
Abstract
Indoxyl sulfate (IS) is a protein-bound uremic toxin that accumulates in patients with declining kidney function. Although generally thought of as a consequence of declining kidney function, emerging evidence demonstrates direct cytotoxic role of IS on endothelial cells and cardiomyocytes, largely through the expression of pro-inflammatory and pro-fibrotic factors. The direct toxicity of IS on human kidney proximal tubular epithelial cells (PTECs) remains a matter of debate. The current study explored the effect of IS on primary cultures of human PTECs and HK-2, an immortalized human PTEC line. Pathologically relevant concentrations of IS induced apoptosis and increased the expression of the proapoptotic molecule Bax in both cell types. IS impaired mitochondrial metabolic activity and induced cellular hypertrophy. Furthermore, statistically significant upregulation of pro-fibrotic (transforming growth factor-β, fibronectin) and pro-inflammatory molecules (interleukin-6, interleukin-8, and tumor necrosis factor-α) in response to IS was observed. Albumin had no influence on the toxicity of IS. The results of this study suggest that IS directly induced a pro-inflammatory and pro-fibrotic phenotype in proximal tubular cells. In light of the associated apoptosis, hypertrophy, and metabolic dysfunction, this study demonstrates that IS may play a role in the progression of chronic kidney disease.
The removal of nitrogenous waste products by the kidney is an essential function, and a failure of this is associated with metabolite accumulation and various deleterious outcomes that are characteristic of uremic syndrome (Meyer and Hostetter 2007). A potential contributor to deleterious outcomes is the accumulation of uremic toxins (toxic metabolites) which may have direct or indirect cytotoxic effects (Meyer and Hostetter 2007). Patients with end-stage kidney disease require kidney replacement therapy, predominantly dialysis, to reduce serum concentrations of nitrogenous waste; a limitation is that albumin-bound solutes are poorly removed by conventional methods (Lesaffer et al. 2000; Bammens et al. 2003).
Indoxyl sulfate (IS) is a representative albumin-bound uremic toxin that has gained attention because of an inability to be readily dialyzed due to high albumin affinity (Lesaffer et al. 2000) and its presumed clinical associations with chronic kidney disease (CKD) progression (Wu et al. 2011). IS is an end product of dietary tryptophan metabolism. L-tryptophan is cleaved to indole in the colon, through the action of bacterial tryptophanase enzymes, and enters the portal-venous circulation. Indole is subsequently oxidized and sulfated in the liver, forming IS, which then enters systemic circulation (Ellis et al. 2016; Vanholder and De Smet 1999). IS is taken up by organic anion transporters (OATs) at the basolateral membrane of proximal tubular cells and excreted into urine via active tubular secretion (Deguchi et al. 2002). When kidney function declines, IS accumulates in serum due to impaired clearance. The serum levels of IS in dialysis patients have been reported to be more than 50 times higher than those with normal renal function (Niwa and Ise 1994; Itoh et al. 2012).
Elevated serum IS was once thought of only as a consequence of declining renal function; however, emerging studies reveal a direct pathogenic role for IS in kidney disease. IS induces the expression of pro-fibrotic molecules such as transforming growth factor-β1 (TGF-β1) and α-smooth muscle actin (Miyazaki et al. 1997a, 1997b; Shimizu et al. 2011) and pro-inflammatory molecules such as monocyte chemotactic protein-1 (Sun, Hsu, and Wu 2013; Tan et al. 2017). In experimental models of CKD, IS contributes to tubulointerstitial fibrosis and glomerular sclerosis (Niwa and Ise 1994), activates renin-angiotensin-aldosterone (Sun, Chang, and Wu 2012), induces cell senescence (Shimizu et al. 2010), perpetuates epithelial-to-mesenchymal transition (Jurgensmeier et al. 1998; Sun, Chang, and Wu 2012), enhances reactive oxygen species (ROS), and impairs anti-oxidative systems in the kidney (Gelasco and Raymond 2006; Owada et al. 2008; Tan et al. 2017). Excessive ROS production activates nuclear factor-κB and p53, which are thought to suppress proliferation and induce senescence in proximal tubular cells (Motojima et al. 2003; Shimizu et al. 2011). Excessive IS in serum in turn impairs the basolateral membrane of renal proximal tubular cells (Taki et al. 2006), which could limit the transport capabilities of OATs and cause a feedback loop that will sustain an increased circulatory level of IS. The end result of these signaling networks is tubular atrophy and renal fibrosis.
Despite this emerging evidence, the direct toxicity, if any, of IS on human proximal tubular epithelial cells (PTECs) is not well established. Tubulointerstitial fibrosis is the result of persistent loss of renal tubular epithelial cells and an associated increase in fibroblasts and components of the extracellular matrix (Mene and Amore 1998; Zhu et al. 2016). Apoptosis not only contributes to tubular cell loss but also to renal fibrosis though the activation of pro-inflammatory and pro-fibrotic factors (Thomas et al. 1998; Yang et al. 2001; Johnson and DiPietro 2013; Mei et al. 2017). There are two well-known apoptotic pathways: extrinsic and intrinsic, both regulated by the Bcl2 family of molecules (Ola, Nawaz, and Ahsan 2011). While the extrinsic pathway is mostly initiated by death ligands, the intrinsic pathway, also known as the mitochondrial pathway, is largely initiated by various exogenous stimuli including toxins and xenobiotics (Ola, Nawaz, and Ahsan 2011). The mitochondrial pathway of apoptosis involves the release and translocation of the cytosolic proapoptotic molecule Bax to mitochondrial membranes where it induces mitochondrial depolarization, opening of the mitochondrial membrane transition pores, and eventual apoptosis (Ola, Nawaz, and Ahsan 2011). Thus, if IS is toxic to human PTEC and if the mechanism of toxicity is apoptosis, then an intrinsic pathway of apoptosis could be anticipated. Consequently, changes in mitochondrial function and integrity would be expected. To this end, IS has been shown to impair mitochondrial metabolic activity and induce oxidative stress (Mutsaers et al. 2013; Lee et al. 2015). In this study, we explored the direct toxicity of clinically plausible concentrations of IS in primary human kidney PTEC and HK-2 cells, an immortalized human PTEC line.
