
Abstract
Critical limb ischemia (CLI) represents the severest manifestation of peripheral arterial disease and is a major unmet medical need. This disease occurs when the arterial blood supply within the limb fails to meet the metabolic demands of the resting muscle or tissue, resulting in chronic ischemic rest pain and/or tissue necrosis. Human mesenchymal stromal cells, termed hMSCs, represent an exciting therapeutic modality for the treatment of this disease due to their immunomodulatory and tissue reparative functions. The aim of the study was to assess the preclinical toxicity profile of human bone marrow–derived MSCs in support of their use as a treatment for CLI. A 3-month toxicity study was carried out under good laboratory practices in immunodeficient mice who received, intramuscularly, a single dose of 3 × 105 (approximately 15 × 106 cells/kg) hMSCs manufactured under good manufacturing practices. No significant changes in body weight, food consumption, clinical signs, or histopathological changes were observed in the hMSC-treated mice in comparison to the controls. These results highlight that the administration of hMSCs during the 3-month study period was well tolerated and not associated with any test item–related tumors. This data set supported the initiation of a phase 1b first in human study in “no option” for revascularization patients with CLI.
Atherosclerotic peripheral arterial disease (PAD) represents one of the most prevalent noncommunicable diseases in the world today. Critical limb ischemia (CLI), the most advanced form of PAD, is associated with high morbidity and mortality. CLI does not symbolize a single pathophysiologic process but is caused by a multitude of different pathogenic mechanisms such as atherosclerosis, arteritides, hypercoagulable states, cardioembolisms, and lower limb graft bypass failure (Adam et al. 2005; Gupta and Losordo 2011). CLI is a disease that presents in patients with great heterogeneity but ultimately occurs when the arterial blood supply within the limb fails to meet the metabolic demands of the resting muscle or tissue, resulting in chronic ischemic rest pain and/or tissue necrosis, often leading to amputation (Gupta and Losordo 2011). Furthermore, CLI is associated with an alarmingly high fatality rate as a result of ischemic cardiovascular events with higher mortality rates similar to, if not worse than, other serious medical conditions (Armstrong, Wrobel, and Robbins 2007). With over 200 million estimated to be suffering from PAD and with reports documenting an increased prevalence of CLI, it is almost a certainty that this disease burden will continue to increase in the absence of preventative measures (Fowkes et al. 2013; Hirsch & Duval 2013)—impacting negatively on both global health and the economy.
Mesenchymal stromal cell (MSC) is the most widely studied cell therapy (CT) and the closest to becoming clinically translated to CLI patients. Although the full mechanism of action of MSC therapy remains unclear, the current view is that MSCs act as growth factor/cytokine factories that produce bioactive molecules that largely contribute to immunomodulatory and anti-inflammatory functions while also providing trophic support through the construction of a regenerative microenvironment (Caplan 2008). By inhibiting T-cell function via affecting antigen presentation and T-cell progenitor expansion, MSCs help protect the injury site from immune surveillance, therefore preventing autoimmune sensitization to the injured tissue (Caplan 2008). In conjunction with their immunomodulatory functions, the activated MSC can inhibit apoptosis in ischemic tissues and stimulate the mitosis of tissue resident progenitors whose progeny is to reconstruct the area of tissue damage (Caplan 2008). MSCs have also been reported to exert antimicrobial effects (Caplan and Correa 2011). In addition to their immunomodulatory effects and trophic activities, there is evidence for endothelial and myogenic differentiation of MSCs (Oswald et al. 2004; Gailli, Vitale, and Vaccarezz 2014). Taking into consideration all of the above, MSCs represent an attractive therapeutic option for patients with CLI.
Collateral blood vessel development remains the therapeutic and prognostic determinant in CLI patients. The damage in the limbs due to ischemia in CLI patients is complex, and tissue repair will require multiple different mechanisms. Thus far, the use of single agents such as the delivery of growth factors and genes has failed to demonstrate efficacy in pivotal human trials (Kitrou et al. 2017). To address this unmet medical need, investigators have explored the administration of MSCs via multiple different routes, and the preclinical data has been very promising. Several groups have shown that MSCs can improve perfusion and augment recovery in mice with hind limb ischemia (Kinnaird et al. 2004; Gremmels et al. 2014; Smadja et al. 2012; Leroux et al. 2010; Moon et al., 2006). Bortolotti et al. (2015) compared the therapeutic properties of three MSC populations and showed that each MSC type provided therapeutic benefit after hind limb ischemia. However, they showed that MSCs derived from the bone marrow were the most effective at restoring tissue perfusion. The superior therapeutic efficacy of bone marrow–derived MSCs in comparison to the other MSC types was associated with increased cytokine/growth factor production such as TGF-β, PDGF-β, MMP9, longer engraftment, and increased smooth muscle migration (Bortolotti et al. 2015). Several other groups including our own have shown that MSCs secrete large amounts of angiogenic cytokines and growth factors that provide trophic support for neoendothelium, aid in endothelial precursor cell recruitment and extracellular matrix remodeling, and promote neovascularization by augmenting angiogenesis and arteriogenesis (Kwon et al. 2014; Smadja et al. 2012; Hung et al. 2007; Brewster et al. 2017; Gremmels et al. 2014).
The complex nature of CT products requires great effort in order to ensure the successful translation from bench to bedside. Every step in the development of a CT product requires complete scientific rigor from manufacturing processes to preclinical testing to clinical administration (O’Brien et al. 2015). While establishing efficacy is critical to CT product development, the successful implementation of these therapies will also heavily rely on resolving potential safety concerns both preclinically and clinically.
Clinical testing using CT products in Europe is regulated under the Clinical Trial Directive and requires national clinical trial applications. We have been developing intramuscular (IM) delivery of human bone marrow–derived MSCs as an experimental treatment for CLI and as part of our investigative medicinal product (IMP) application process we met with the national competent regulatory authority.
It is suggested that the safety of a CT product is based on the product’s attributes. Due to the heterogeneity of CT products in terms of manufacturing process, cell type used, and mechanism of action, safety evaluations are carried out on “case-by-case” basis (US Food and Drug Administration [FDA] 2013; European Medicines Agency [EMA] 2008, 2011, 2013). Based on regulatory guidelines, it is recommended that preclinical toxicity and biodistribution assessments using the intended good manufacturing practices (GMP) grade CT product are carried out under good laboratory practices (GLP) prior to submitting an application for clinical testing (US FDA 2013; EMA 2008). It therefore was evident to us that despite the preclinical and clinical data demonstrating safety of human MSCs (hMSCs; Gupta et al. 2017; Ramot et al. 2009; Guess et al. 2017; Creane et al. 2017; Yun et al. 2016; Schmuck et al. 2016; Rengasamy et al. 2016; Bura et al. 2014; Braun et al. 2016; Chen et al. 2015; Wang et al. 2012; Macisaac et al. 2012) that preclinical toxicity and biodistribution studies would be required to assess the safety of our GMP CT product. It was also clear that the product should be administered in a similar manner to the proposed clinical route, which in our case was IM.
Herein we present the results of our preclinical toxicity study that was submitted to the Health Products Regulatory Authority (HPRA) in Ireland as part of an IMP application to support the clinical testing of our hMSC product in “no option” for revascularization CLI patients. This study assessed key safety concerns surrounding local or systemic toxicities of GMP grade MSCs, extrapolated to represent a dose 15 times greater than the proposed clinical dose and administered via the intended clinical route.
