
Abstract
Skeletal muscle (SKM) injury or myopathy results in structural or functional defects in SKMs that can be caused by variety of factors such as (1) genetic, (2) drug-induced, (3) disease progression (cachexia), or (4) aging (sarcopenia). Creatine kinase (CK) and aspartate transaminase (AST) activity assays have been routinely used as SKM injury biomarkers, but they lack sensitivity and tissue specificity. In collaboration with the Predictive Safety Testing Consortium, we evaluated the diagnostic performance of a muscle injury biomarker panel (MIP) compared to CK and AST and their correlation with the histology scores across 34 different rat studies. The MIP panel included the analytes skeletal troponin I, myosin light chain 3, fatty acid binding protein 3, and a CK mass (versus activity) assay. The area under the receiver operator characteristic curve for MIP panel ranged from 0.82 to 0.91 as compared to 0.71 and 0.82 for CK and AST activity assays, respectively. Because the MIP biomarkers outperformed the routine biomarkers, the European Medicines Agency and U.S. Food and Drug Administration posted Letters of Support encouraging further study of these analytes and acknowledged the utility of the MIP panel. Ongoing efforts are directed toward the application of the MIP panel biomarkers in clinical studies and regulatory qualification.
Keywords
Myopathies can be caused by a variety of disorders characterized by either primary structural or functional impairment of skeletal muscle (SKM). Myopathies can be drug induced or inherited/genetic based. Nutritional myopathies can occur as a result of acute, critical illness (Burnham, Moss, and Ziegler 2005). In animals, nutritional myodegeneration can occur that has been associated with diets deficient in selenium and/or vitamin E. Myopathy symptoms can include muscle weakness, fatigue, exercise intolerance, cramps and myalgias, contractures, myotonia, and myoglobinuria (Mestrovic 2017; Sieb and Gillessen 2003).
Drug-induced myopathies can be a result of direct or indirect drug exposure. Examples of drugs that can have a direct myotoxic effect are alcohol, cocaine, glucocorticoids, lipid-lowering drugs, antimalarials, and zidovudine (anti-viral for HIV). Indirect muscle damage can occur as a result of ischemic muscle compression due to drug-induced coma, diuretic-induced hypokalemia, alcohol withdrawal hyperkinetic states (delirium tremens or seizures), or cocaine-use-related hyperthermia. D-penicillamine and TNFα inhibitors used in the treatment of rheumatoid arthritis can cause immunologically induced inflammatory myopathy (Valiyil and Christopher-Stine 2010).
Inherited or genetic-based myopathies are conditions that result in the impairment of muscle function. The disease can have a direct effect on muscle function, being a pathology of muscle (e.g., Duchene or Limb-girdle muscular dystrophies), or indirect effect, being a pathology of nerves or neuromuscular junctions (i.e., amyotrophic lateral sclerosis, myasthenia gravis, or spinal muscular atrophy).
Multiple methods can be used to diagnosis myopathies and each has limitations. Subject self-reporting of clinical symptoms or history can be ambiguous and subjective in nature with differences in terminology and, clearly, is not an option in preclinical models. Muscle biopsies with histopathology would be considered a reference method but is invasive, requires skilled technical resources, and often unnecessary for diagnosis, though an important tool during drug development. Several imaging modalities are available (e.g., ultrasound, computed tomography scans, magnetic resonance imaging, magnetic resonance spectroscopy, and positron emission tomography). Some of these imaging techniques are not yet widely available and outcomes can be technique dependent (Schiffenbauer 2014). Functional assays such as electromyogram and mobility assessments may be difficult to administer and interpret due to subject cooperation and motivation. Functional assays have limited utility in the diagnosis of most drug-induced myopathies.
Laboratory diagnostic tests using blood or urine samples are frequently included in myopathy diagnoses. These tests are readily available, provide quantitative, broadly accepted results, and are relatively inexpensive to perform. The conventional or “classic” biomarkers of SKM injury, creatine kinase (CK), and aspartate transaminase (AST) activity assays lack tissue specificity and sensitivity (Castro and Gourley 2012; Keltz, Khan, and Mann 2013). CK and AST activity assays work well for detecting higher grade myopathies but are poor predictors of low-grade histopathology findings (Dabby et al. 2006). A number of newer SKM injury biomarkers and platforms are being evaluated (e.g., proteomics, genomic, or miRNA), but the characterizations of these methods are incomplete and their utility unproven.