Materials and Method
Cell Culture
HK-2 cells were obtained from American Type Culture Collection (Rockville, MD) and grown in a humidified atmosphere of 5% CO2 in air at 37°C in DMEM/F12 with 10% fetal bovine serum (FBS) and antibiotics (50 U/mL penicillin, 50 mg/mL streptomycin). Primary cultures of human PTEC were obtained from the kidneys of consenting patients undergoing radical nephrectomy at the Princess Alexandra Hospital, Brisbane, Australia (ethics approval no. HREC/12/QPAH/125). The indication for nephrectomy was a renal mass suspicious of malignancy. Cells were derived only from patients with preserved kidney function, histologically-normal cortical parenchyma was sampled from regions as distal as possible to the tumor (Ellis et al. 2017). PTECs were prepared, plated, and grown in 5% CO2 at 37°C in serum-free defined medium (Dulbecco’s Modified Eagle Medium F-12 [DMEM/F12], supplemented with 10 μg/mL insulin, 5 μg/mL transferrin, and 5 ng/mL selenium, with antibiotics) as per our previous publication (Vesey et al. 2009).
General Protocol for Experiments
Cells were grown and maintained as mentioned above. On the day of the experiments, cells were seeded in appropriate cell culture dishes and incubated for 24 hr. The cells were washed twice in serum-free culture medium (DMEM/F12) and incubated in serum-free medium for further 24 hr. The serum-free medium was then removed from the cells, and the cells were treated with various concentration of IS in culture medium containing 1% FBS. The cells and culture media were analyzed for various parameters after 48 hr of treatment. Each major cell morphometry experiment was repeated at least in triplicate. Three separate cultures of PTECs derived from three different patients were used for all experiments involving primary cells.
MTT Assay for Metabolic Activity
The cells were seeded at a density of 5 × 103 cells per well of 96-well plates (total culture volume was 100 µL per well) and treated with various concentrations of IS. MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide; thiazolyl blue; 10 µL from a stock of 5 mg/mL in phosphate buffered saline) was added to each well and incubated for 90 min. MTT is converted to water-insoluble purple formazan crystals by mitochondrial enzymes, thereby acting as a surrogate for metabolic activity. The culture medium was removed, the purple crystals were dissolved in dimethylsulfoxide, and absorbance was determined spectrophotometrically at 570 nm with a background correction at 690 nm. Absorbance was normalized to untreated control and analyzed using a one-way analysis of variance (ANOVA) with Dunnett’s multiple comparisons test.
Assessment of Apoptosis and Hypertrophy
Cells were cultured on coverslips in 24-well culture plates at a density of 5 × 104 cell per well and treated with IS (200 or 400 μM). After 48 hr, the culture medium was removed and the cells were fixed with 4% formalin and stained with hematoxylin and eosin for microscopy as per routine methodology. To determine apoptosis morphologically, five 400× fields of view per coverslip were randomly selected. Total cell number and the number of apoptotic cells were determined using morphological criteria: (i) shrunken eosinophilic cells with condensed, marginated nuclear chromatin and intact cell membrane; (ii) discrete apoptotic bodies compromising large, dense, pyknotic nuclear fragments usually surrounded by a narrow eosinophilic cytoplasmic rim; and (iii) clusters of small apoptotic bodies (assessed as a single apoptotic occurrence). Cells with nuclear features of apoptosis not showing the characteristics of secondary necrosis (swollen cytoplasm) known to occur in cell culture were also counted as apoptosis (Gobe 2009). Counts were collated for each coverslip; percentage apoptosis was analyzed using a one-way ANOVA with Dunnett’s multiple comparisons test. Analysis of cell and nuclear size was undertaken using NIS Elements Software version 2.0 (Nikon Instruments, Melville, NY). Images were captured using a Nikon Eclipse 50i bright field microscope and DSFi1 color camera (Nikon Australia, Sydney) at 400× magnification. Cells from five representative fields of view per coverslip were assessed. Investigators were blinded to treatment group allocation to reduce selection bias. Cell and nuclear size were analyses using a repeated measures ANOVA.
Immunofluorescence
HK-2 cells were cultured as above, and coverslips were fixed in 100% methanol. Immunofluorescence staining for β-galactosidase was undertaken per standard protocols (primary antibody and Alexa-Fluor® 488 green fluorescent tag supplied by Invitrogen). Negative controls were prepared without the antibody.
Gene Expression Studies
Cells grown to approximately 90% confluence in 6-well plates were treated with IS. Forty-eight hours later, total RNA was isolated using PureLink RNA Mini Kit (Life technologies, Carlsbad, CA) as per the manufacturer’s protocol and quantified with Nano drop (Thermoscientific, Waltham, MA). cDNA was synthesized using a high capacity cDNA Reverse Transcription Kit (Life Technologies) as described before (Thakor et al. 2017). In brief, 1 µg of RNA in an RT-PCR reaction mixture (2 µL 10× buffer, 0.8 µL dNTP, 2 µL r-hex, 1 µL enzyme, water to 20 µL per reaction) was subjected to the following PCR conditions: 25°C 10 min, 37°C 60 min, 37°C 60 min, and 80°C for 5 min followed by holding at 4°C. The cDNA was diluted to 50 µL with 30 µL of RNAse-free water. Fully validated TaqMan Gene Expression Assay (Life Technologies) for transforming growth factor beta (TGF-β; Hs004276620_m1), Bax (Hs00180269_m1), Bcl2 (Hs00236808_m1), interleukin-6 (IL-6; Hs00985639_m1), interleukin-8 (IL-8; Hs00174103_m1), tumor necrosis factor α (TNF-α; Hs01113624_g1), and GLB1 (Hs01035168_m1) was used with the SensiFAST™ Probe No-ROX Kit (Bioline, London, UK) in a LightCycler 480 (Roche Applied Science, Penzberg, Germany) to determine relative gene expression by the comparative Ct method. The TATA box binding protein (Hs00427620_m1; Life Technologies) was used as an internal control and for normalization.
Western Blotting of Culture Medium to Measure Fibronectin Release
In brief, cells grown in 6-well plates were treated with IS, and at 48 hr of treatment, the culture medium was removed and centrifuged at 13,000 rpm at 4°C for 15 min to remove any cell debris. The proteins (20 µg) were resolved in 12% BIS-TRIS gels and electrotransferred into polyvinylidene fluoride membranes (Millipore Corporation, Burlington, MA). Culture medium (15 μL) was mixed with sample buffer and reducing agents and added to the gel. Equal loading of proteins was confirmed by staining the membrane with Ponceau S solution. Standard Western blotting procedures were followed, and the signals were detected by Super Signal West Pico Chemiluminescent Substrate (Pierce, Rockford, IL). The differences in intensities of the signals were analyzed by ImageJ 1.48v software (National Institutes of Health, Bethesda, MD).