Method
Manufacturing Process of the Bone Marrow–derived MSCs
HMSCs were isolated from adult bone marrow and cultured expanded in accordance with local ethical approval and GMP regulations. Upon receipt, the bone marrow aspirate was washed with Dulbecco’s Phosphate Buffered Saline (DPBS) and centrifuged at 900 g. A 4% acetic acid wash was performed on a sample of the marrow to lyse the red blood cells and enable an accurate mononuclear cell (MNC) count. MNCs plated at 40 to 50 million per 175 cm2 were cultured with complete medium (αMEM supplemented with 10% selected fetal bovine serum) in 5% carbon dioxide (CO2) at 37°C. On day 3, a media top up was performed. On day 5, the cultures were washed with DPBS to remove nonadherent cells, and fresh complete medium was added to each flask. When colonies were large and nonoverlapping, the adherent cells were washed with DPBS and detached from the culture plastic with 0.25% trypsin/ethylenediaminetetraacetic acid (EDTA). The dissociated cells were centrifuged at 400 g for 5 min. The resultant pellet was resuspended in complete fresh medium and the cellular yield determined. hMSCs were further subcultured in triple flasks through two passages (Figure 1).
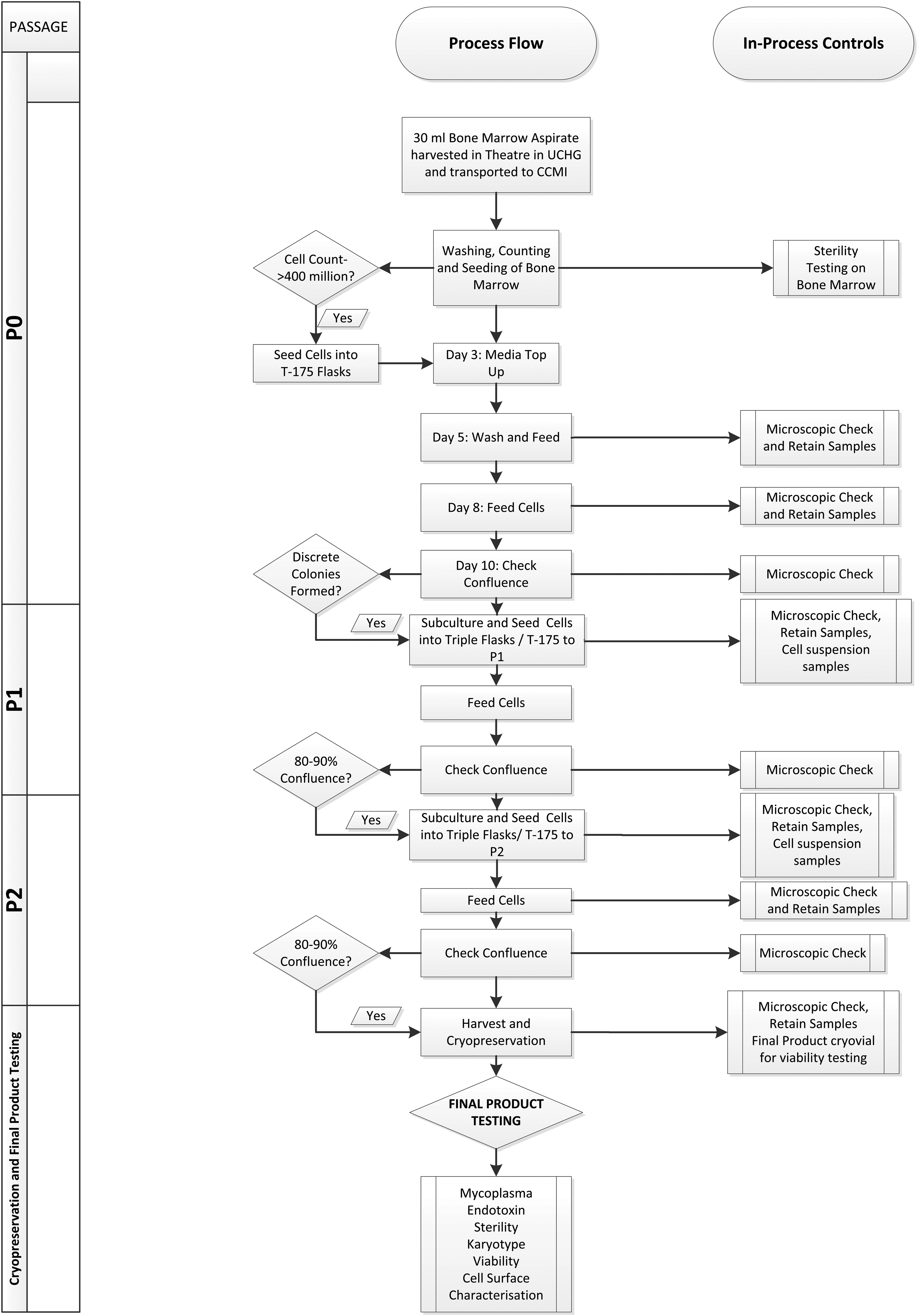
Process flow for the GMP manufacturing of hMSCs for the 3-month toxicity study. GMP = good manufacturing practice; hMSCs = human mesenchymal stromal cells.
The cells were harvested and suspended in a final formulation of 10% dimethyl sulfoxide and 4.5% human serum albumin. The hMSCs were filled into 1.8 ml cryovials at a final dosage of 2 × 106 cells/ml and cryopreserved at −150°C storage following controlled rate freezing at −1°C/min from room temperature to −80°C.
Following cryopreservation, the hMSCs product was tested for mycoplasma, endotoxin, sterility, and viability. The cells also underwent karyology and immunophenotyping.
For immunophenotype characterization, the cells were stained with antibodies for MSC-positive markers CD73, CD90, and CD105 and for negative markers CD45, CD34, CD11b, CD19, and HLA-DR. The immune phenotype test specifications were set as ≥90% for all positive markers and ≤5% for all negative markers.
Upon meeting the release criteria, the hMSCs were then shipped to Charles River Laboratories Preclinical Services, Tranent (PCS-EDI), Edinburgh, EH33 2NE, UK, on dry ice where they were received and stored in liquid nitrogen until the initiation of the toxicity study.
Animals, Treatments, and Experimental Procedures
Animal care and administration of the hMSCs were conducted in Charles River preclinical services, a GLP-certified site. Approval was obtained under the Animal Scientific Procedures Act 1986 by the Home Office in Scotland before the initiation of the study.
Male and female BALB/c nude mice (Hsd-Foxn1nu ) were obtained from Harlan Ltd. (Oxon, United Kingdom) and maintained on a Teklad Rodent Diet 2919. The diet and water was provided ad libitum except during designated procedures. During the acclimation period and study duration, animals were housed in a limited access rodent facility and kept in groups of 2 or 3 per cage in appropriately sized polycarbonate/polypropylene cages with stainless steel grid tops and solid bottoms. Each cage was fitted with a filter top and had sterilized white wood shavings. However, the bedding material was changed during the study to help reduce eye irritation in the mice. The mice were allowed a approximately 2-week acclimation period to the Charles River facility conditions (19°C to 23°C, 40–85% relative humidity, and a 12-hr light/dark cycle) prior to inclusion in the study.
hMSC Preparation
hMSCs were thawed and prepared immediately before injection. The cells were removed from liquid nitrogen, thawed in a 37°C water bath, and transferred directly into tubes containing 4 ml of the saline vehicle. The cryovial was washed once to ensure that all cells were removed. A cell count was performed using the trypan blue (Sigma Aldrich) exclusion method. A cell suspension containing a total number of 3 × 105 cells/ml was transferred into 15 ml conical tubes and centrifuged at 400 g for 5 min at room temperature. The supernatant from the centrifuged cells was discarded, and the pellet of hMSCs was resuspended in 150 μl of saline vehicle. The cell suspension was mixed well and transferred to a sterile cryovial. The cell suspension was then aspirated into three insulin syringes each of which contained 50 µl. The hMSCs were administered to the animals within 2 hr of resuspension in saline.
hMSC Transplantation
hMSCs were administered at a dose of 3 × 105 cells in 150 μl per animal. The total volume was divided between 3 injection sites (50 μl per site), two in the thigh and one in the calf on the right leg. Each injection was administered over approximately 1 to 3 sec. The control animals received 3 injections of 150 μl of saline in a similar manner. Animals in each group were subjected to termination at 3 months after the hMSC administration.