There is a clear need for sensitive, specific, and qualified biomarkers that can be used for the diagnosis of SKM injury. Through the Critical Path Institute’s (C-Path) Predictive Safety Testing Consortium (PSTC), a “novel” immunoassay-based muscle injury biomarker panel (MIP) was evaluated that includes the analytes skeletal troponin I (sTnI), myosin light chain 3 (Myl3), fatty acid–binding protein 3 (FABP3), and creatine kinase measured by a mass assay (CKm). The analytes were measured using serum and/or plasma samples and commercially available reagent systems.
sTnI is the only SKM-specific protein biomarker included in the MIP panel. Myl3, FABP3, and CKm are present in SKM and myocardial tissue as well as other organ tissues. sTnI is a component of myofilaments and can exist as 2 isoforms: slow-twitch (Type I) or fast-twitch (Type II) SKM fibers. Myl3 is also a component of myofilaments. It is found predominantly in slow-twitch SKM and in cardiac muscle. FABP3 is a cytosolic lipid transport protein and is abundant in SKM and cardiac muscle but is also present in brain, liver, and small intestine. CK is a cytosolic enzyme involved in ATP utilization and generation and consists of 2 subunits: M (muscle type) and B (brain type). The CK subunits can combine to form 3 different isoenzymes: CK-MM, CK-MB, and CK-BB. SKM expresses predominantly the CK-MM isoenzyme with low concentrations of CK-MB. Myocardial tissue expresses approximately 70% CK-MM and 25% to 30% CK-MB (Apple et al. 1984). The CK-BB isoenzyme is predominantly expressed in brain and smooth muscle tissue. The antibodies utilized in the MIP panel CKm immunoassay target the CK-MM isoenzyme but will also react with the CK-MB isoform.
C-Path’s PSTC Biomarker Qualification
C-Path’s PSTC was formed in 2006 and brings together pharmaceutical companies and academia to share, validate, and qualify innovative safety testing methods under advisement of the U.S. Food and Drug Administration (FDA), the European Medicines Agency (EMA), and Japanese Pharmaceutical and Medical Devices Agency; see Figure 1. A goal of PSTC’s safety biomarker validation and qualification process is to gain industry and regulatory agency agreement regarding the reliability of novel biomarkers that would support their use in preclinical and clinical drug development applications to detect and/or monitor drug-induced organ injury.
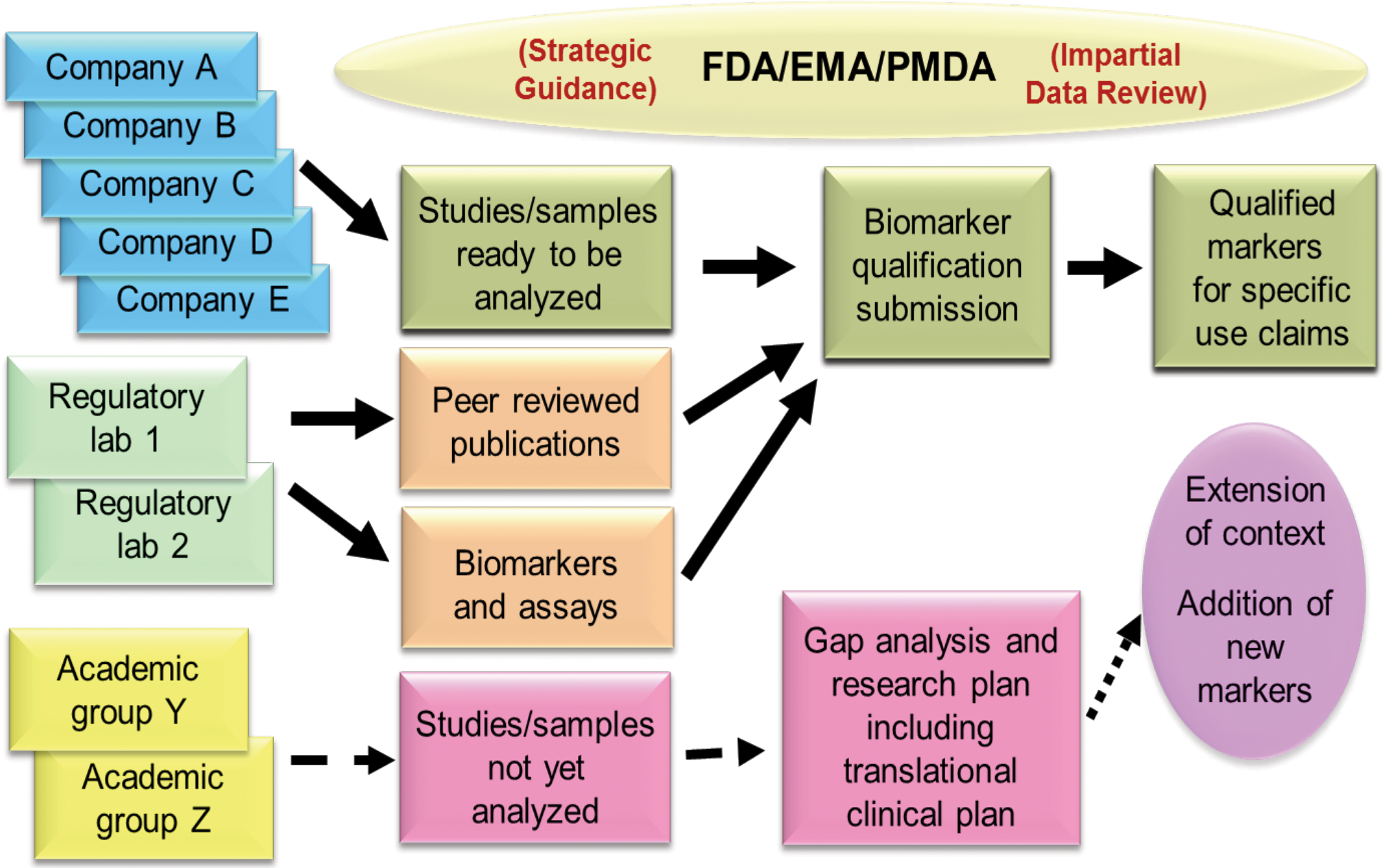
C-path’s Predictive Safety Testing Consortium biomarker qualification process.