Statistical Analyses
Statistical analysis was undertaken using Stata 14 (StataCorp, College Station, TX) and GraphPad Prism 7 (GraphPad Software, La Jolla, CA). Two-sided α was set at .05.
Results
IS Decreased the Metabolic Activity of HK-2 and PTEC
To establish an ideal concentration of IS for the experiments, HK-2 cells were treated with various concentrations of IS (0, 50, 100, 150, 200, 400, and 800 µM). After 48 hr of incubation, IS, up to a concentration of 150 µM, did not induce a statistically significant change in the metabolic activity. Observation of the culture plates under the microscope after 24 hr of incubation showed that IS above 400 µM precipitated in the culture medium and crystals of IS could be observed on top of the cells. Therefore, for further experiments, 200 and 400 µM of IS were selected. Both concentrations induced a statistically significant decrease in metabolic activity of HK-2 (Figure 1A) and PTEC (Figure 1B) after 48 hr of incubation. To study the effect of albumin concentration in the culture medium, IS (200 µM) was dissolved in culture medium containing various concentrations of FBS (0%, 1%, and 10%). After 48 hr, when compared with the untreated control (HK-2 cells), IS induced a statistically significant decline in metabolic activity in all three groups; however, no differences among the FBS groups treated with IS were observed (Figure 1C). Similar results were observed when FBS was substituted with albumin (data not shown). These findings show that the concentration of albumin has no effect on metabolic derangements induced by IS.
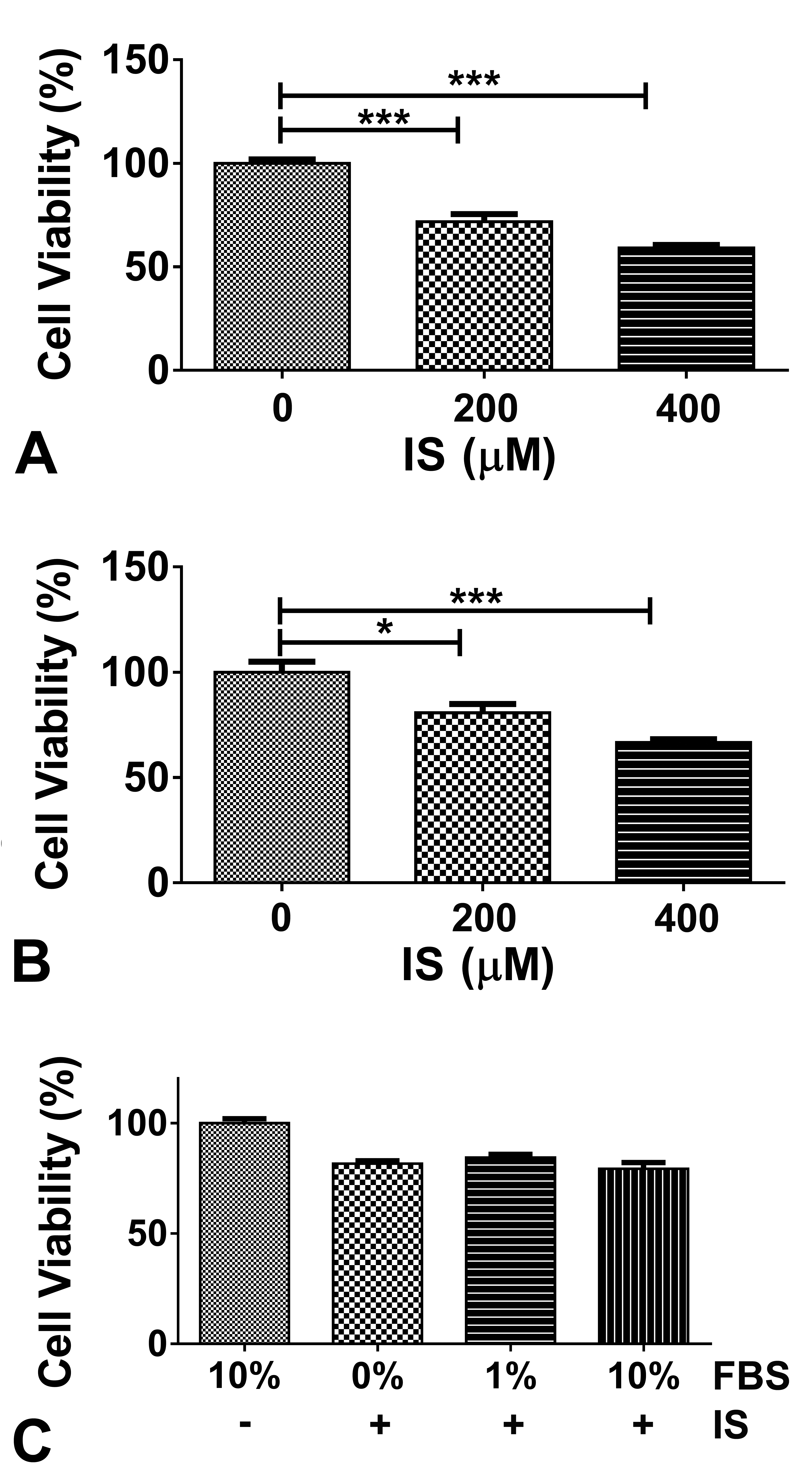
Indoxyl sulfate (IS) induced a decrease in metabolic activity. Primary proximal tubule cells (PTEC) and HK-2 cells were incubated with IS for 48 hr. An MTT assay was performed as a measure of metabolic activity. IS induced a statistically significant reduction in metabolic activity of HK-2 (A) and PTEC (B). To study the effect of albumin, IS (200 µM) was dissolved in various concentrations of fetal bovine serum and HK-2 cells were treated for 48 hr. MTT assay showed that the concentration of albumin had no effect on IS-induced changes in metabolic activity (C). FBS = fetal bovine serum. *p < .05. ***p < .001.
