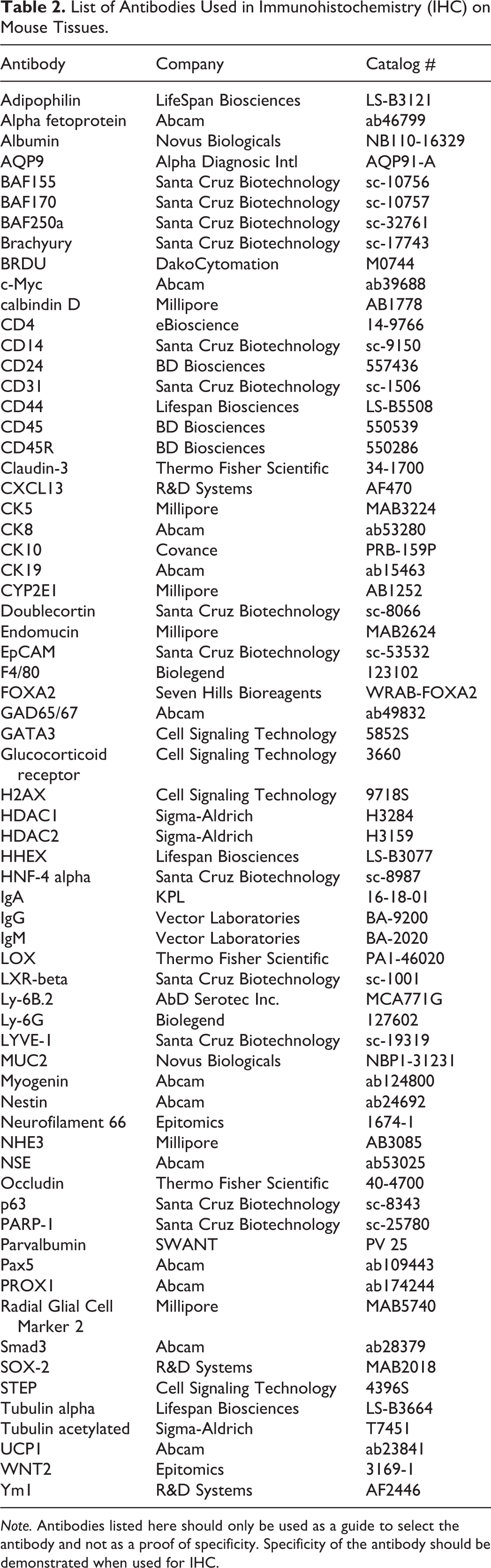
Note. Antibodies listed here should only be used as a guide to select the antibody and not as a proof of specificity. Specificity of the antibody should be demonstrated when used for IHC.
List of Antibodies Used in Immunohistochemistry (IHC) on Rat Tissues.
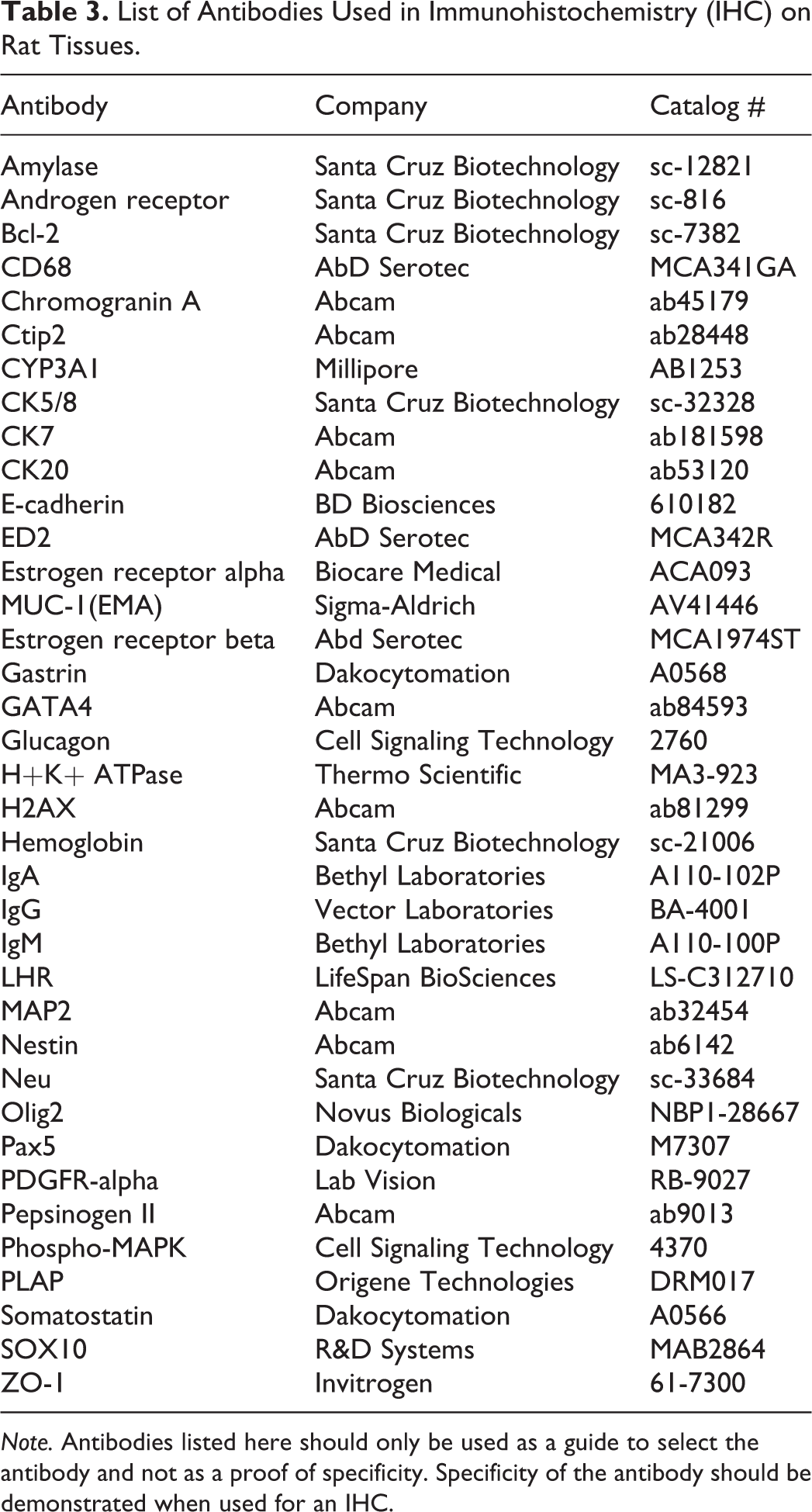
Note. Antibodies listed here should only be used as a guide to select the antibody and not as a proof of specificity. Specificity of the antibody should be demonstrated when used for an IHC.
List of Antibodies That Worked in Immunohistochemistry (IHC) on Both Rat and Mouse Tissues.
Note. Antibodies listed here should only be used as a guide to select the antibody and not as a proof of specificity. Specificity of the antibody should be demonstrated when used for an IHC.
Change of lot number
Commercially available antibodies when purchased at different times can have a different lot number referring to the production of antibodies. It is very critical to record the lot number each time an antibody is purchased. Since the quality of antibody can vary between different lots, it is important to include this information in publications. This will assist in reproducing the published results. Within each laboratory, when a new lot of antibody is received, stained tissue sections should be evaluated for nonspecific staining. When nonspecific staining is present, the antibody should be titrated to determine the optimal dilution to be used in the staining. The following two examples will illustrate the latter. CD3 is a pan T-cell marker which is extensively used to identify T cells in tissue sections. Our laboratory has been using a polyclonal CD3 antibody (Abcam; catalog # ab5690) at a dilution of 1:100 to get specific staining of T-cells in mouse spleen (Figure 10A). Initial staining with a new lot of antibody used at a dilution of 1:100 produced significant nonspecific staining in non-T-cell regions of spleen (Figure 