Safety biomarker qualification requires providing sufficient evidence that links the biomarkers to a biological finding or clinical end point. The evidence provides a specific context of use (COU) for the biomarkers (i.e., how the biomarkers could be used), and qualification is an indication of regulatory acceptance of the biomarker for the specific COU.
Considerations for Evaluating and Qualifying Safety Biomarkers
Several key considerations must be made when evaluating and qualifying safety biomarkers. The biomarker must display dose-related responses consistent with histopathology findings and demonstrate improved performance relative to conventional biomarkers. The biomarker response should be consistent across mechanistically different compounds, gender, strains, and species. The analytical reagents used for measurement should be well characterized, readily available, and assay performance sufficiently validated.
It is important to understand the mechanistic and biologic response of the biomarker to injury and relevance to toxicity. Biomarker sensitivity and specificity must be demonstrated. The biomarker response needs to be characterized to toxicities in the target tissue as well as other tissues. It is also important to understand the biomarker response to pharmacologic effects in the target tissue without toxicity.
PSTC MIP Biomarkers: FDA Biomarker Letter of Support
PSTC formed the Skeletal Muscle Working Group (SKMWG) in 2010 with the goal to identify and qualify safety biomarkers of drug-induced SKM injury in rats. Initial steps included evaluation and selection of analytes for inclusion in an SKM injury biomarker panel to be qualified. The analytes selected for PSTC SKM injury panel were sTnI, Myl3, FABP3, and CKm. Commercially available reagents from several vendors were evaluated, and reagent systems from Meso Scale Discovery (Rockville, MD, USA), Muscle Injury Panel 1 (rat; cat. K15181C), and Muscle Injury Panel 2 (rat, cat K15180C) were selected for biomarker measurements. A cross-site assay validation was performed using a single sample set that was distributed to 4 PSTC SKMWG member companies. The data were compiled into an assay validation report.
A total of 34 rat toxicity studies were conducted or sponsored by PSTC member companies, and the validated reagent systems were used to analyze samples from the studies. Eighteen of the studies were designed to evaluate biomarker sensitivity using compounds inducing SKM degeneration and/or necrosis. Biomarker specificity was evaluated from 16 studies using compounds designed to induce injury in tissues other than SKM including 5 liver studies, 5 kidney studies, 2 inducing both liver and kidney injury, 3 gastrointestinal studies, and 1 vascular injury study. Submitted studies utilized approximately 8- to 10-week-old male and/or female Sprague-Dawley CD (SD), Fischer 344, or Wistar Han WI rats. One included study assessed biomarker performance in older, approximately 1-year-old SD rats. A histopathology lexicon was developed to assure consistent evaluation of the tissues examined. All studies were reviewed and approved by each company’s Institutional Animal Use and Care Committee. Summary and details of the findings were published (Burch et al. 2016).
U.S. FDA and EMA Biomarker Letters of Support Received in 2015
PSTC MIP biomarker qualification efforts demonstrated that all analytes included in the novel SKM injury panel, sTnI, Myl3, FABP3, and CKm, outperformed the established SKM injury biomarkers CK and AST activity assays (see area under the receiver operator characteristic data, Figure 2). These findings resulted in the U.S. FDA and EMA issuing biomarker Letters of Support which encouraged the further study and use of these biomarkers in research, nonclinical, and early clinical drug-development programs to detect and monitor for SKM injury in an exploratory context (European Medicines Agency 2015; U.S. FDA 2015).