IS Induced Apoptosis in HK-2 Cells and PTEC
To study the mode of the death, apoptosis was evaluated morphologically (Gobe 2009). Other modes of cell death (e.g., necrosis) were not observed. IS induced a statistically significant increase in apoptosis of both HK-2 cells (Figure 2A) and PTEC (Figure 2B). To further confirm these morphological findings at a molecular level, the expression of apoptosis-regulatory molecules was studied by qPCR. The expression of the antiapoptotic molecule Bcl2 was not significantly altered in HK-2 (Figure 2C) or PTEC (Figure 2D) by incubation with IS. However, there was a dose-dependent increase in the expression of the proapoptotic molecule Bax in HK-2 (Figure 2E) and PTEC (Figure 2F) exposed to IS.
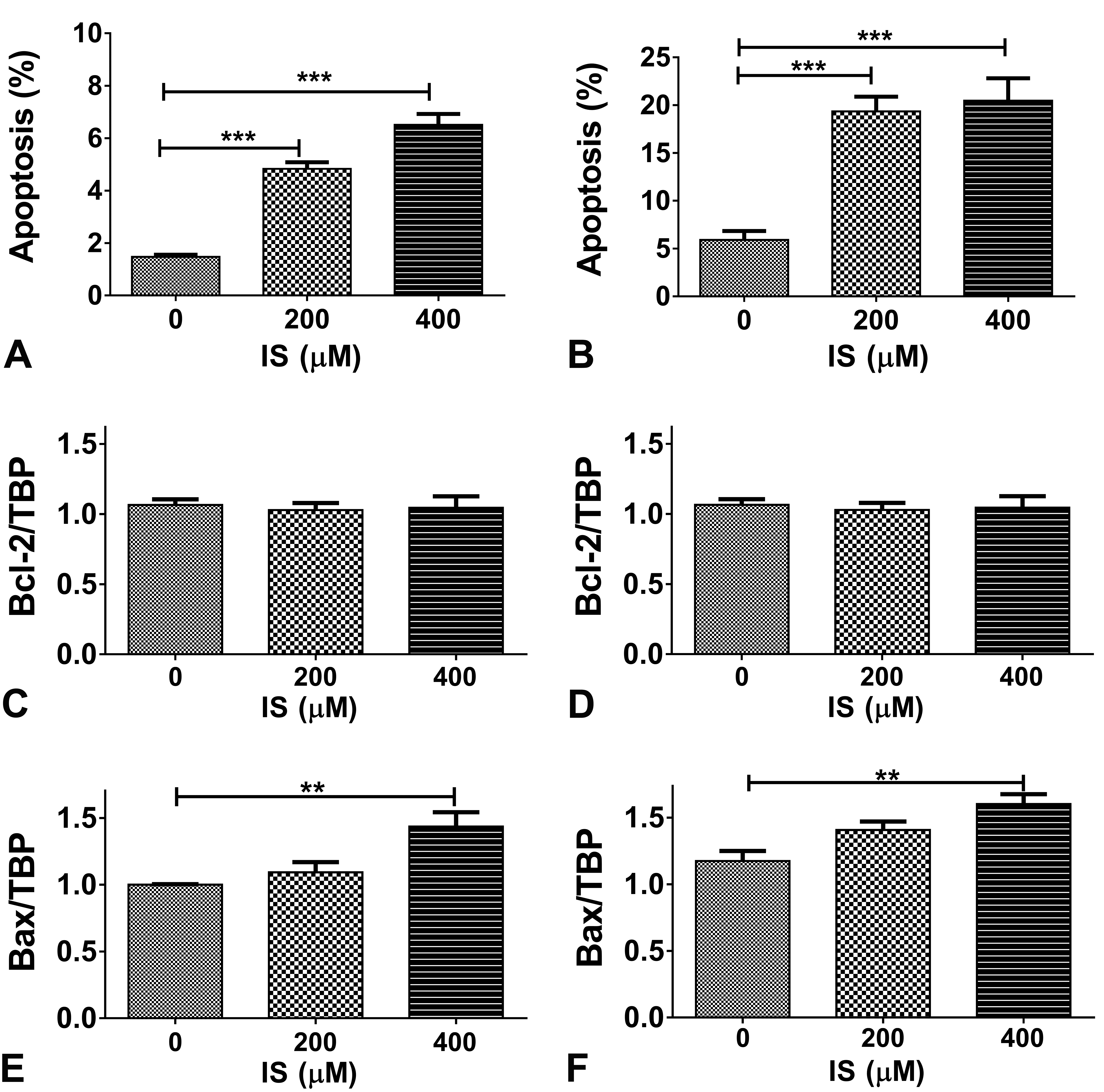
Indoxyl sulfate (IS) induced apoptosis in renal tubular cells. Morphological assessment of cells after 48 hr of incubation with IS showed notable apoptosis in HK-2 (A) and primary cells (PTEC; B). Gene expression studies by qPCR showed no changes in the antiapoptotic molecule Bcl2 (C and D), whereas the proapoptotic molecule Bax was significantly increased in HK-2 (E) and PTEC (F). **p < .01. ***p < .001. PTEC = primary proximal tubule cells.
IS Induced Hypertrophy of Viable Cells
After identifying apoptosis as a mechanism of cell loss in response to IS, the fate of the remaining viable cells was analyzed. IS induced notable hypertrophy in both cell types. Morphometry showed that, in both HK-2 cells and PTEC, IS induced a statistically significant increase in cell and nuclear size (Figures 3 and 4). To distinguish between hypertrophy and senescence, immunofluorescence was performed for senescence-associated β-galactosidase; no differences in expression were detected between in HK2 cells treated with 200 μM IS and controls (Online Supplementary Figure 1). To confirm this finding, GLB1 was assessed using qPCR in HK2 cells and PTEC; no differences were observed between controls and groups treated with 200 μM IS (Online Supplementary Figure 2).