Toxicology Study Design
The study included 8 male and 8 female mice per group (Table 1). Animals in each group were subjected to study termination at 3 months after hMSC dosing. Animals were weighed and randomly assigned to the 2 treatment groups (Table 1).
Experimental Study Design—3-month Toxicity Study.
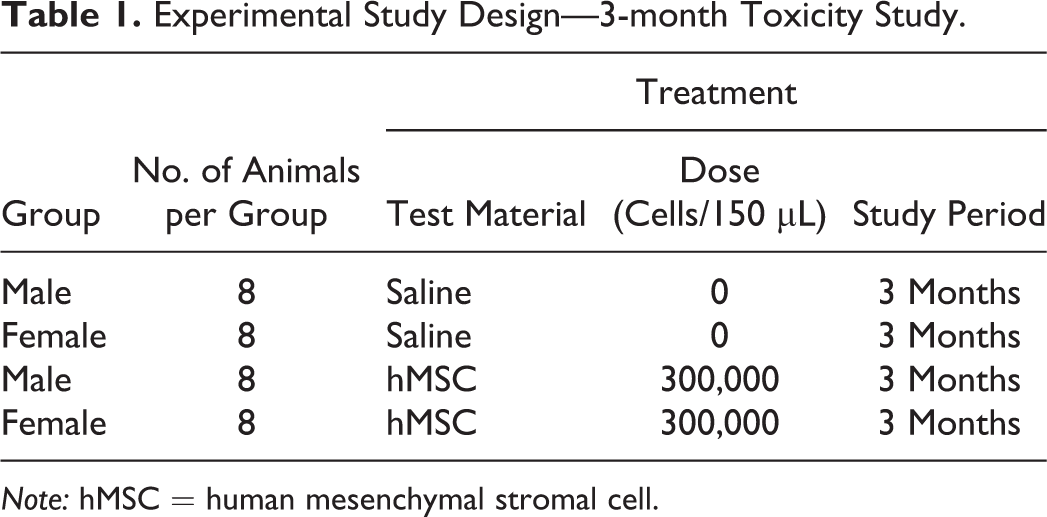
Note: hMSC = human mesenchymal stromal cell.
In parallel with our preclinical toxicity study, animals were dosed for our biodistribution study (N = 8 per group). Animals for both biodistribution and toxicity studies were combined for the clinical observations (N = 16 per group) and body weight measurements (N = 16 per group).
Food Consumption
Food consumption was quantitatively measured once weekly throughout the study. Measurements were started at −1 week, and the last food consumption was carried out prior to the scheduled study termination. Food consumption measurements were not obtained from 8 animals in error. For this reason, food consumption measurements were performed only on N = 6 animals per group.
Hematology and Biochemistry
Blood was collected from the vena cava at necropsy. Blood samples were collected in EDTA-coated tubes for hematology and lithium heparin tubes for clinical chemistry. Animals were euthanized via CO2 asphyxiation followed by exsanguination. Blood samples were obtained via the vena cava using a hypodermic needle and a plastic syringe. Animals were fasted for 4 hr prior to their scheduled necropsy.
Necropsy
Animals were subjected to a complete necropsy examination, which included evaluation of the carcass and musculoskeletal system; all external surfaces and orifices; cranial cavity and external surfaces of the brain; and thoracic, abdominal, and pelvic cavities with their associated organs and tissues.
Representative samples from the following tissues and organs were collected and fixed in 10% neutral buffered formalin: administration site (right leg with the foot attached), aorta, femur, sternum, bone marrow (femur and sternum), brain, cervix, epididymides, eyes, gallbladder, adrenal glands, Harderian glands, lacrimal glands, mammary glands, pituitary glands, salivary glands, seminal vesicles, thyroid gland, gut-associated lymphoid tissue, heart, kidneys, caecum, colon, rectum, liver, lungs, mandibular lymph node, mesenteric lymph node, inguinal lymph node, skeletal muscle (left leg), nasal cavity, sciatic nerve (left leg), oesophagus, ovaries, oviducts, pancreas, skin, duodenum, ileum, jejunum, spinal cord, stomach, thymus, tongue, trachea, ureters, urinary bladder, uterus, and vagina. The optic nerves were fixed in Davidson’s fixative and the testes in modified Davidson’s fixative. In addition, any organ/tissue with gross macroscopic change(s) was collected, recorded, and fixed in 10% neutral-buffered formalin.
Tissues were trimmed, embedded with paraffin, sectioned, mounted on glass slides, and stained with hematoxylin and eosin (H&E). Bone marrow smears were stained with May Grünwald Giemsa staining. The injection sites (right thigh and calf musculature) were removed after whole leg fixation, and the samples were labeled from 1 to 5. The thigh muscle was cut into 3 equal parts and labeled from 1 to 3. Injection site 1 = proximal thigh; injection site 2 = middle thigh; injection site 3 = distal thigh. The calf muscle was also dissected and cut into 2 equal pieces and labeled 4 and 5. Injection site 4 = proximal calf; injection site 5 = distal calf. Each injection site from the right leg was embedded in separate paraffin wax blocks. A single 5-μm section was cut from each block, stained with H&E, and evaluated microscopically.
The muscle from the left limb served as an internal control for each mouse. Briefly, the thigh muscle was dissected and labeled from 1 to 3 as above. A single 5-μm section obtained from injection site 2 (middle thigh) was then cut, stained with H&E, and evaluated microscopically.
Lesion Grading
Histopathological changes such as infiltration, inflammation, degeneration, atrophy, hyperplasia, hemorrhaging, plasmacytosis, and edema were scored using a semiquantitative grading of five grades (0 to 4); 0 = no visible lesion, 1 = minimal change, 2 = mild change, 3 = moderate change, and 4 = marked change. All histopathology was carried out under GLP by a board-certified veterinary pathologist.
Body and Organ weights
Males and females were randomized separately, and body weights were measured at randomization, on the day of injection and once weekly thereafter for the study duration. The group body weights of the animals were compared to ensure homogeneity.
Organ weights were taken at scheduled necropsy after blood sampling from all the surviving animals. Paired organs were reported together, and the terminal body weights were used for organ weight analysis.
Statistical Analysis
Body weights, food consumption, hematology, and clinical chemistry (females only due to insufficient data in the male animals) were analyzed for homogeneity of variance using the “F Max” test. The group variances were homogenous; therefore, a parametric analysis of variance was used, and pairwise comparisons were made using Fisher’s F protected LSD method via Student’s t test.
Results
Unscheduled Death
One animal was found dead in its cage prior to terminal necropsy. The death occurred in the saline-treated group and was not test article related. Nonetheless, the animal was subject to necropsy, and the full tissue list was obtained. There were no gross or microscopic findings to explain this death.
Clinical Observations
There were no test item–related clinical signs post dose or throughout the 3-month study period (Table 2). Both hMSCs and saline-treated animals demonstrated transient inflammation (swollen eyes) of the eyes. This issue was due to the type of bedding used in the cages, and as a result, both the bedding and nestle material were changed. Other occasional observations included skin redness (hMSC and vehicle males) and overgrown toenails (hMSC and vehicle females).
Incidence of Clinical Observations in hMSCs and Vehicle Control over the 3-month Study Period.