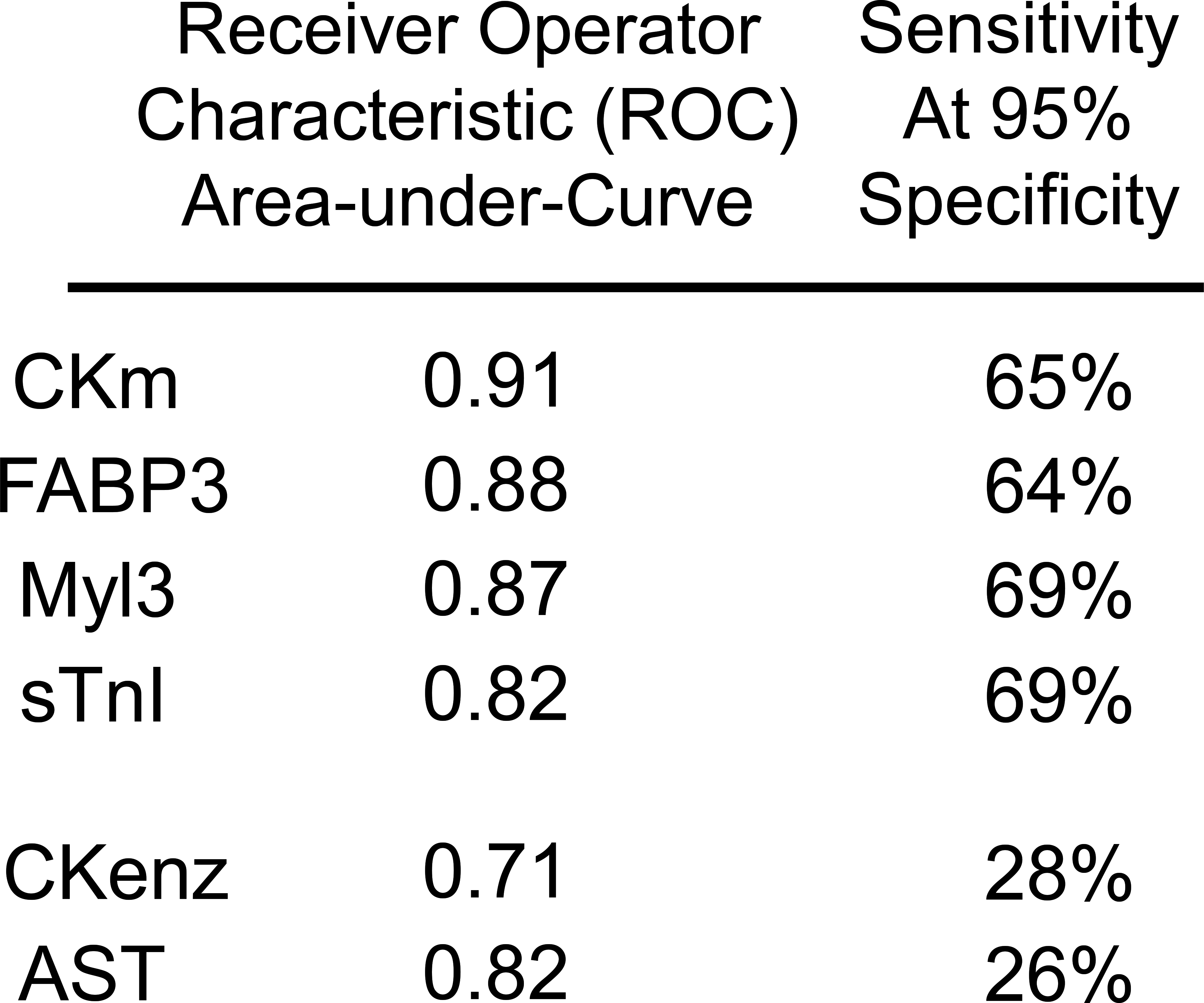
Predictive Safety Testing Consortium biomarker qualification, assay sensitivity and specificity, receiver operator characteristic results based on 34 rat toxicity studies contributed by member companies; 18 studies to assess biomarker sensitivity (skeletal muscle [SKM] injury) and 16 studies to assess assay specificity (non-SKM injury).
PSTC Muscle Injury Panel Biomarker Performance
Example 1: Biomarkers Kinetics
CK and AST activity and the MIP biomarkers release and clearance dynamics were evaluated by inducing acute, localized SKM injury utilizing Marcaine (bupivacaine hydrochloride injection, USP, 0.5%, NDC 0409-1610-50, M615, Hospira, Inc., Lake Forest, IL). Marcaine is a local anesthetic with myotoxic properties via perturbation of Ca2+ homeostasis. All animals received intramuscular injections of 0.3-ml Marcaine in the tibialis anterior of both right and left hind limbs. Terminal blood samples were collected pretreatment or at timed intervals of 2, 4, 7, or 24 hr posttreatment. All samples were assayed for the MIP analytes, sTnI, Myl3, and FABP3, and the established SKM biomarkers, CK and AST activity.
The project goal was to compare the kinetics of biomarker release and clearance following acute drug-induced SKM injury. The timing of biomarker release and clearance following acute SKM injury was similar between the MIP analytes, sTnI, Myl3, and FABP3 and CK and AST activity assays (see Figure 3). All biomarkers were elevated at 2 hr postinjury. Peak concentrations occurred between 4 and 7 hr postinjury except FABP3 that reached peak concentrations at 2 hr. All biomarkers trended toward baseline concentrations at 24 hr with AST, sTnI, and Myl3 remaining above baseline concentrations.