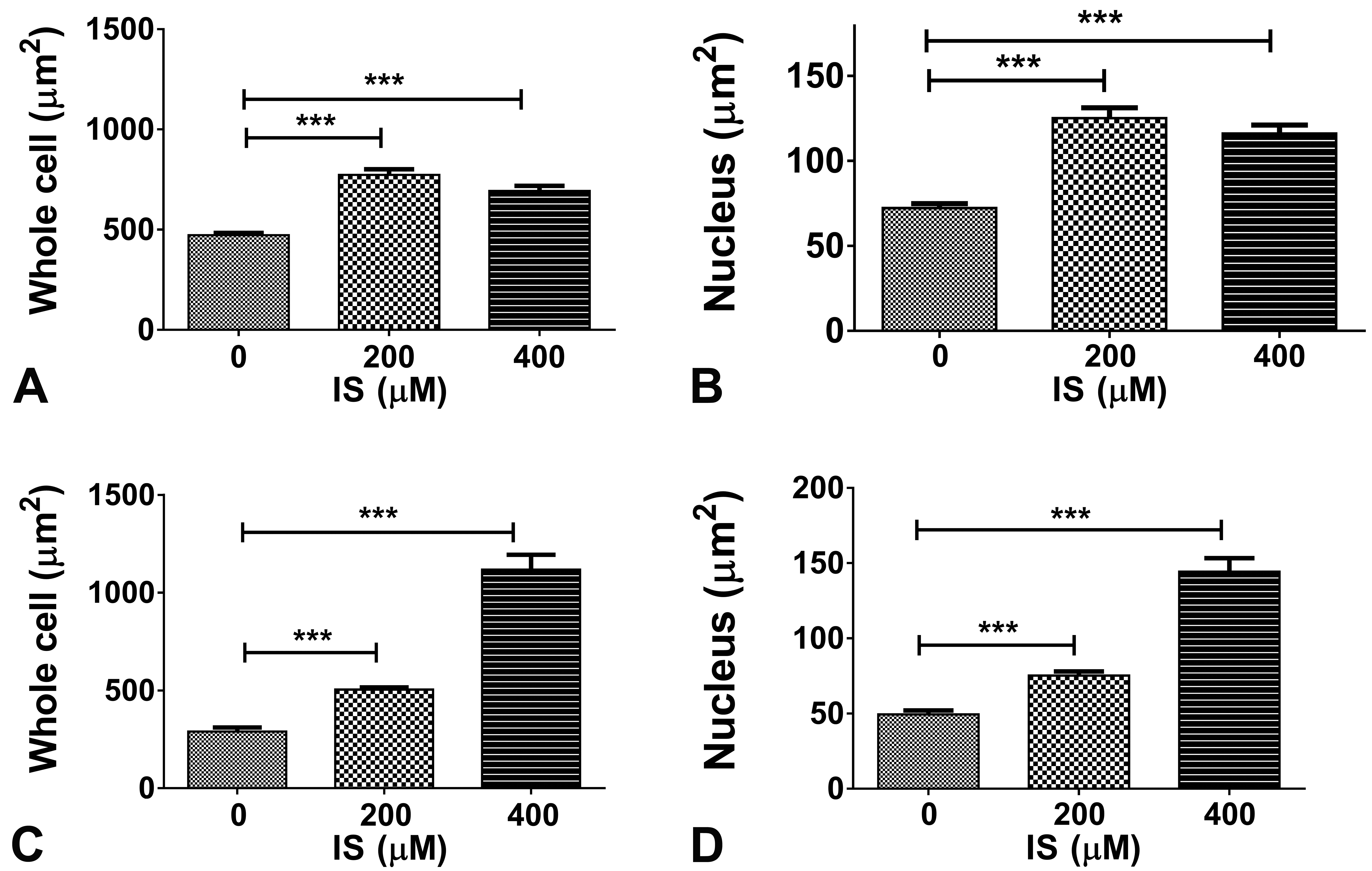
Indoxyl sulfate (IS) induced hypertrophy. After 48 hr of incubation with IS, the cells were stained with hematoxylin and eosin, and differences in cell size were measured using NIS Elements Software. There was statistically significant increase in both cell (A) and nuclear (B) size in HK-2 cells, and in cell (C) and nuclear (D) size in primary proximal tubule epithelial cells. Experiments were repeated three times. ***p < .001.
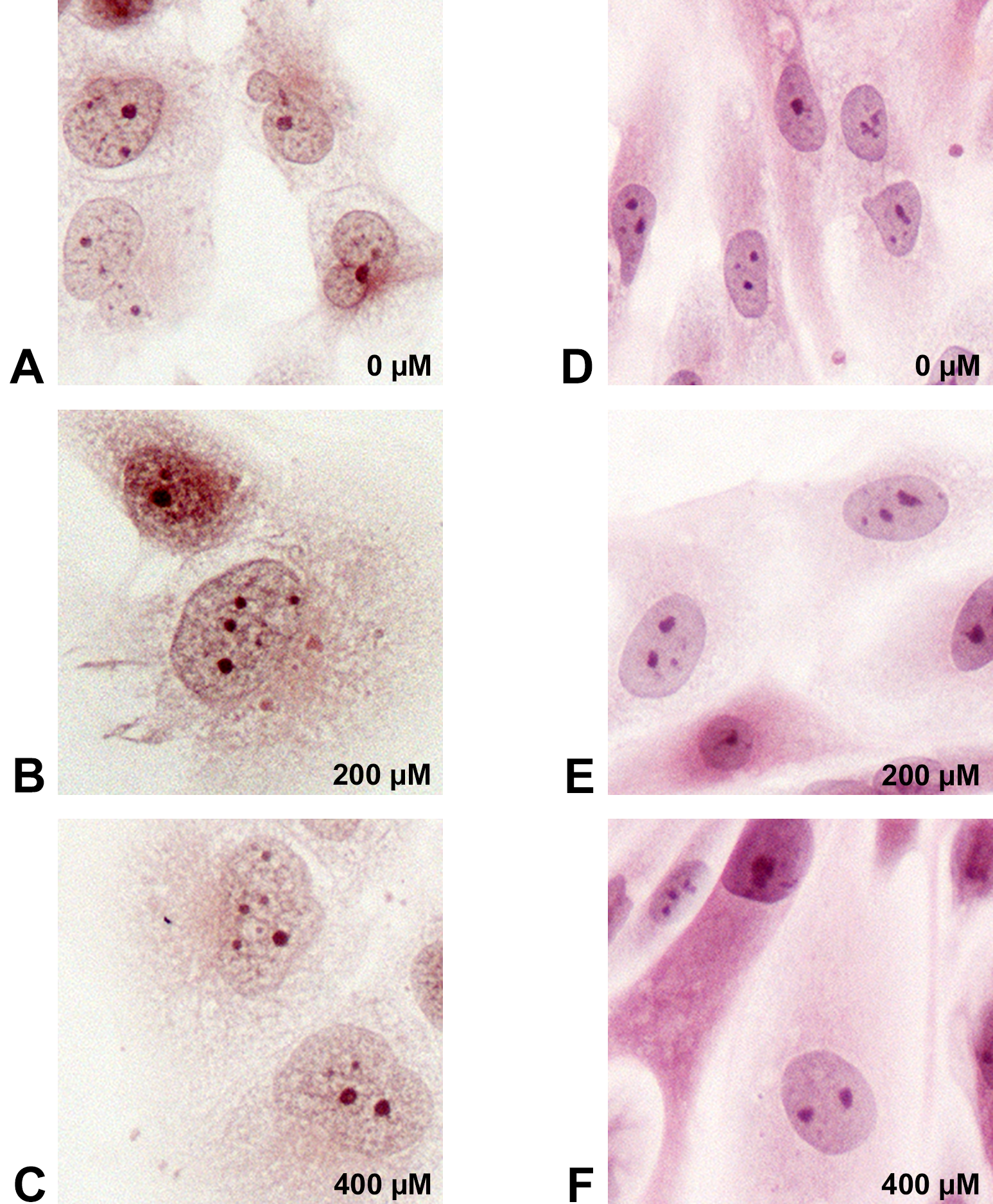
Micrographs demonstrating hypertrophy following indoxyl sulphate (IS) exposure. After 48 hr of incubation with IS at 0, 200, or 400 µM, the cells were stained with hematoxylin and eosin. Example micrographs are shown for HK-2 cells (A–C) and primary proximal tubule epithelial cells (D–F).