10B). The staining protocol using the new lot was again optimized and had to be used at a 1:1,000 dilution to obtain specific staining of T-cells (Figure 10C). The protein concentration of both was reported to be 0.2 mg/ml. Antibodies to cytokeratin-18 can be used to specifically label uterine endometrial and glandular epithelium (Figure 10D). Routinely, sections are stained at a dilution of 1:25 (Santa Cruz Biotechnology, Cat# sc-51582, Lot#H2714). When the same dilution was used for a new lot of antibody (#K0116), nonspecific staining was present (Figure 10E). The protein concentration (0.2 mg/ml) between the two lots was the same. To obtain specific staining, the antibody had to be titrated again, and the appropriate dilution was 1:250 (Figure 10F). Although the protein concentration is the same, specific antibodies can vary between lots due to variability in immune response. Every time there is a change in lot numbers, the appropriate dilution should be determined again. If the sample size to be stained for an experiment is known, enough antibody from one particular lot should be purchased and used before expiration date to avoid complications associated with change of lots.
(A)–(F) demonstrate how using antibodies from different lots can affect staining results. (A) Section of spleen stained with a CD3 antibody shows specific staining in T-cells. (B) Section of the spleen stained with CD3 antibody from a different lot has significant nonspecific staining (asterisks). (C) The CD3 antibody used in (B) was diluted 10 times more to obtain specific staining in the spleen. (D) A section of uterus stained with a cytokeratin 18 antibody shows specific staining in the glandular epithelium (arrows). (E) Section of the uterus stained with the cytokeratin antibody from a different lot produces substantial nonspecific staining in the stroma and myometrium (asterisks). (F) The antibody used in (E) was diluted 10 times more to obtain specific staining in uterus.