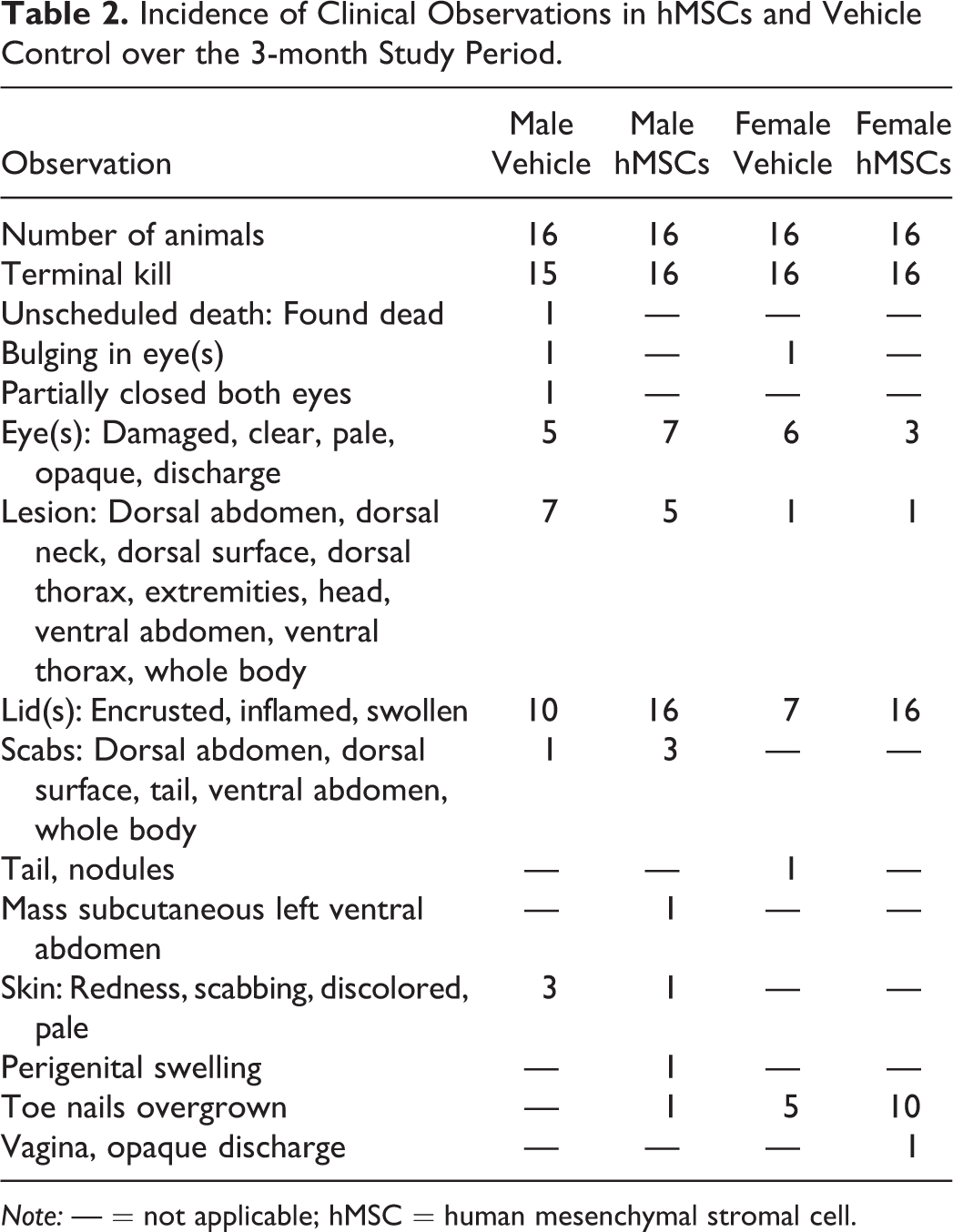
Note: — = not applicable; hMSC = human mesenchymal stromal cell.
Food Consumption
Food consumption measurements of the hMSCs-treated groups were similar to those of the vehicle control group throughout the 3-month study duration (Table 3).
Group Mean Values of Food Consumption in hMSCs and Vehicle Control Nude Mice over the 3-month Study Period.
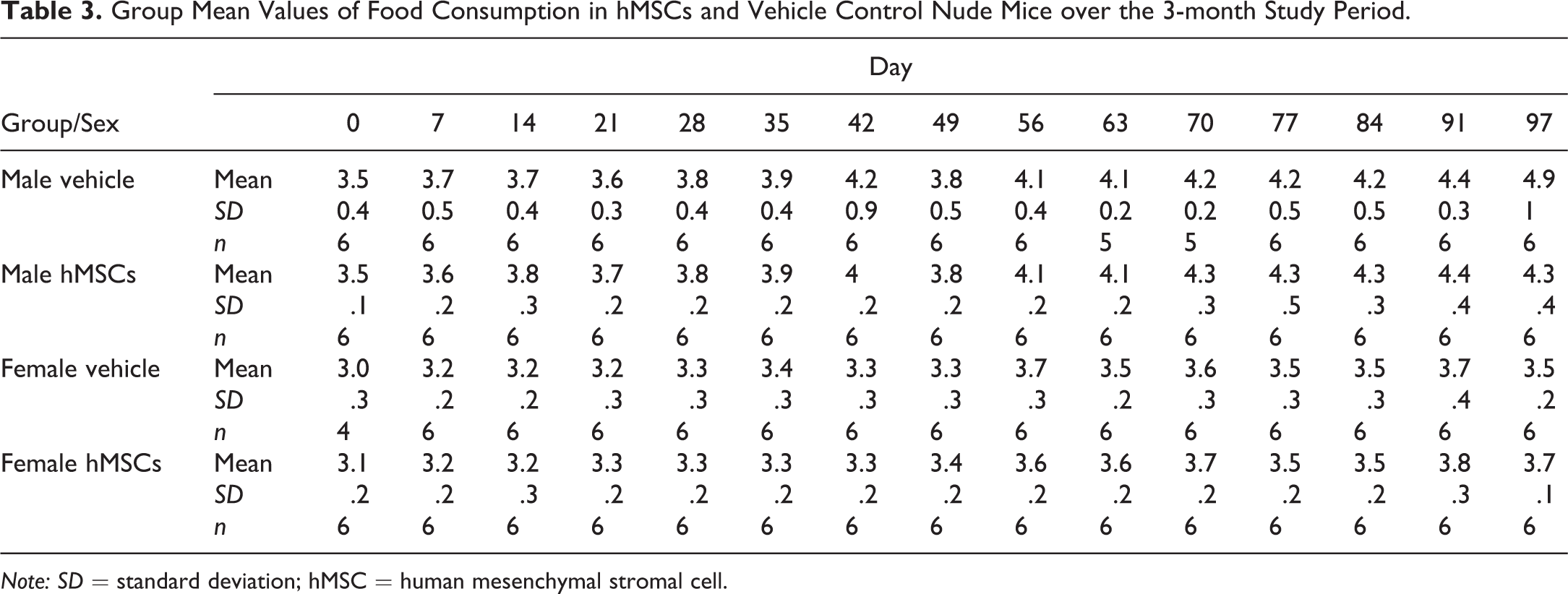
Note: SD = standard deviation; hMSC = human mesenchymal stromal cell.
Body Weights
There were no test item–related findings on body weight gain and final body weight over the study period in either male or female animals (Table 4). Although the group mean body weights were increased in hMSC-treated males, compared with control males (Table 4), for most of the study, this observation was considered incidental due to the fact that hMSC-injected mice body weights were slightly greater than control mice values from pretrial (day −7).
Group Mean Body Weights in hMSCs and Vehicle Control Nude Mice over the 3-month Study Period.

Note: Statistical difference from vehicle mean expressed as p values. SD = standard deviation; hMSC = human mesenchymal stromal cell.
Significantly different from vehicle: *p < .05, **p < .01.
Organ Weights
Absolute organ weights are presented in Table 5. One statistically significant difference versus the control was noted. A decrease (p < .01) in the mean lung weight was noted in the hMSC-treated male when compared to the control.