IS Induced an Increase in Pro-fibrotic and Pro-inflammatory Molecules
IS induced a statistically significant increase in the expression of the pro-fibrotic molecule TGF-β in HK-2 (Figure 5A) and PTEC (Figure 5B). The level of secreted fibronectin was also significantly increased in response to IS in PTEC (Figure 5C). Furthermore, IS also increased the expression of the pro-inflammatory molecules IL-6 (Figure 6A and B), IL-8 (Figure 6D and E), and TNF-α (Figure 6E and F) in both cell types (left panel HK-2 and right panel PTEC).
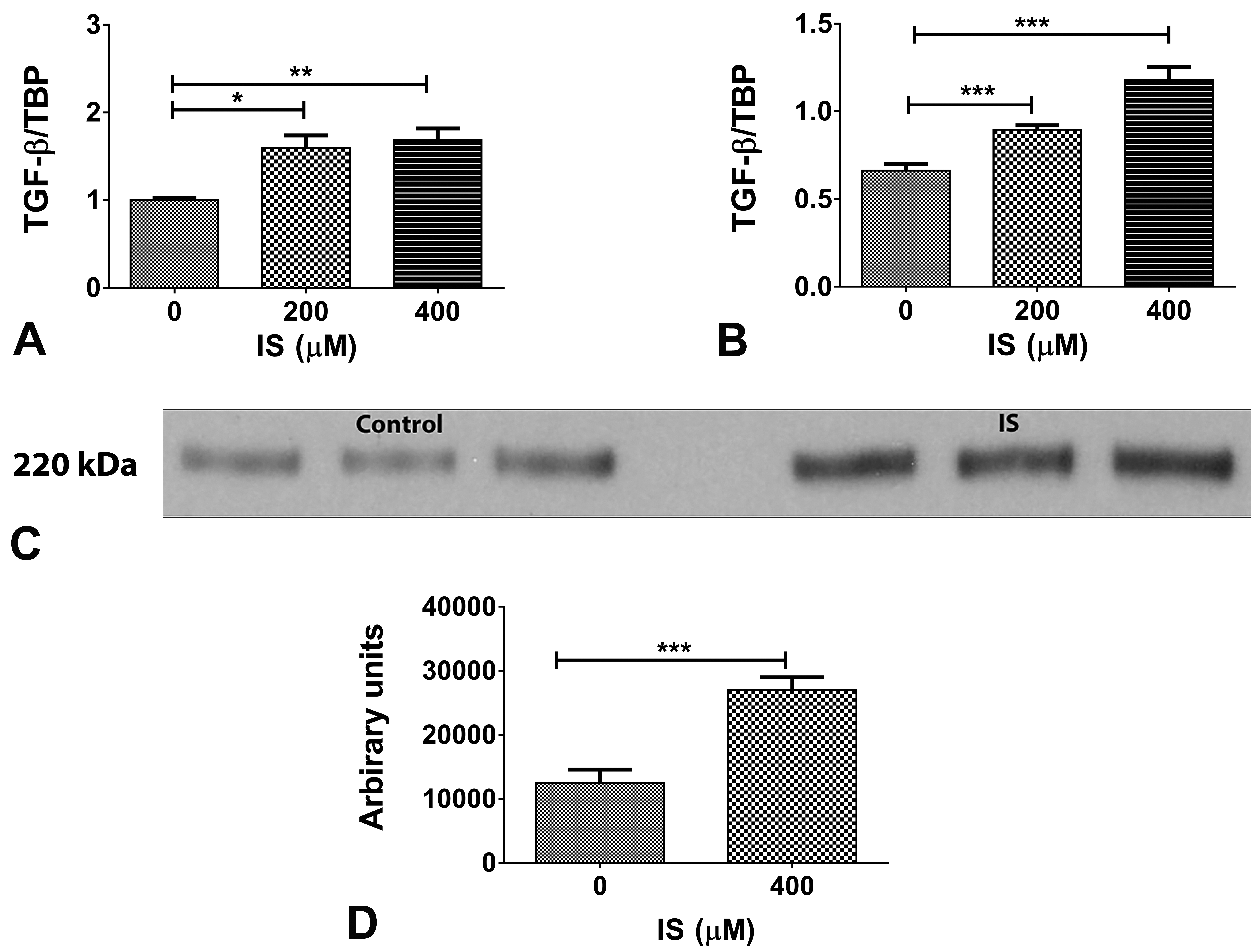
Indoxyl sulfate (IS) induced the expression and release of pro-fibrotic factors. qPCR showed that IS induced a statistically significant increase in the expression of the pro-fibrotic molecule TGF-β in HK-2 (A) and PTEC (B). IS induced statistically significant increases in fibronectin secretion by PTECs. Cells were treated for 48 hr with 400 µM IS (C and D). *p < .05. **p < .01. ***p < .001. TGF-β = transforming growth factor-β. PTEC = primary proximal tubule cells.