Nonspecific binding in certain cell types
In some instances, certain cells are known to stain nonspecifically with certain or most antibodies. This information can either be obtained by referring to published literature (Ward and Rehg 2014) or through experience. Here, some examples involving neutrophils, mast cells, and plasma cells are highlighted. The example in Figure 11A represents nonspecific staining for Ki67 in neutrophils. Proliferating cells in all phases of cell cycle except G0 can be stained using Ki67 antibody (Scholzen and Gerdes 2000). Since neutrophils are terminally differentiated cells, staining of Ki67 in neutrophils should be interpreted as nonspecific. Based on the IHC principles, and knowledge on sources of nonspecific staining, various methods to overcome this issue have been tried in our laboratory without success. In such instances, it is important to ignore the nonspecific staining at evaluation. In a situation where the laboratory is only asked to perform the staining and not the evaluation, this issue should be explained to the investigators, so that they do not include nonspecifically stained cells while counting the number of positive cells in tissue sections. This awareness is all the more critical when the stained tissues are used for automated image analysis, so that all the brown cells are not counted as positive.
Nonspecific staining in a variety of cell types. (A) Nonspecific staining of neutrophils (arrows) by Ki67 antibody in a section of mouse lung. (B) Nonspecific staining of mast cells in a section of rat uterus. (C) Section of mouse intestine stained with a bromodeoxyuridine (BrdU) mouse monoclonal antibody. The nuclear staining in the crypt epithelial cells is specific (arrows). However, cytoplasmic staining in the plasma cells (arrowheads) in the lamina propria is nonspecific. (D) A section of the same intestine in (C) stained by omitting primary antibody. Notice cytoplasmic staining in plasma cells (arrowheads) even in the absence of primary BrdU antibody. This results from using mouse monoclonal antibody on mouse tissues. Various methods have been described to overcome this issue. (E) and (F) provide two such examples. In (E) and (F), sections of intestine are stained for BrdU using a mouse-on-mouse horseradish peroxidase (HRP) polymer and a mouse on mouse HRP kit, respectively. In both, there is reduction in the nonspecific cytoplasmic staining of plasma cells (arrowheads). However, the staining for BrdU is also substantially reduced (arrows). Such a reduction can be an issue when the antigen of interest is not abundant in tissue.
Cytoplasmic mast cell staining is nonspecific in most instances. Many antibodies nonspecifically stain mast cells (Figure 11B) and various methods have been described to overcome nonspecific staining in these cells (Bussolati and Gugliotta 1983; Harley, Gruffydd-Jones, and Day 2002; Ward and Rehg 2014). We have tried the different methods described in the literature, including the nonbiotin-based method. However, we continue to experience nonspecific staining in mast cells. In the majority of the instances, awareness of nonspecific mast cell staining is sufficient for accurate interpretation if mast cells are not the cell of interest. Rat tissues and some mouse tissues contain very large number of mast cells, and the cytoplasmic staining in mast cells should be considered nonspecific unless proven otherwise by other methods.
Plasma cells are also known to yield nonspecific staining (Ward and Rehg 2014; Ramos-Vara and Miller 2014). This is common when the primary antibody is an unconjugated mouse antibody, which necessitates the use of anti-mouse antibody as a secondary antibody. Since the secondary antibody is directed against mouse immunoglobulin, it specifically binds with plasma cells rich in immunoglobulins (Figure 11C and D). This is not problematic when the protein of interest is localized to the nucleus, as plasma cell staining will be cytoplasmic. However, it becomes an issue when the protein of interest is localized to cytoplasm. Trained pathologists are able to exclude staining in plasma cells from evaluation; however, this will be a challenge for untrained eyes. One method employed to avoid nonspecific staining is by using a primary antibody raised in a species other than mouse. Methods designed to eliminate nonspecific staining while using primary mouse antibody on mouse tissues have been described (Ramos-Vara 2005). However, in our experience, none of the described methods completely eliminate nonspecific background (Figure 11E and F).
Nonspecific Staining: Sources and Solutions
Apart from the variables explained above, there are some additional causes of nonspecific staining. Technical errors, such as allowing the section to dry during a staining procedure, can result in nonspecific staining. In Figure 12, a uterine section is stained with CD31, which labels endothelial cells. Epithelial cells, endometrial stromal cells, and smooth muscle cells should have no staining and serve as internal negative controls. When the section is allowed to dry during staining, antibodies bind nonspecifically and result in nonspecific staining in most of the cells (Figure 12A). This problem is effectively corrected if the tissue is not allowed to dry during staining (Figure 12B).
Technical error as a source of nonspecific staining. (A) Section of uterus allowed to dry during immunohistochemical staining with a CD31 antibody. As a result, instead of staining only the endothelium, nonspecific staining was present throughout the section. (B). Tissue not allowed to dry stained with CD31 antibody resulted in a specific staining of endothelial cells (arrows). Epithelium, stroma, and myometrium (asterisks) that serve as internal negative controls are not stained.
Serum used for blocking nonspecific interactions could be another source of technical error. For blocking, it is a common practice to use serum from the species in which the secondary antibody is raised. If the incorrect serum is used for blocking, such as one from the species in which the primary antibody is raised (as opposed to secondary antibody), it will result in nonspecific staining throughout the tissue. This is not a common problem. However, this can occur with individuals who are relatively new to performing IHC.
Other sources of nonspecific staining result from endogenous peroxidase or AP activity, hydrophobic, ionic and electrostatic interactions, binding of antibodies to Fc receptors on various cells, endogenous biotin, and nonspecific binding of avidin. Various strategies have also been described to prevent these (Ramos-Vara 2005; Taylor, Shi, and Barr 2011).