Absolute Organ Weights and Organ to Body Weight Ratio of Male and Female Nude Mice Following the Injection of hMSCs or Vehicle Control.
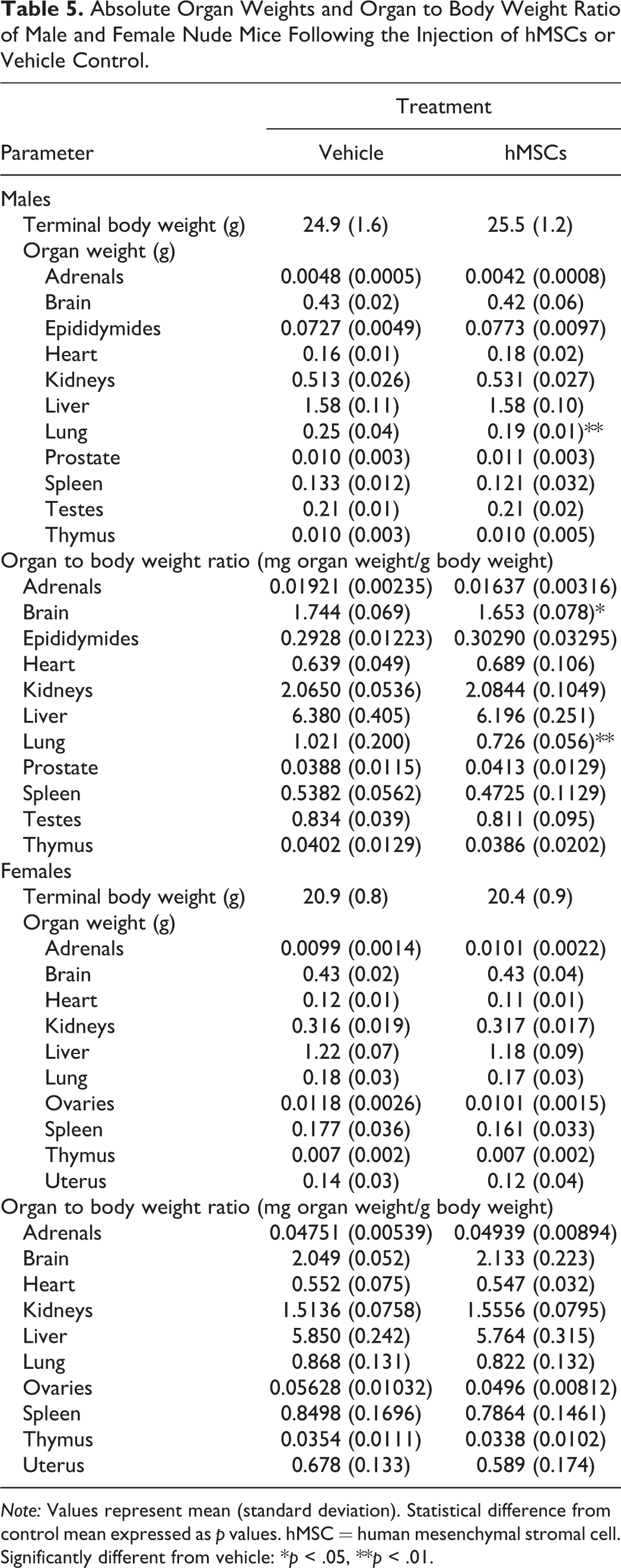
Note: Values represent mean (standard deviation). Statistical difference from control mean expressed as p values. hMSC = human mesenchymal stromal cell.
Significantly different from vehicle: *p < .05, **p < .01.
Organ weight as % body weight is also presented in Table 5. Two statistically significant differences versus the control were noted. A decrease (p < .05) in the mean brain weight and a decrease (p < .01) in the mean lung weight were noted in the hMSC-treated male when compared to the control.
Hematology and Biochemistry
Due to issues surrounding the blood collection from mice and sample quality, some parameters could not be read in the hematology and biochemistry in both the treatment and the control mice. Missing data were due to insufficient sample collection or clotting of the sample. Furthermore, coagulation testing was not performed due to technical difficulties in obtaining sufficient volumes of mouse plasma in this study.
In the samples that were analyzed, there were no changes in hematology evaluations attributable to the administration of hMSCs in the female groups (Table 6). The sample size for the male animals (N = 2) was too low, and therefore, an effect of hMSC treatment could not be determined. Clinical chemistry evaluations indicated a 2-fold increase in plasma creatine phosphokinase after 3 months in male and female hMSC-injected mice in comparison to the control mice (Table 7). As the sample size was too small for statistical analysis, an hMSC treatment–related effect could not be determined. No treatment-related effects were observed in any of the other clinical chemistry parameters measured.
Selected Hematology Analyses of Male and Female Nude Mice Following the Injection of hMSCs or Vehicle Control.
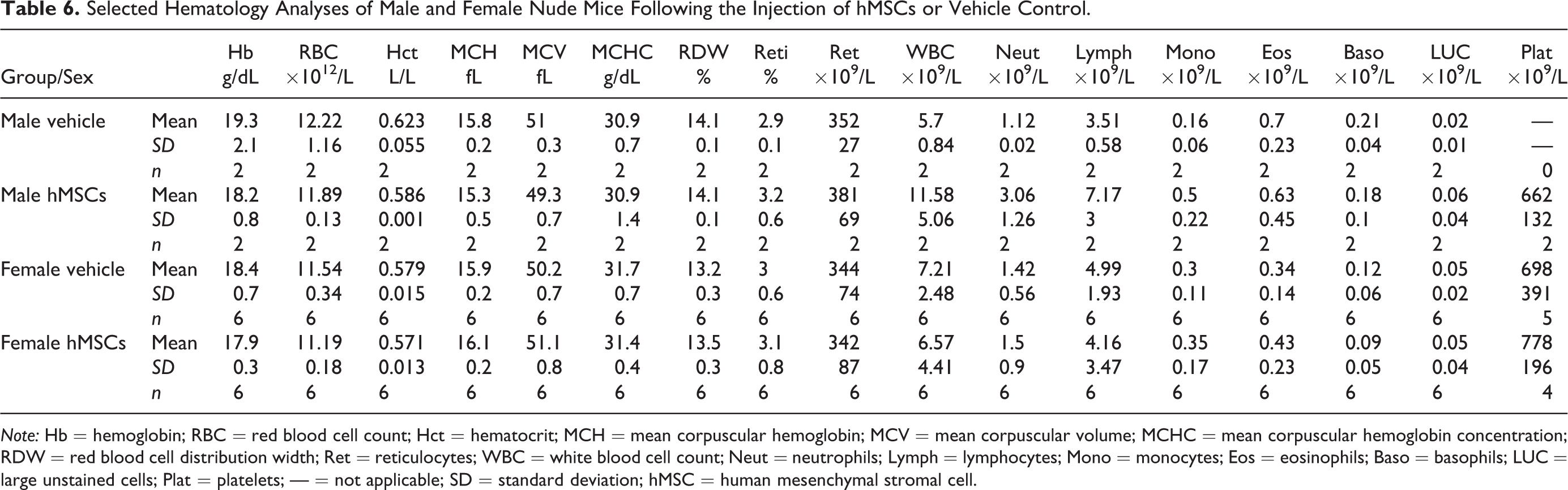
Note: Hb = hemoglobin; RBC = red blood cell count; Hct = hematocrit; MCH = mean corpuscular hemoglobin; MCV = mean corpuscular volume; MCHC = mean corpuscular hemoglobin concentration; RDW = red blood cell distribution width; Ret = reticulocytes; WBC = white blood cell count; Neut = neutrophils; Lymph = lymphocytes; Mono = monocytes; Eos = eosinophils; Baso = basophils; LUC = large unstained cells; Plat = platelets; — = not applicable; SD = standard deviation; hMSC = human mesenchymal stromal cell.