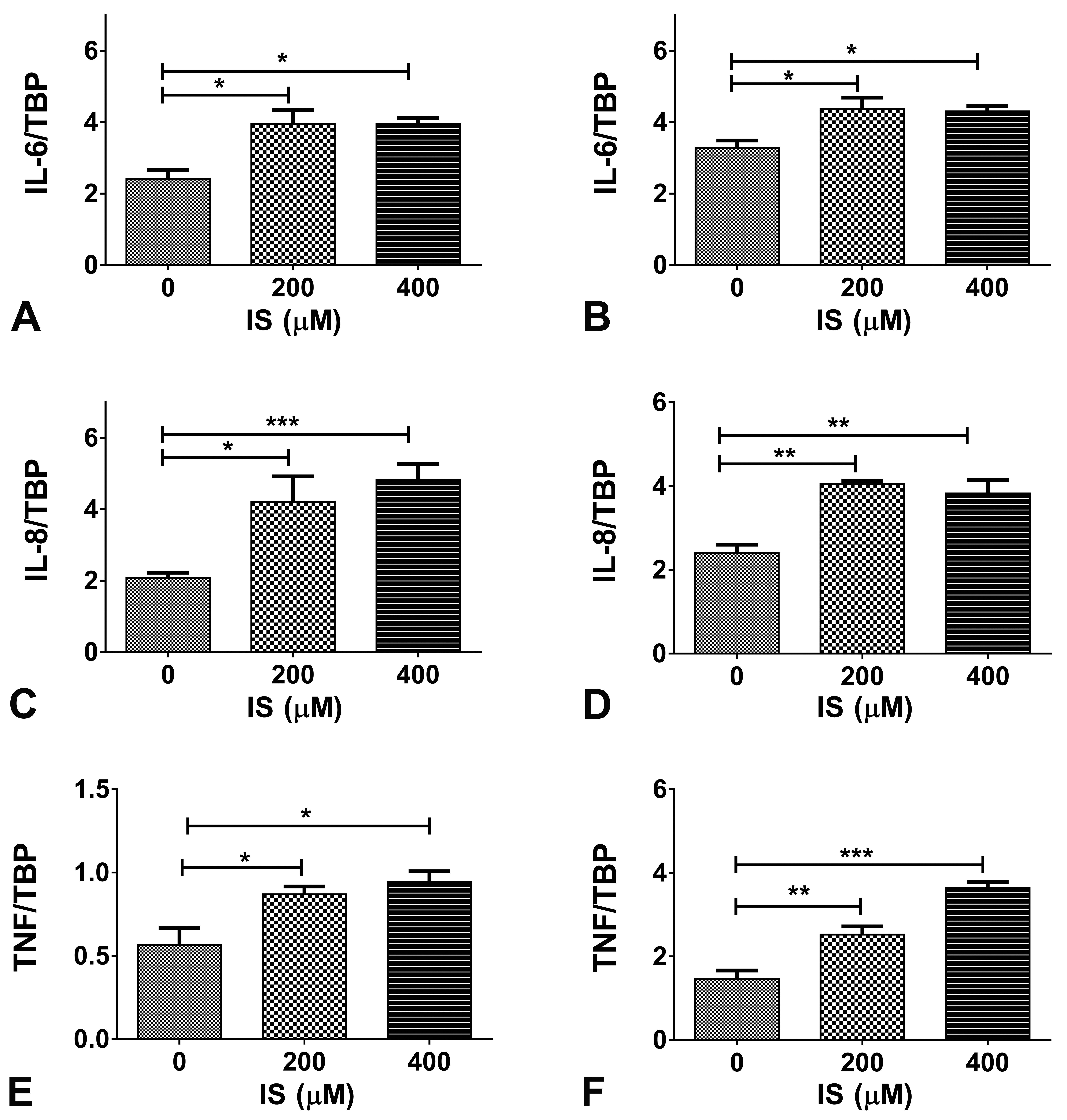
Indoxyl sulfate induced the expression of pro-inflammatory molecules. Gene expression studies showed statistically significant increases in the expression of pro-inflammatory molecules interleukin-6, interleukin-8, and tumor necrosis factor in HK-2 (A, C, and E) and primary proximal tubule cells (B, D, and E). *p < .05. **p < .01. ***p < .001.
Discussion
The two most common criticisms of in vitro studies of IS are the use of concentrations that are not representative of human pathophysiology and the lack of adequate albumin concentrations in the culture media (Vanholder et al. 2003; Hammarlund-Udenaes 2010; Duranton et al. 2012). First, we sought to address these two issues. Pertinent to concentration of IS, the total circulatory concentration of IS in uremic patients continues to be a matter of debate. In a 2003 review, Vanholder et al. based on the findings of Niwa and Ise (1994) reported a physiologic serum IS concentration of 0.6 ± 5.4 mg/L and a mean value of 53.0 ± 91.5 mg/L (with the highest ever reported value as 236 mg/L). Subsequent updates in 2012 and 2014 report physiologic serum IS concentrations of 0.53 ± 0.29 mg/L and a highest mean uremic value of 44.5 ± 15.3 mg/L (Ujhelyi et al. 2006; Lee et al. 2010; Duranton et al. 2012; Vanholder et al. 2014). It should be pointed out that the updated values are based on patients who have been on peritoneal dialysis for at least six months prior to the measurements (Lee et al. 2010). In CKD patients with preserved residual renal function, serum IS values are expected to be considerably lower (Ellis et al. 2016). Nonetheless, based on these values, the concentration of 200 and 400 µM used in the present study equates to a clinically plausible concentration of 42.6 and 85.2 mg/L, respectively. Regarding albumin concentration in the culture media, the circulatory level of plasma albumin in humans ranges from 35 to 50 mg/mL. Adding such a high concentration of albumin per milliliter of culture medium is toxic to human proximal tubules (Erkan, Devarajan, and Schwartz 2007). The typical concentration of albumin in FBS is 20 to 36 mg/mL, which, at 10%, equates to 2.0 to 3.6 mg/mL. In our experiments, the final concentration of albumin, FBS at 1%, was between 0.20 and 0.36 mg/mL. Our results indicate that the toxicity of IS is similar in the presence of 0%, 1%, or 10% FBS or albumin. Thus, at least in vitro, neither albumin nor FBS appears to influence the toxicity of IS in terms of cell viability measured by MTT. Therefore, for further studies, we used the lower limit of albumin. Under these experimental conditions, IS induced apoptosis, hypertrophy, mitochondrial dysfunction, and various pro-fibrotic and inflammatory molecules.