Although endogenous peroxidase has been described as a source of nonspecific staining, it can be easily avoided by incubating the tissue sections in 0.3% to 3% hydrogen peroxide solution. A quick method to determine the presence of endogenous peroxidase in tissue sections is by directly incubating the sections with an HRP-chromogen (after deparaffinization and rehydration). Color development indicates that there is endogenous activity in the tissue section. In Figure 13, tissues known to be rich in endogenous peroxidase were incubated with chromogen. There is no staining in any of these tissues. Only red blood cells demonstrate some color. Therefore, endogenous peroxidase may not be a major issue when dealing with nonspecific staining during IHC method development.
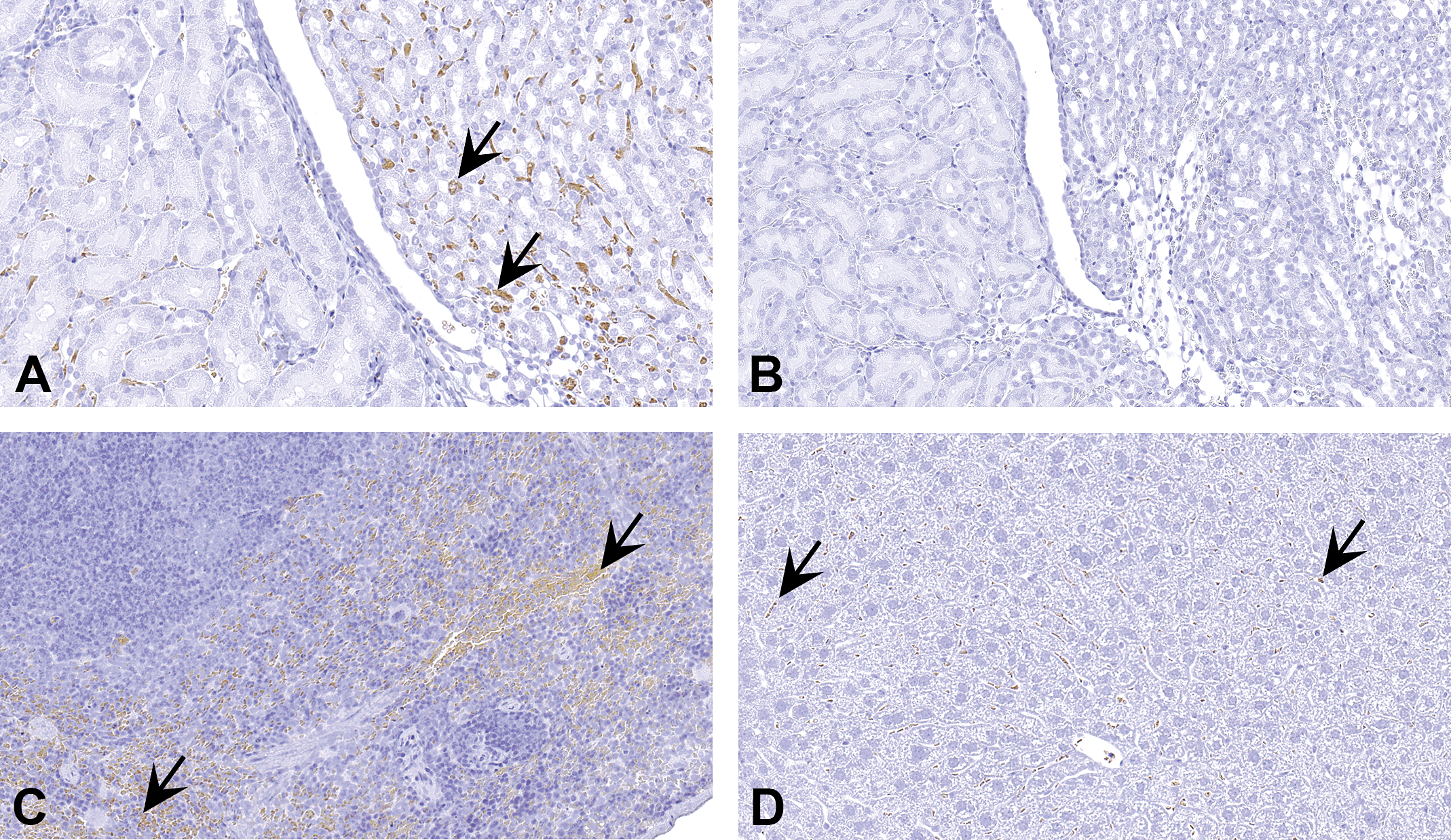
Endogenous peroxidase is not a major source of nonspecific staining in tissues. Sections of a kidney incubated with chromogen without (A) and with (B) preincubation in hydrogen peroxide. Only weak nonspecific staining is present in red blood cells (arrow) in (A) compared to no staining in (B). Similarly, staining sections of spleen (C) and liver (D) without preincubation in hydrogen peroxide produces weak nonspecific staining in red blood cells (arrows).