Selected Biochemistry Analyses of Male and Female Nude Mice Following the Injection of hMSCs or Vehicle Control.
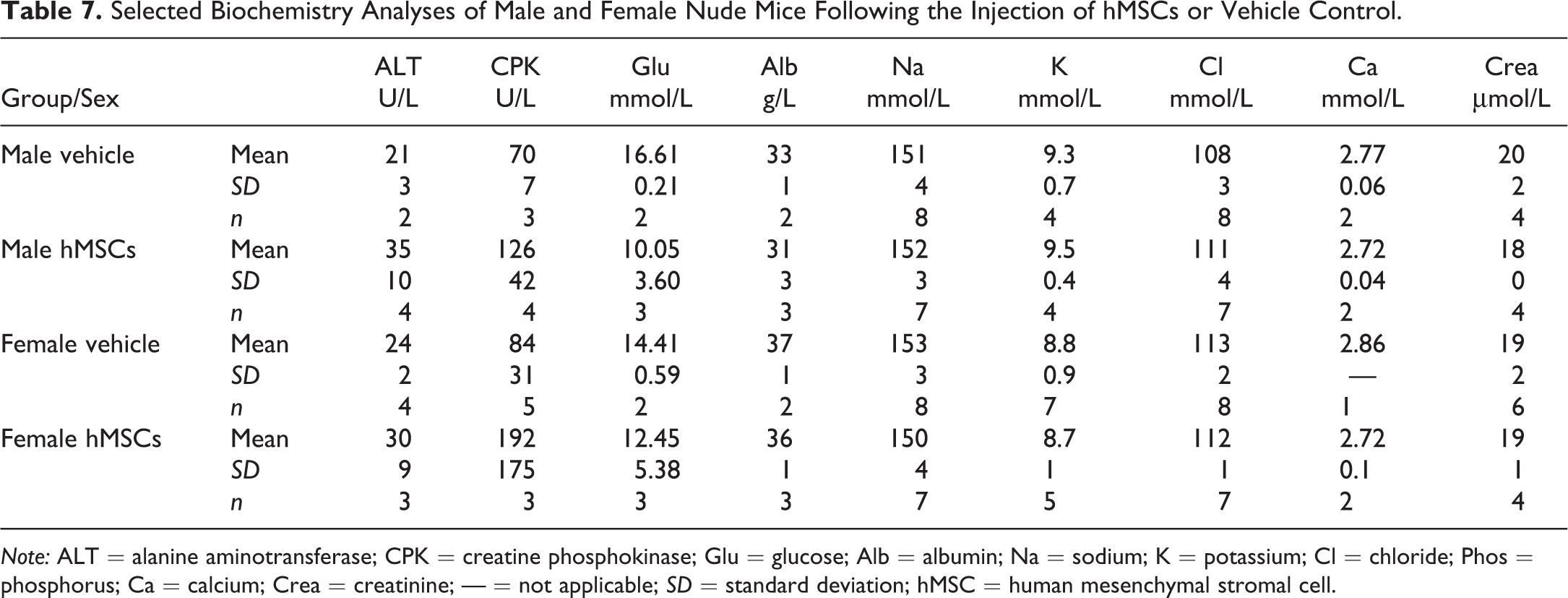
Note: ALT = alanine aminotransferase; CPK = creatine phosphokinase; Glu = glucose; Alb = albumin; Na = sodium; K = potassium; Cl = chloride; Phos = phosphorus; Ca = calcium; Crea = creatinine; — = not applicable; SD = standard deviation; hMSC = human mesenchymal stromal cell.
Macroscopic and Histopathological Findings
Macroscopic findings at necropsy are summarized in Table 8. One animal in the male hMSCs group developed a subcutaneous mass that was determined histologically as an abscess of the preputial gland and was deemed nontreatment related. Dark foci and mild to moderate discoloration consistent with hemorrhaging was observed in the lungs of both male and female treated and control animals (Table 8).
Macroscopic Findings at Necropsy.
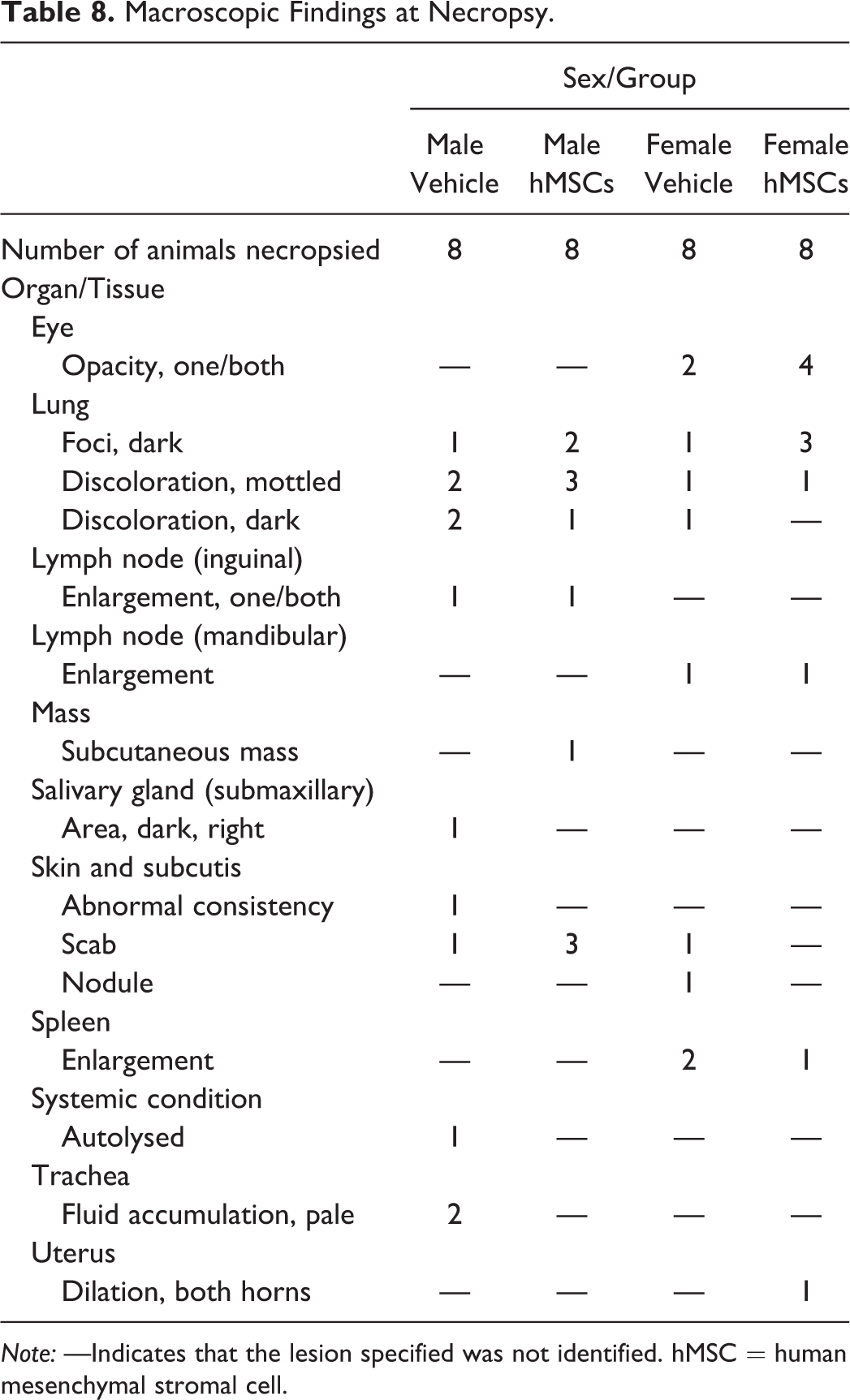
Note: —Indicates that the lesion specified was not identified. hMSC = human mesenchymal stromal cell.