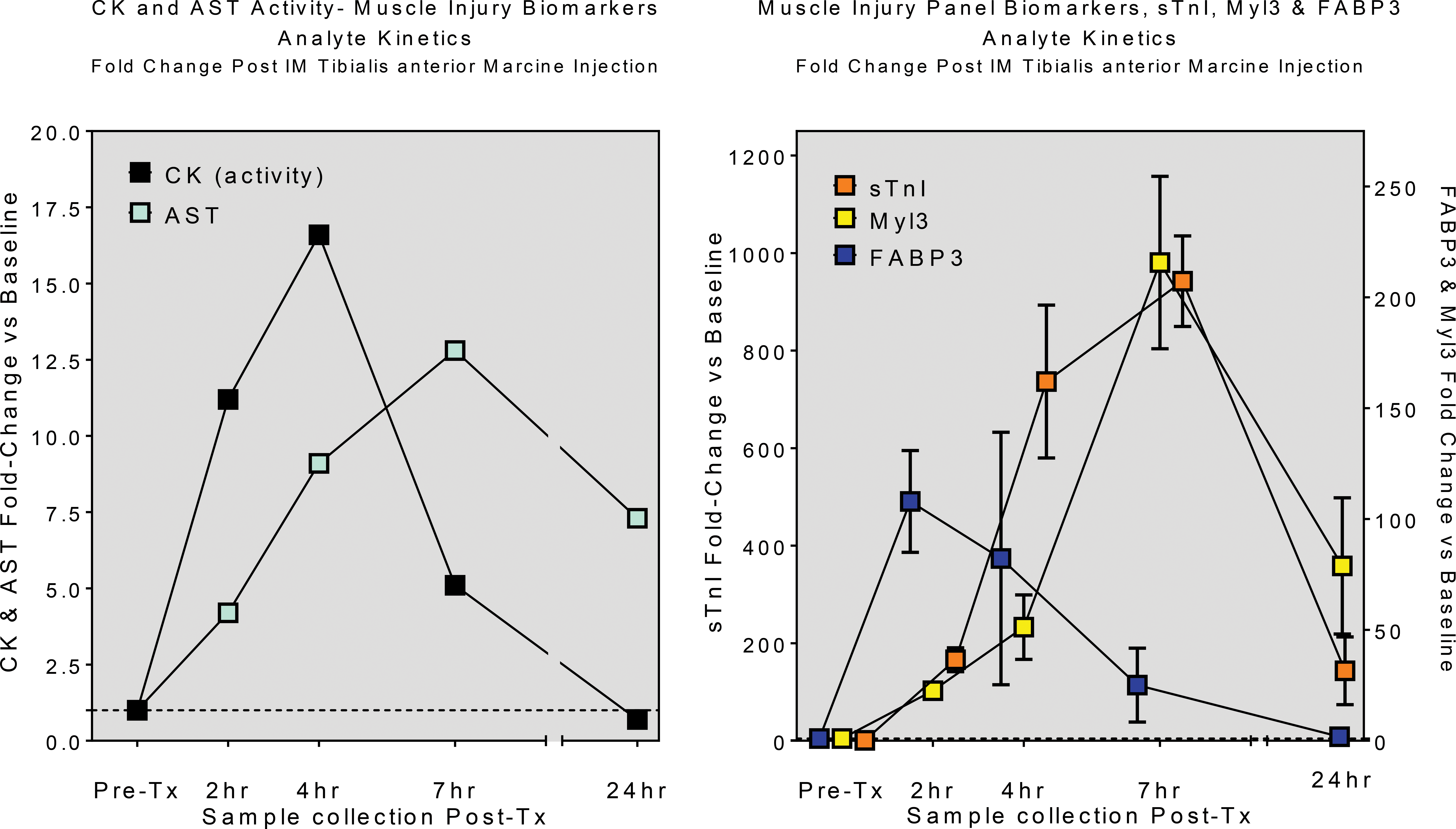
Established skeletal muscle (SKM) injury biomarkers, creatine kinase (CK) and aspartate transaminase (AST) activity, and muscle injury biomarker panel (MIP) biomarkers demonstrate similar release and clearance kinetics following acute intramuscular Marcaine-induced SKM injury. The MIP biomarkers demonstrate enhanced dynamic range with greater fold-change increases over baseline values than CK and AST suggesting improved sensitivity to detect SKM injury.
Based on fold-change differences from baseline concentrations, the MIP biomarkers demonstrated an enhanced dynamic range, which suggests improved sensitivity to detect SKM injury versus the CK and AST activity biomarkers. The MIP biomarkers had maximum fold-changes from baseline values for sTnI (942×), Myl3 (215×), and FABP3 (108×) versus the enzyme activity biomarkers, CK (16.6×) and AST (12.8×).