For other causes of nonspecific staining such as hydrophobic interactions, ionic and electrostatic interactions, and nonspecific binding of primary antibodies to Fc receptors, blocking with serum, bovine serum albumin, and various other agents are widely practiced. However, recently, the need for blocking in IHC has been questioned in a report demonstrating that nonspecific staining did not occur in 25 different tissues using 46 different antibodies when blocking was omitted (Buchwalow et al. 2011). Our laboratory findings support this report. For example, Figure 14 illustrates the lack of nonspecific IHC staining in sections of the spleen, intestine, and brain performed with and without blocking. We have also noted that blocking for endogenous biotin is not required in many instances. Based on the published literature and our experience, it appears that blocking with serum or other agents or blocking for biotin is not an essential step in IHC. Therefore, when nonspecific staining is observed, the focus should be more on the quality of antibody and tissue itself in addressing the problem. It is best practice to check the need for blocking at the method development stage. If it is not required, omitting the blocking step will save time, money, and eliminate one another variable.
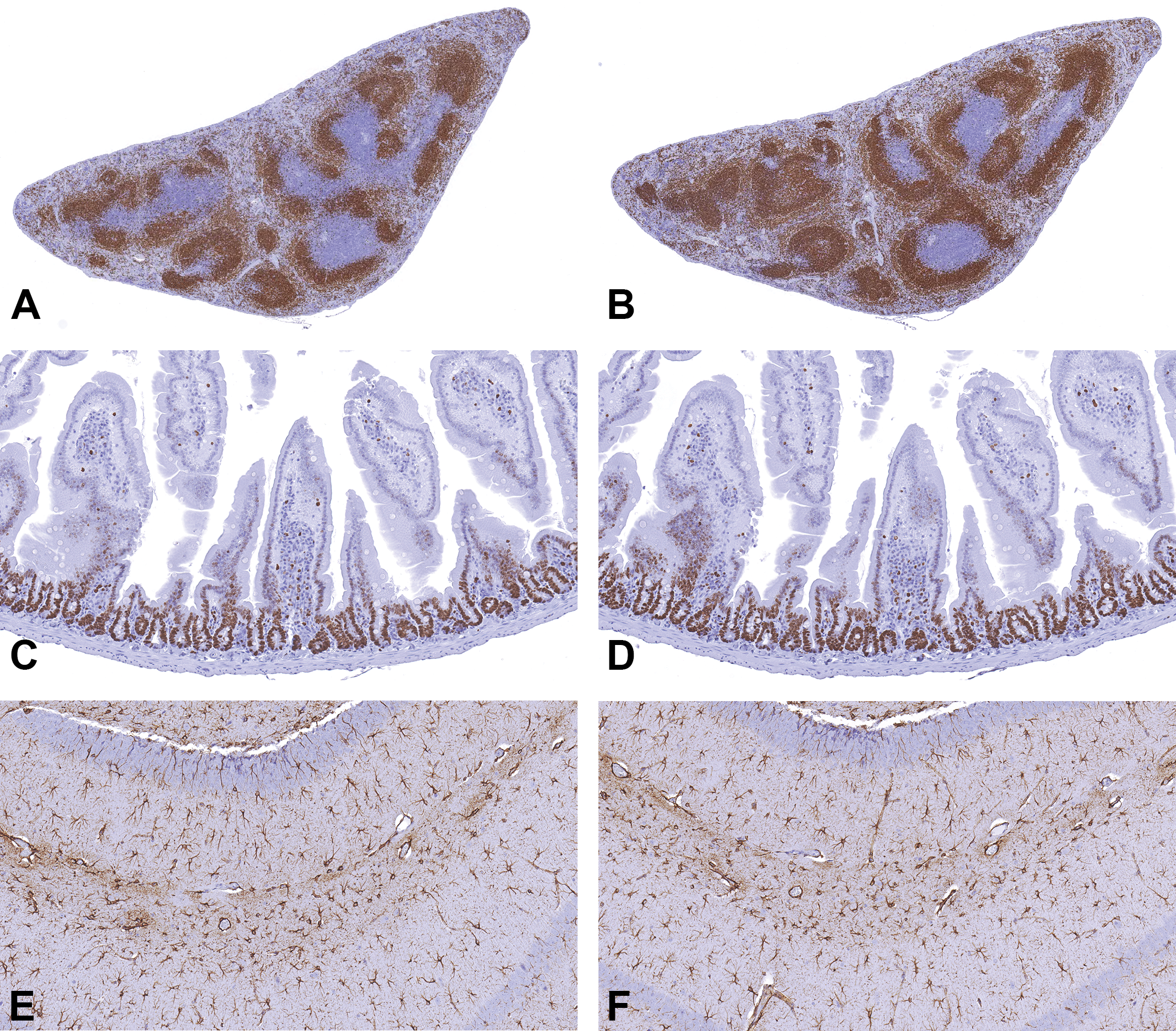
Blocking with serum or other agents is not critical in immunohistochemistry on formalin fixed paraffin embedded tissues. Sections of mouse spleen (A and B), intestine (C and D), and brain (E and F) are stained with paired box protein 5, Ki67, and glial fibrillary acidic protein, respectively. Sections in (A), (C), and (E) are stained after blocking with 10% normal serum, and sections in (B), (D), and (F) are stained without blocking with serum, before incubating with the primary antibody. Staining is similar in sections stained with or without blocking. There is no nonspecific staining when blocking with serum is omitted.