No treatment-related effects were observed in any histological slides analyzed (Tables 9 and 10). Any microscopic findings observed were considered incidental of the nature that would be commonly observed in this age and strain of nude mouse. The findings were of similar frequency and severity in both control and treated animals and were therefore considered unrelated to the administration of the hMSCs (Tables 9 and 10).
Histopathology Findings in the Male Nude Mice Following the Injection of hMSCs or Vehicle Control.
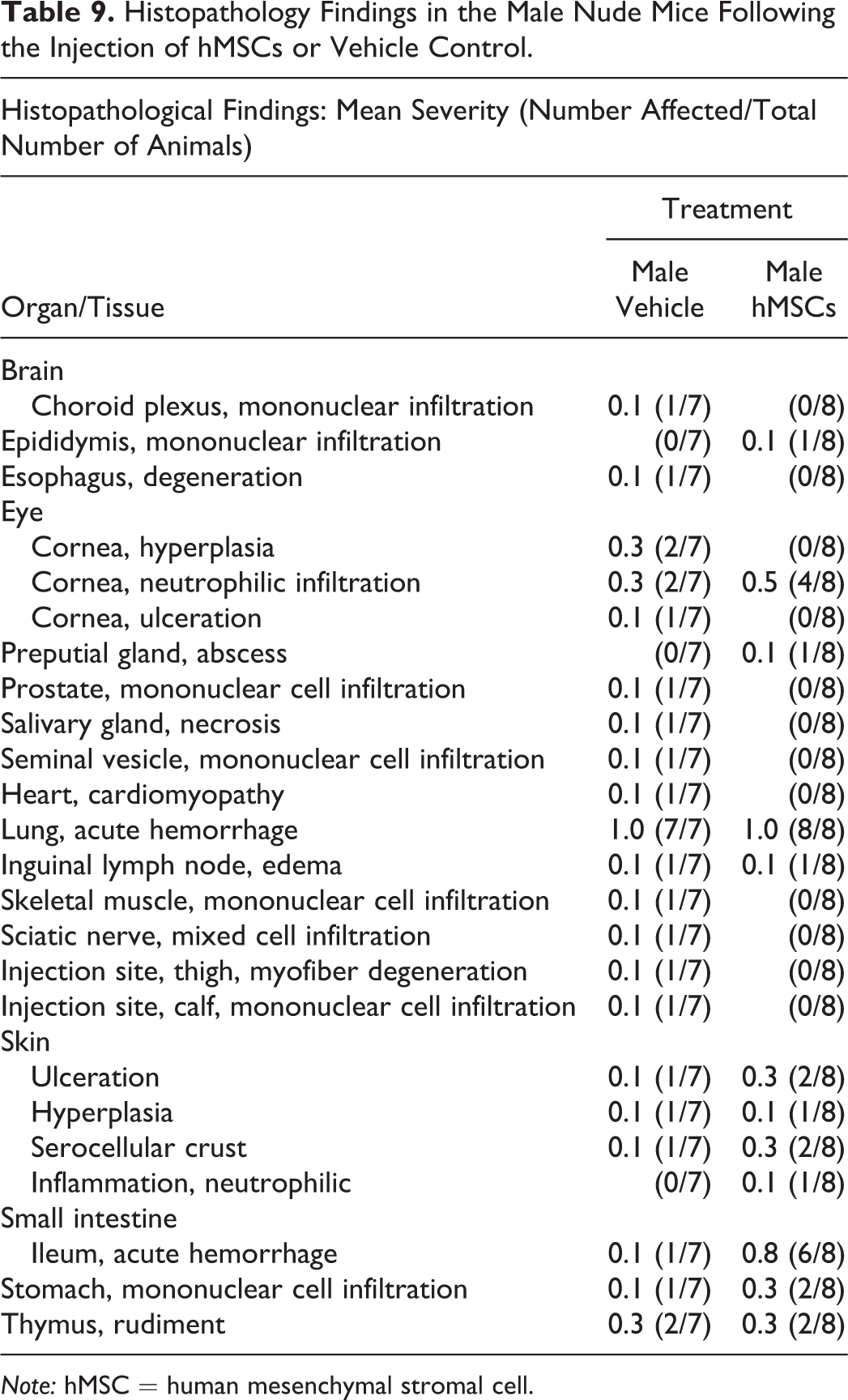
Note: hMSC = human mesenchymal stromal cell.
Histopathology Findings in the Female Nude Mice Following the Injection of hMSCs or Vehicle Control.
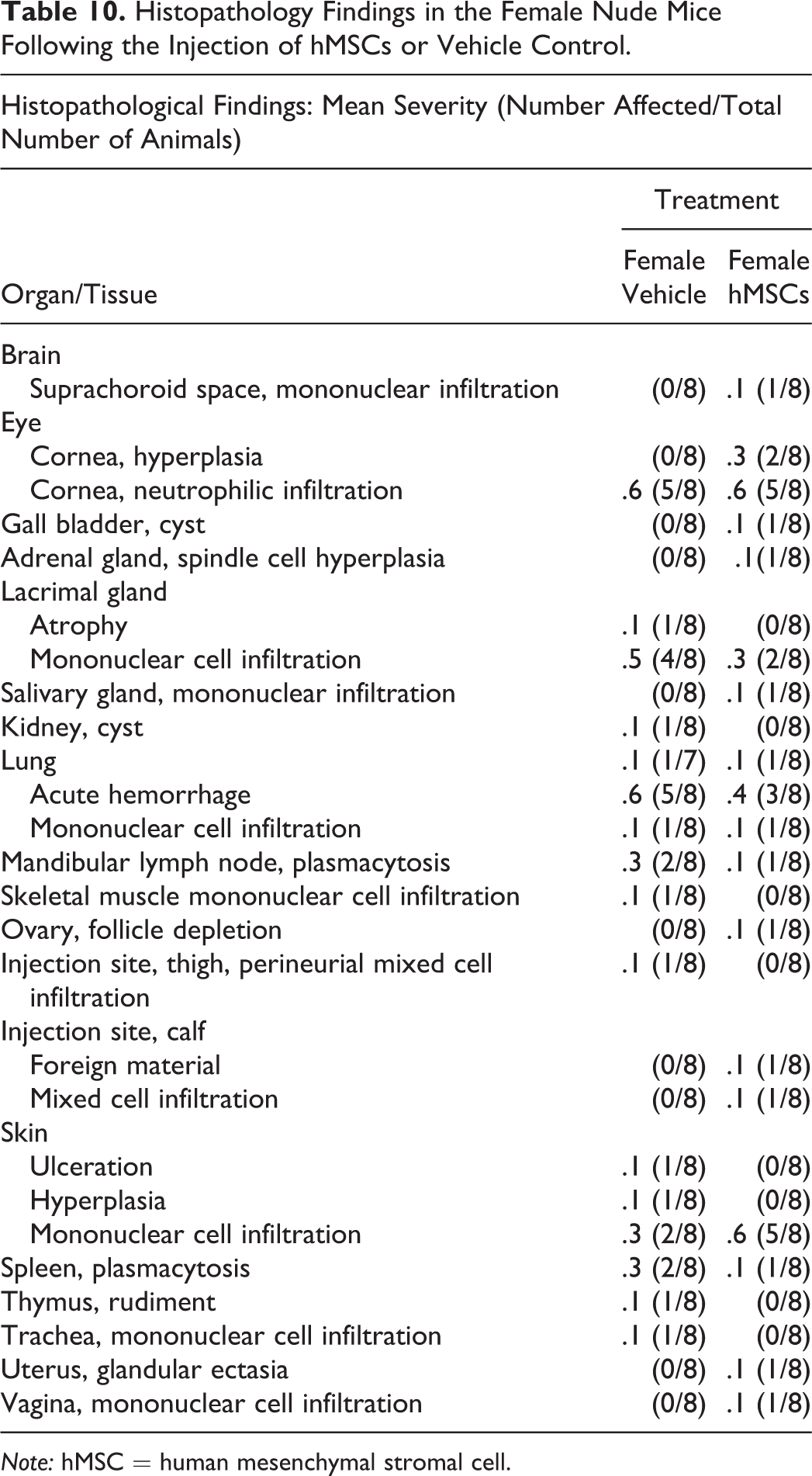
Note: hMSC = human mesenchymal stromal cell.