Example 2: CK Activity versus the MIP Biomarker, CK Mass Assay
The performance of the CKm assay versus CK activity assay was evaluated in serum samples collected from 2 dogs following isoproterenol-induced myocardial injury. Samples were collected pretreatment, 4, 7, 24, and 48 hr posttreatment. The analyte assay results displayed similar release and clearance kinetics. Peak values for both assays were observed 7 hr posttreatment. Peak CKm values increased 6.3× and 9.7× above baseline values, while CK activity changes were 2.2× and 3.1× above pretreatment values. The CKm assay’s large fold-change increases from baseline concentrations compared to the CK activity assay results demonstrate an enhanced dynamic range, which suggests an improved sensitivity to detect changes in CK enzyme concentrations.
Example 3: Sensitivity and Specificity of MIP Biomarkers to Detect Tetramethyl p-phenylenediamine (TMPD)-induced SKM Injury or Acetaminophen (APAP)-induced Liver Injury
An investigative rat study was run using animals treated with a single dose of the muscle-specific toxicant, TMPD at 1 mg/kg or 5 mg/kg or with APAP at a hepatotoxic dose (1,400 mg/kg). The study used 2 age-groups of SD rats: “young” 8 to 10 weeks old and “aged” 1-year-old rats. On study day 2, the main study group animals were sacrificed and blood and tissue samples were collected (n = 10 per group for aged rats, n = 5 per group for young rats). A subset of the 1-year-old rats were assigned to a recovery group (n = 5 rats per group) and were sacrificed on study day 9. No young rats were assigned to a recovery group.
The blood samples collected at necropsy via the vena cava were processed to serum and stored frozen at −80°C until assayed for the MIP biomarkers and an enzyme activity panel that included the analytes CK, AST, alanine aminotransferase, alkaline phosphatase, and glutamate dehydrogenase. Tissues collected at necropsy for microscopic examination included soleus and quadriceps muscles plus heart, liver, and kidney. Microscopic tissue injury was graded on a scale of 0–4 using the PSTC histopathology lexicon.
At study day 2, no microscopic SKM injury was noted in the 1 mg/kg TMPD-treated young rats. In the 1 mg/kg TMPD-treated aged rat group, grade 1 (minimal) SKM microscopic injury was present in 4 of the 10 rats. In the 5 mg/kg TMPD treatment groups, 5 of the 5 young rats and 8 of the 10 aged rats had grade 1 or grade 2 (mild) microscopic SKM injury. When microscopic SKM was present, the predominant finding was myocyte degeneration/ necrosis. At recovery, study day 9, 1 of the 5 aged rats in the 1 mg/kg TMPD group and 1 of the 5 aged rats in the 5 mg/kg TMPD group had microscopic SKM findings of grade 1 inflammation. No test article–related changes were observed in the liver, heart, and kidney in any of the TMPD-treated rats.
Liver injury was observed in all young and aged APAP-treated rats. Study day 2 liver microscopic findings ranged from minimal to moderate centrilobular hepatocyte necrosis and minimal to mild diffuse hepatocellular vacuolation. At study day 9, mild diffuse hepatocellular vacuolation was observed in 1 of 5 of the APAP-treated aged rats. No APAP-related changes were observed in the heart, kidney, soleus, and quadriceps muscles.
The sensitivity and specificity of the MIP biomarkers, CK, and AST assays to detect SKM or liver injury was based on agreement between the interpretation of the MIP or enzyme serum biomarker concentration, that is, the biomarker concentration being “elevated” or within normal limits, and the observed tissue microscopic findings, that is, tissue injury being present or absent.
The criteria used to interpret a serum biomarker concentration as elevated and an indicator of detected tissue injury were when the biomarker concentration was greater than the upper limit of normal value of this laboratory’s internally established, species-specific reference ranges.