Use of Appropriate Controls in IHC
Controls are an essential and integral part of IHC. The IHC results are meaningless if not interpreted in the context of appropriate controls. (Burry 2011). Appropriate use of controls assures that reagents and methods were correctly applied to obtain the expected results. Controls can be broadly categorized into primary antibody control, secondary antibody control, and label control.
Primary antibody controls are used to determine whether the antibody is specific for an antigen. This can be accomplished by the following different approaches: Using a knockout mouse, transfected cells expressing a protein of interest, and cell line lacking the protein of interest. While using a knockout animal, care has to be taken not to use functional knockout tissues as the protein of interest can still be present in the tissue. Incubating the primary antibody with peptide or antigen before using it in the IHC reaction. Although many researchers consider this to be a very good measure of antibody specificity, it is not a great control by itself. It will only prove that antibody is specific to the antigen that it was raised against. After preincubation with peptide or purified antigen, since there is no primary antibody remaining for the actual reaction, staining will not be seen. Additionally, it prevents the antibody from binding to any possible cross-reactive protein/s present in the tissue of interest (Holmseth et al. 2012). Therefore, the absence of staining in sections stained with preabsorbed control section does not prove antibody specificity. This may result in misinterpretation. Using preimmune serum, nonspecific immunoglobulins or isotype matched normal immunoglobulins for negative controls. Since most antibodies are purchased commercially, preimmune serum is not available for most of these antibodies. Instead of primary antibody, purified normal immunoglobulins or serum from the species in which the primary antibody is raised is the most commonly used control. This will allow the user to determine whether the immunoglobulins from the species in which the primary antibody is raised are binding specifically or nonspecifically. If the primary antibody is specific, there should not be any staining when the purified normal immunoglobulins or serum is used instead of primary antibody. Substituting primary antibody with a buffer instead of using purified normal immunoglobulins or serum is not an acceptable control as nonspecific binding cannot be determined in the absence of any immunoglobulins. Use of Western blotting to determine the specificity of the antibody. This is very valuable for determining the specificity when the antibody recognizes the correct molecular weight protein in tissue lysate rather than the pure antigen.
Secondary antibody controls are used to demonstrate that the secondary antibody binds only to the primary antibody used. In the absence of primary antibody, there should not be any binding, and hence, no colored product should be present. The easiest method to test the specificity of secondary antibody is by replacing primary antibody with buffer during the staining.
The labeling control should include omission of the enzyme-labeled antibody/reagent. In the absence of the enzyme, there should not be any color development. If color develops without the presence of enzyme-labeled antibody/reagent, it indicates the presence of endogenous enzymes.
While performing IHC, appropriate supportive evidence should be provided for any antibody used in the experiment. If evidence is not part of the experiment in which the antibody is used, citing literature where a particular antibody is shown to be very specific is acceptable. In case of novel proteins, multiple controls described above should be included to show the specificity of the antibody. In addition, supporting the results of IHC by other methods will make the interpretation stronger. Irrespective of the type of controls used, thorough knowledge on the protein of interest will be very important for meaningful interpretation. Whenever possible, the tissue chosen for staining should include some internal positive and negative controls. Some examples of the use of internal positive and negative tissue controls are shown in Figure 15.
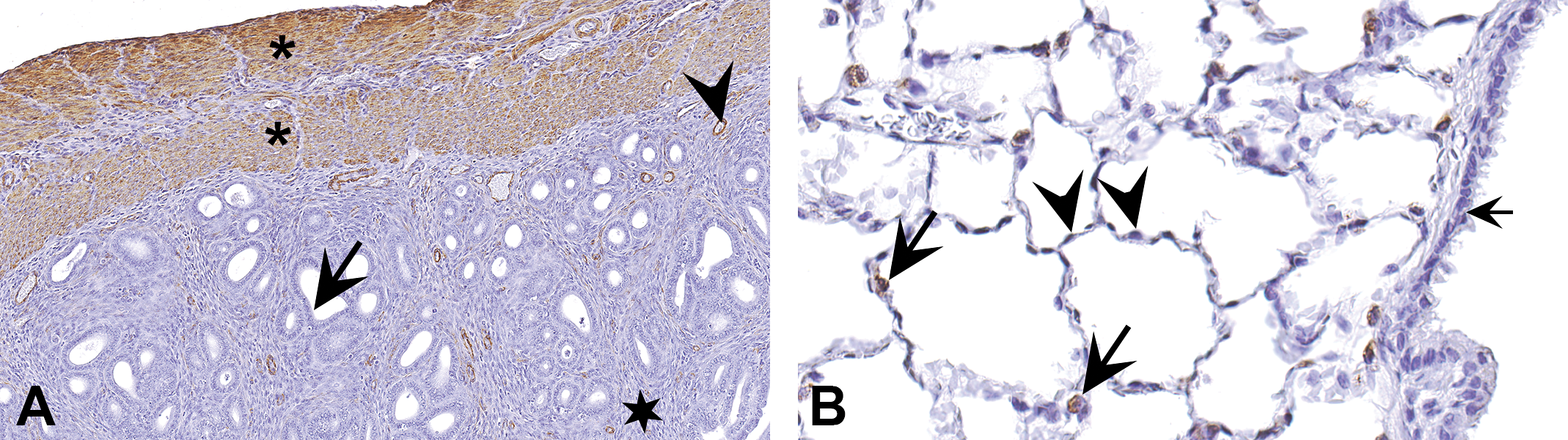
Examples of internal controls in immunohistochemistry. (A) Section of a rat uterus stained with smooth muscle actin. Since smooth muscle actin is known to be expressed by smooth muscle cells (in myometrium and around blood vessels) and not by epithelium or endometrial stroma, they serve as internal positive and negative controls, respectively. In this section, smooth muscle cells in the myometrium (asterisks) and around the blood vessels (arrowhead) are stained. The epithelium (arrow) and endometrial stroma (star) are not stained. Therefore, the observed staining can be interpreted with confidence as specific. (B) Similarly, in a section of mouse lung stained with surfactant protein C (SPC; B), type II alveolar epithelium serves as an internal positive control and type I alveolar epithelium and bronchiolar epithelium serve as internal negative controls. In this section, only type II cells (arrows) are stained; type I cells (arrowheads) and bronchiolar epithelium (short arrow) are not stained. Based on these, staining for SPC can be interpreted as specific.