Discussion
Recent clinical studies have demonstrated that the administration of MSCs to patients with CLI is safe (Das et al. 2013; Gupta et al. 2013, 2017; Lasala et al. 2011; Lu et al. 2011). Given these results and a wealth of encouraging preclinical data, MSCs have become the most clinically relevant CT for testing in CLI patients. From a regulatory perspective, safety concerns represent the largest barrier to the translation of MSCs from a laboratory-based therapeutic to a successful clinical product. Toxicities due to novel delivery techniques, acute immunogenicity, and tumor formation are the main toxicity end points of concern for both investigators and regulatory authorities.
There are a number of distinct issues from a toxicity perspective that need to be considered in the context of CT. Many of these are based on the fact that the CT is a living product, and thus, issues such as aberrant differentiation and malignant potential are matters of concern. In addition, species issues need to be considered, as the human CT product may result in xenogeneic responses when delivered to animals with an intact immune system. Thus, if human cells are to be tested, which is often the preference of the regulatory authorities as this represents the final intended human CT product, it may be necessary to test the product in immunodeficient animals. This also has disadvantages, as the human subjects who will eventually be enrolled in clinical trials will have an intact immune system. This will have distinct relevance in the context of either autologous or allogeneic transplantation.
The results from this study demonstrated that the administration of 3 × 105 (15 times the maximal proposed clinical dose extrapolated based on a weight-adjusted basis) via the intended clinical route of administration did not result in any adverse clinical observations, local or systemic toxicities. We chose this study protocol after extensive interactions were held with the regulatory authority prior to the submission of the IMP application. It was clear to us that in spite of the widespread use of MSCs in clinical trials for multiple indications globally, the regulatory body in Ireland would require toxicity and biodistribution studies, testing the GMP-manufactured CT product. This was necessary, as products used in previous preclinical and clinical studies may differ from the processes used by our GMP facility to manufacture the CT product tested in this study. Manufacturing processes across GMP facilities are heterogeneous and may differ in the following ways: cell source, tissue processing after harvest, bioreactor used, culture media (serum vs. serum free media), growth factors, and culture conditions (normoxia vs. hypoxia).
We chose to test our GMP-manufactured CT product in BALB/c nude mice, as these mice are immunodeficient and provide an environment that is permissive to human cell survival with the avoidance of confounding xenogeneic effects (Braid et al. 2018; Ra et al. 2011; Macisaac et al. 2012; Creane et al. 2017; Vilalta et al. 2008). This is important to assess the risk of malignant transformation, as a xenogeneic immune response may lead to rapid clearance. The overall aim in the toxicity study was to demonstrate the safety and tolerability of the hMSC product via the IM route in this model. Both aspects of this model enabled the evaluation of safety concerns surrounding the administration of our CT product, for example, local and systemic toxicity.
Several preclinical parameters were evaluated in order to investigate the safety and tolerability of hMSC administration. Firstly, our data showed no impact of the proposed CT product on the general well-being of the animal. All animals showed consistent body weight (Table 4) and food consumption (Table 3) measurements across the hMSC and saline-treated mice. Furthermore, no test item–related clinical signs after the hMSC administration at any time throughout the 3-month study duration were observed as measured by the clinical parameters set in this study (Table 2). In spite of the blood sampling and processing issues with the male treated animals, no hematologic abnormalities were observed in the female animals. Female animals that received hMSCs presented with a similar white blood cell count, red blood cell count, hemoglobin concentration, hematocrit, mean corpuscular volume, mean corpuscular hemoglobin, and mean corpuscular hemoglobin concentration to that of the control animals (Table 6). No abnormal readings were observed in the selected serum chemistry tests performed (Table 7). Although the sample size was too low to perform statistical analysis, a 2-fold increase in CPK was observed only in the hMSC-treated animals. However, it is possible that the increase observed within these samples may have been a result of the sample collection process. It has been reported that incompletely filled heparinized tubes can influence CPK values (Lippi et al. 2012).
Local toxicities at the injection site such as altered tissue function, tumor formation, and cell differentiation to unwanted cell types at the administration sites may be caused by the CT product interacting adversely with host tissue environment or the degradation of the product itself or its components. Furthermore, the migration of the CT product outside the target tissue represents a major safety risk and may lead to systemic toxicities such as immune-mediated toxicities or ectopic tissue formation in the distant organs. In order to assess local and systemic toxicities, extensive histopathological analysis was performed on the injection site and selected organs (Tables 9 and 10). The histopathological analysis conducted in this study did not uncover any tumors, ectopic tissue formation, or major inflammatory response that was associated with our MSC product. Although differences were noted in the absolute organ and organ to body weight ratios in the lungs of the hMSC-treated groups, these data when evaluated with the histopathology data and taking into consideration the method of euthanasia were deemed an expected finding (Fawell, Thomson, and Cooke 1972; Burkholder et al. 2010).
As determining the biodistributive fate of a CT product after administration is an important part of characterizing its safety profile, we designed in parallel with our toxicity study a preclinical biodistribution study. The biodistribution and retention of the IM-delivered hMSCs was measured using a highly sensitive quantitative polymerase chain reaction–based detection human DNA via human Alu sequences (Creane et al. 2017). In support of the absence of findings in the histopathological data, the results of the biodistribution study demonstrated that hDNA was detected in small quantities only at the injection site and not in the distant organs of the mice (Creane et al. 2017).
In view of the reported findings and under the conditions of this study, we can report that the administration of GMP grade hMSCs via IM injection at a single dose of 3 × 105 cells per mouse (approximately 15 million cell/kg) was well tolerated with no indication of any malignancy systemically or locally.
Based on the toxicity data presented in this article in accordance with the biodistribution study (Creane et al. 2017), historical efficacy data, and data from the literature, the risk benefit of the GMP grade hMSC product was considered acceptable and cleared by HPRA to initiate a phase 1b study in no option for revascularization patients with CLI.
In this study, we have emphasized the special features that need to be considered in the toxicity assessment of a CT product to aid in the translational process such as the need for studies performed under GLP using products manufactured under GMP, issues related to the testing of a human CT product in an immunodeficient model, dose considerations, route of administration, and important toxicity end points. As publications on the safety assessment of human bone marrow–derived MSCs via the IM route of administration are rare (Rengasamy et al. 2016), we believe that this article is an important contribution to the very limited toxicology database of CT products.
Footnotes
Author Contribution
Authors contributed to conception or design (MC, TO); data acquisition, analysis, or interpretation (MC, MM, AD, CS, TO); drafting the manuscript (MC); and critically revising the manuscript (MC, MM, AD, CS, TO). All authors gave final approval and agreed to be accountable for all aspects of work in ensuring that questions relating to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Declaration of Conflicting Interests
Professor Timothy O'Brien is a founder, director, and equity holder in Orbsen Therapeutics (Galway, Ireland). The other authors declare that they have no conflict of interest.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: this work was supported by Science Foundation Ireland (SFI) Strategic Research Cluster Grant No. SFI:09/SRC B1794, European Regional Development Fund and SFI-Health Research Board Grant No. TRA 201115.