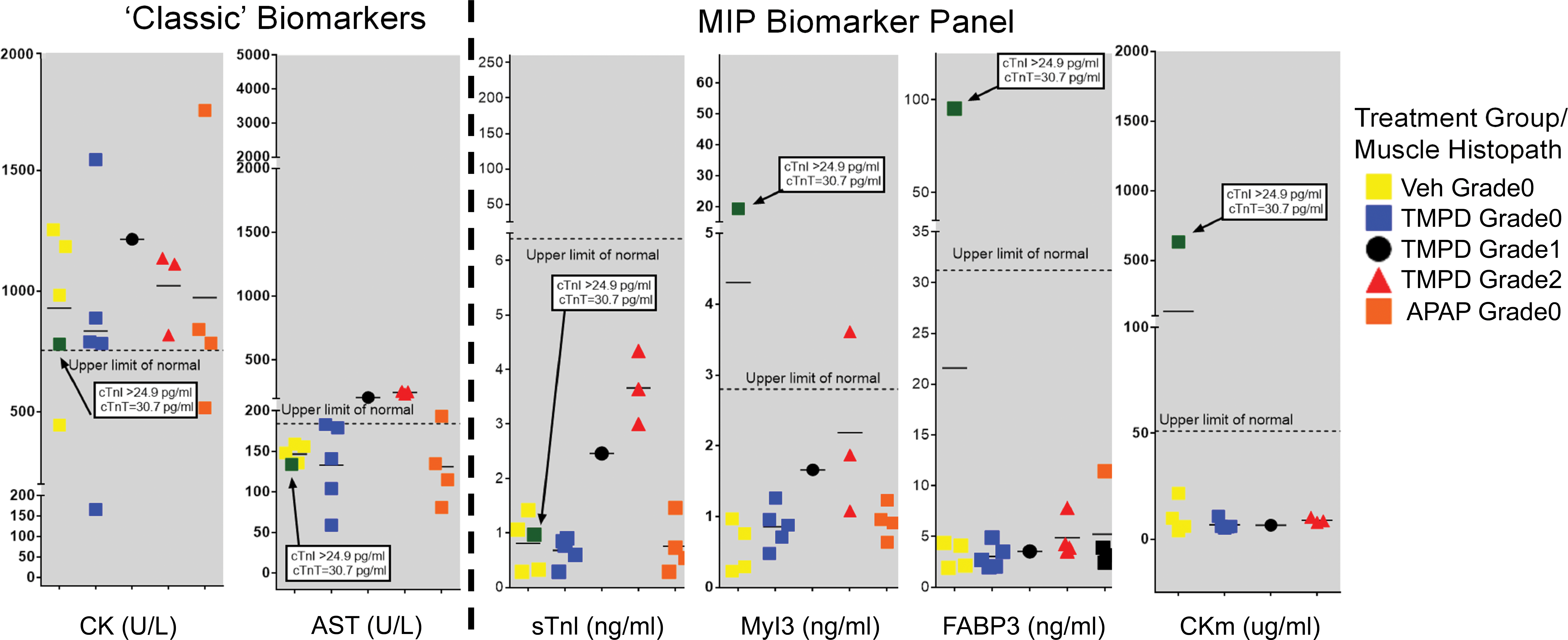
Study day 2 biomarker data from the combined young and aged rat groups with their SKM tissue microscopic findings are displayed in Figure 4. The CK activity results did not discriminate between vehicle treated, the TMPD-treated rats with or without histopathologic SKM injury, or the APAP-treated rats having microscopic liver injury, but no SKM injury. TMPD-treated rats without SKM injury were correctly identified by the AST activity assay with concentrations within normal limits in 7 of the 8 rats, and the MIP biomarkers, sTnI, Myl3, FABP3, and CKm were within normal limits and correctly identified 8 of the 8 rats. The TMPD-treated rats with SKM injury, both the AST activity assay and the MIP biomarker panel analytes correctly identified 5 of the 8 rats with grade 1 SKM injury, except FABP3 which identified 4 of the 8 rats with grade 1 SKM injury. AST and the MIP biomarkers detected 4 of the 4 rats with grade 2 TMPD-induced SKM injury.
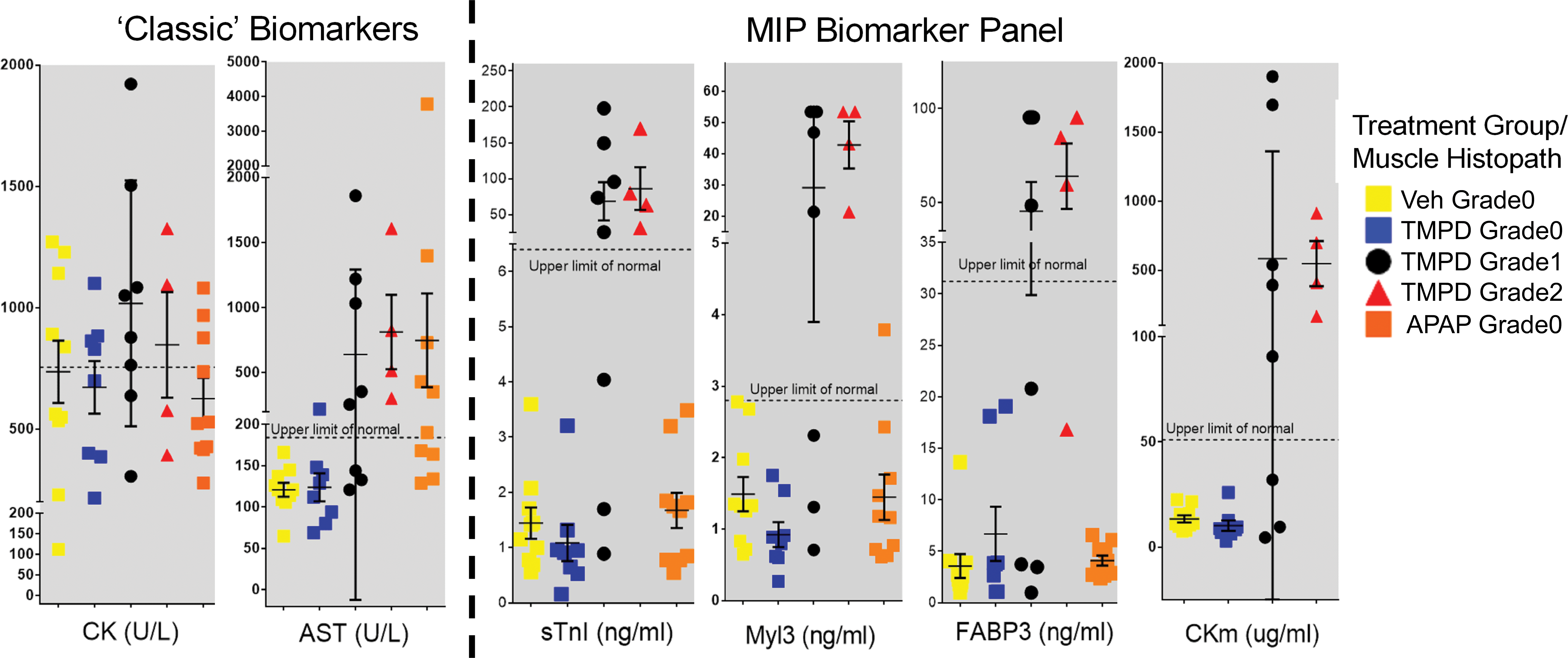
Study Day 2 skeletal muscle (SKM) biomarker data of combined “young” and “aged” rats with SKM histopathology findings following a single dose of tetramethyl p-phenylenediamine (TMPD) or acetaminophen (APAP). No significant differences in creatine kinase activity were observed between the treatment groups or rats based on SKM histopathology observations. Aspartate transaminase (AST) activity and the muscle injury biomarker panel (MIP) biomarkers perform similarly to identify SKM injury based on SKM histopathology observations. Elevated AST concentrations in TMPD-treated rats with SKM injury and APAP-treated rats with liver injury indicate the inability of AST to discriminate SKM and liver injury. The MIP biomarkers specifically detected SKM injury and were within normal reference range limits in the APAP- induced liver injury rats. The MIP biomarkers demonstrated larger fold-change increases than AST in rats with SKM injury suggesting enhanced sensitivity of the MIP biomarkers to detect SKM injury.
In rats with APAP-induced liver injury and no SKM injury, AST activity was elevated in 6 of the 10 rats. The MIP biomarkers were not elevated in any of the APAP-treated rats. These findings demonstrate the ability of the MIP biomarker analytes to discriminate between TMPD-induced SKM and APAP-induced liver injury.