How to Approach IHC Method Optimization for an Antibody
When IHC methods optimization or development is needed for an antibody, a systematic approach will save time, energy, and money. First, the objectives should be clearly defined. If it is a novel protein, IHC should either be used as a supplementary tool to support the hypothesis generated by other tools or the findings of the IHC should be complemented using other tools such as Western blots, flow cytometry, in situ hybridization, immunoprecipitation, and so on. If it is not a novel protein, a thorough understanding of the protein biology with regard to expression and cellular localization will be critical in determining the specificity of the antibody.
Second, it is important to know the fixative that was used and the duration of fixation. This information is very helpful when IHC is unsuccessful. In most laboratories, antibodies are commercially obtained based on vendor-provided information. An antibody is best selected on the basis of published literature or discussions with a person or persons with experience and expertise in IHC. When using published literature, the investigator should critically review the IHC results to form his or her own conclusions with regard to specificity of an antibody.
Third, since the outcome in IHC can vary based on the type of AR used, starting with one particular dilution of antibody (based on manufacturer’s recommendation or based on literature) along with different ARs and without any AR is recommended. At this stage, if there is any specific staining, the optimal dilution should be determined for the primary antibody in the next step. If there is no staining, increasing the antibody concentration or increasing the incubation time can be tried. If this also fails, it is very unlikely that the antibody will work, and a different antibody should be tried.
Finally, after optimization of primary antibody dilution, the staining should be repeated using a polymer-based system and at least one nonpolymer-based system (such as ABC method) to determine the optimal method. If results are the same between both the systems, we prefer the polymer-based system, as it shortens the overall time required to perform IHC. With the optimized method, it is advisable to stain only a few samples before staining a large number of samples. This is especially essential when the method is optimized using a different tissue and not using the samples of interest. This approach to method optimization is summarized in Figure 16.
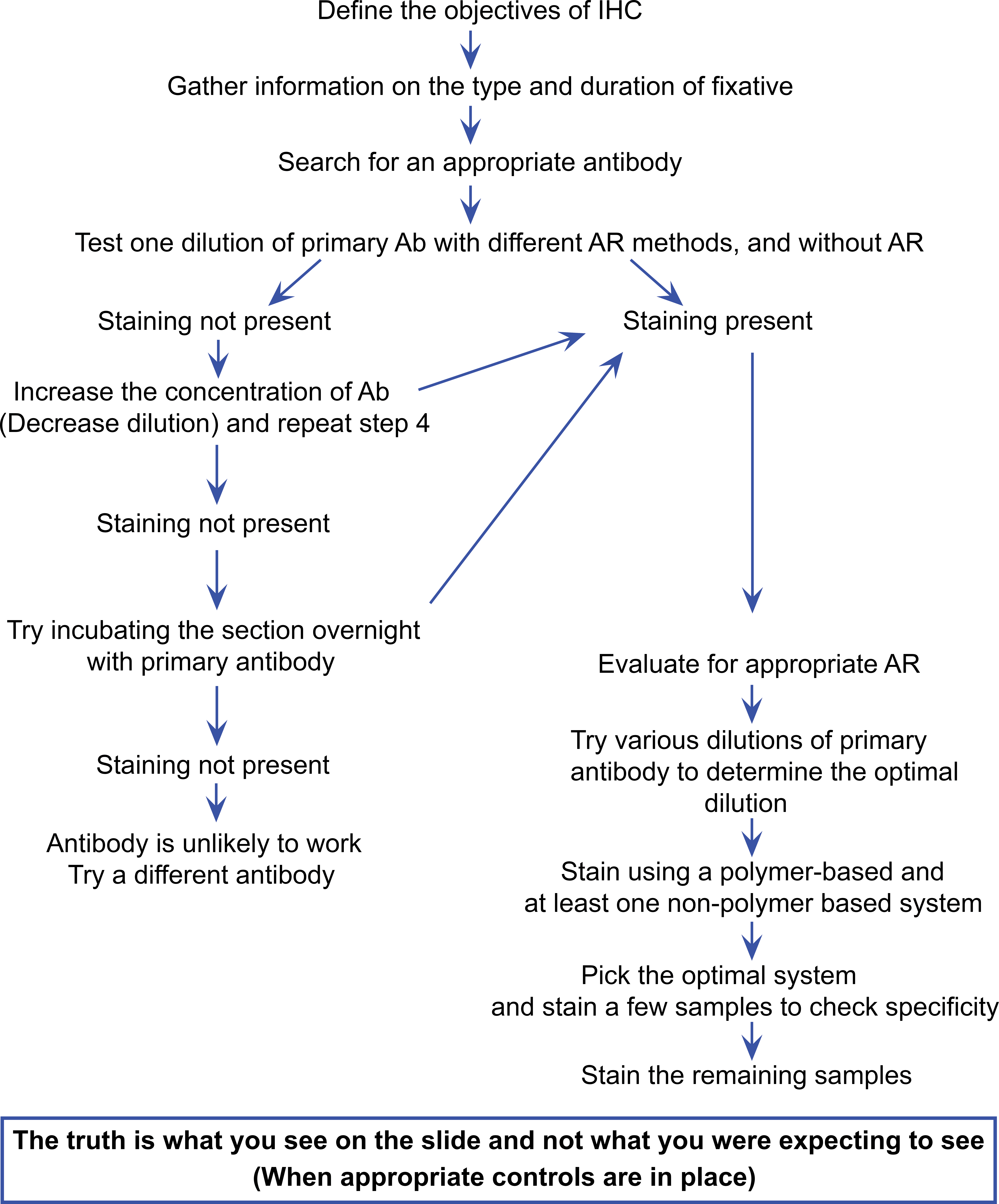
Method optimization for immunohistochemical staining with a new antibody.
What Can Reviewers/Journals Do to Ensure Reproducibility of IHC Results
There is no doubt that reproducibility of IHC results is a major issue in science. Numerous manuscripts have been written with very good proposals to resolve this issue (Marx 2013; Bordeaux et al. 2010; Goldstein et al. 2007; Delpire 2015; Torlakovic, Nielsen, Vyberg et al. 2015, Hewitt et al. 2014; Holmseth et al. 2012; Torlakovic et al. 2014; Torlakovic, Nielsen, Francis et al. 2015). If the journal and reviewers demand the information mentioned in the Table 5 from the researchers, issues of reproducibility and misinterpretations can be resolved.