To the best of our knowledge, this is the first study that shows apoptosis and hypertrophy in response to IS in human primary and immortalized PTECs, although surface-membrane annexin V-positive cells, considered a marker of early apoptosis, in response to IS has been reported in rat renal tubular epithelial cells (NRK-52E cells; Kim et al. 2012). The hypertrophy-inducing effect of IS has been reported in cardiomyocytes (Yang et al. 2015). At the molecular level, there was no changes in the antiapoptotic molecule Bcl2, but the proapoptotic molecule Bax was significantly increased in response to IS. Bax, a cytosolic resident of the proapoptotic Bcl2-family proteins (Oltvai, Milliman, and Korsmeyer 1993; Yin et al. 1994), translocates to mitochondria upon induction of apoptosis (Wolter et al. 1997), where it induces mitochondrial depolarization and disrupts mitochondrial inner transmembrane potential (ΔΨm; Pastorino et al. 1998), through cytochrome c release and caspase activation (Jurgensmeier et al. 1998; Rosse et al. 1998), eventually leading to apoptosis (Zamzami et al. 1995a, 1995b; Zoratti and Szabo 1995; Marchetti et al. 1996; Finucane et al. 1999). The conversion of MTT to purple formazan crystals is mainly the function of mitochondrial succinate dehydrogenase (Wang et al. 1996). Thus, it is reasonable to conclude that an increase in Bax, coupled with a decreased mitochondrial function, is the major mechanism of apoptosis in response to IS in human PTEC.
In addition to apoptosis, IS induced hypertrophy of PTEC. Hypertrophy of the PTEC could be an adaptive mechanism to compensate for tubular cell loss via apoptosis. Such an adaptive hypertrophy, when sustained long enough, could lead to maladaptive hypertrophy and eventual cell death. Compensatory renal hypertrophy increases energy demand per nephron, leaving the cells in a hypermetabolic state with an increased mitochondrial electron transport rate and mitochondrial depolarization that could eventually lead to the opening of the mitochondrial transition pore, mitochondrial damage (Harris, Chan, and Schrier 1988; Hwang et al. 1990; Nath, Croatt, and Hostetter 1990; Shapiro et al. 1994; Benipal and Lash 2011, 2013), and subsequent cell death, possibly via apoptosis. IS-induced mitochondrial dysfunction has been reported in skeletal muscle cells (Nishikawa et al. 2015; Enoki et al. 2017), endothelial cells (Lee et al. 2015), and to a lesser extent in conditionally immortalized renal proximal tubule epithelial cells (Mutsaers et al. 2013). Thus, we propose that IS is a possible mitochondrial toxin for human PTEC.
It is well accepted that, irrespective of the nature of the initial insult, interstitial fibrosis is the final common outcome of CKD, which is characterized by the accumulation of extracellular matrix protein, largely mediated by inflammation and interstitial myofibroblast activation (Wynn and Ramalingam 2012). While the role of apoptosis in cell loss is well established, it is emerging that apoptotic cells per se could contribute to fibrosis either by directly activating pro-fibrotic TGF-β1 or by eliciting inflammatory mediators such as TNF-α and IL-6 (Johnson and DiPietro 2013; Mei et al. 2017). The current in vitro findings demonstrate a direct pro-inflammatory and pro-fibrotic effect of IS on PTEC, as evidenced by increased expression of TGF-β, fibronectin, IL-6, and TNF-α. Irrespective of the effect being direct or indirect, the findings are clinically relevant given that, in a cross-sectional observational cohort study, the serum levels of IS in patients with stages 3 and 4 CKD correlated with levels of IL-6 and TNF-α (Rossi et al. 2014).
In conclusion, using pathologically relevant concentrations of IS, we show evidence for the direct toxicity of IS in human PTEC. The combined action of apoptosis, hypertrophy, mitochondrial dysfunction, inflammation, and fibrosis could contribute to fibrosis or hasten CKD progression. Of particular interest is the null effect of albumin concentration in culture medium on PTEC. The current consensus is that free IS, which is approximately 10% of the albumin-bound portion of IS, is the culprit in inducing any pathological changes. This notion needs to be revisited. If the albumin-bound fraction of IS is found to be toxic, then the therapeutic potential of agents that restore mitochondrial function requires intense research because removal of albumin-bound IS by dialysis continues to be a clinical challenge.
Supplemental Material
Supplemental Material, DS1_TPX_10.1177_0192623318768171 - Indoxyl Sulfate Induces Apoptosis and Hypertrophy in Human Kidney Proximal Tubular Cells
Supplemental Material, DS1_TPX_10.1177_0192623318768171 for Indoxyl Sulfate Induces Apoptosis and Hypertrophy in Human Kidney Proximal Tubular Cells by Robert J. Ellis, David M. Small, Keng Lim Ng, David A. Vesey, Luis Vitetta, Ross S. Francis, Glenda C. Gobe, and Christudas Morais in Toxicologic Pathology
Supplemental Material
Supplemental Material, DS2_TPX_10.1177_0192623318768171 - Indoxyl Sulfate Induces Apoptosis and Hypertrophy in Human Kidney Proximal Tubular Cells
Supplemental Material, DS2_TPX_10.1177_0192623318768171 for Indoxyl Sulfate Induces Apoptosis and Hypertrophy in Human Kidney Proximal Tubular Cells by Robert J. Ellis, David M. Small, Keng Lim Ng, David A. Vesey, Luis Vitetta, Ross S. Francis, Glenda C. Gobe, and Christudas Morais in Toxicologic Pathology
Footnotes
Authors’ Note
Luis Vitetta is an employee of Medlab Clinical, a biotech company which has interest in chronic kidney disease research, relevant to the administration of probiotics as an adjuvant medicine.
Author Contribution
All authors (RE, DS, KN, DV, LV, RF, GG, CM) contributed to conception or design; data acquisition, analysis, or interpretation; drafting the manuscript; and critically revising the manuscript. All authors gave final approval and agreed to be accountable for all aspects of work in ensuring that questions relating to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Robert Ellis was supported by an Australian Government Research Training Stipend while undertaking a portion of this work.
Supplemental Material
Supplementary material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.