Recommendation on the Details to be Included in the Methods in Published Manuscripts with Immunohistochemistry Results.

Conclusion
To summarize, IHC is an invaluable tool in diagnostic and investigative pathology and in science in general. It is very important to use optimally fixed tissue and a good quality antibody. Resolution of background staining should focus more on the quality of tissue and antibody rather than various other interactions such as hydrophobic interactions, ionic and electrostatic interactions, and nonspecific binding of primary antibodies to Fc receptors described as causes of nonspecific staining in the literature (Ramos-Vara 2005). When publishing, it is very important to include details on all the variables that can affect the result and reproducibility of the IHC experiment. When staining for a novel protein for which not much information is available, results should be supported by other methods. The importance of positive and negative controls should be taken seriously and routinely included as part of any IHC staining procedures. There are numerous research antibodies available from 300 different companies (Baker 2015), a fraction of which are considered to be unique (Delpire 2015). Therefore, a methodical thought process and approach, critical review, and validation are essential in choosing and use of an antibody. Marx (2013) highlighted various resources essential in the use of an antibody. Thoughtful publications that can assist in obtaining reproducible results (Bandrowski et al. 2016) have led to the useful web portals such as https://scicrunch.org/resources and http://antibodyregistry.org/. If all the above points are addressed, reproducibility and misinterpretation of IHC results will be less of a problem.
Footnotes
Acknowledgments
The authors would like to thank the staff of the NIEHS/DNTP Immunohistochemistry Core Laboratory for some of the materials used in this article; Quashana Brown for her help in compiling the list of antibodies successfully used in the core laboratory; Eli Ney, NIEHS/DNTP Image Analysis Core, for scanning of slides, Beth Mahler, EPL, for her help with the images, and David Sabio, EPL, for helping with the drawings; and Drs. Ramesh Kovi (EPL, Inc.) and Cynthia Willson (ILS, Inc.) for their critical review of this article.
Author Contribution
Authors contributed to conception or design (KJ); data acquisition, analysis, or interpretation (KJ, HJ, RH, NC); drafting the manuscript (KJ); and critically revising the manuscript (KJ, HJ, RH, NC). All authors gave final approval and agreed to be accountable for all aspects of work in ensuring that questions relating to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the NIH, National Institute of Environmental Health Sciences.