Recovery, study day 9 MIP biomarker concentrations (see Figure 5) were within normal limits in both the TMPD- and APAP-treated rats, except for 1 vehicle-treated rat which had elevated Myl3, FABP3, and CKm results. Follow-up testing on the vehicle group rat with elevated Myl3, FABP3, and CKm values measured elevated cardiac troponin I (cTnI) concentrations, suggesting an unrelated myocardial injury, though no histopathologic myocardial finding was noted. Myl3, FABP3, and CKm proteins are abundant in both SKM and myocardial tissue. The observed MIP biomarkers, Myl3, FABP3, and CKm elevations are likely related to myocardial injury indicated by the cTnI elevation. The sTnI result on this cTnI positive sample was within normal limits, demonstrating the SKM tissue specificity of this analyte. The return of the recovery, study day 9 MIP biomarker concentrations to within normal limits suggests no ongoing tissue damage was occurring from the single-dose TMPD treatment.
The muscle injury biomarker panel biomarker in “aged” rats results collected at recovery study day 9 was within normal limits for both the tetramethyl p-phenylenediamine and acetaminophen-treated rats except for 1 vehicle-treated rat that had elevated Myosin Light Chain 3, Fatty Acid Binding Protein 3, and creatine kinase measured by a mass assay results. Follow-up cardiac troponin I testing on this Vehicle group rat measured elevated Cardiac Troponin I concentrations, suggesting an unrelated myocardial injury.
The 2 age-groups of rats utilized in this project, young and aged, illustrate the impact age has in the response to the SKM toxicant TMPD (see Figure 6). At study day 2, in the 1 mg/kg TMPD treatment groups, both young (n = 5) and aged (n = 10) rats, MIP biomarker concentrations are within normal limits. No abnormal SKM microscopic findings were observed in the 1 mg/kg TMPD-treated young rats and 4 of the 10 aged rats had grade 1 (minimal) myocyte degeneration/necrosis. In the 5 mg/kg TMPD young rat treatment group (n = 5), the MIP biomarkers had no or minimal increases in analyte concentrations and only 1 rat had an abnormal, grade 1 SKM microscopic finding of interstitial inflammation. In the 5 mg/kg TMPD aged rat treatment group (n = 10), the MIP biomarkers, sTnI, Myl3, and CKm were significantly elevated in 9 of the 10 rats while FABP3 was elevated in 7 of the 10 rats. Abnormal SKM microscopic findings were observed 8 of the 10 aged rats with MIP biomarker elevations. The 1 aged rat with MIP biomarker concentrations within normal limits had no abnormal SKM microscopic findings.
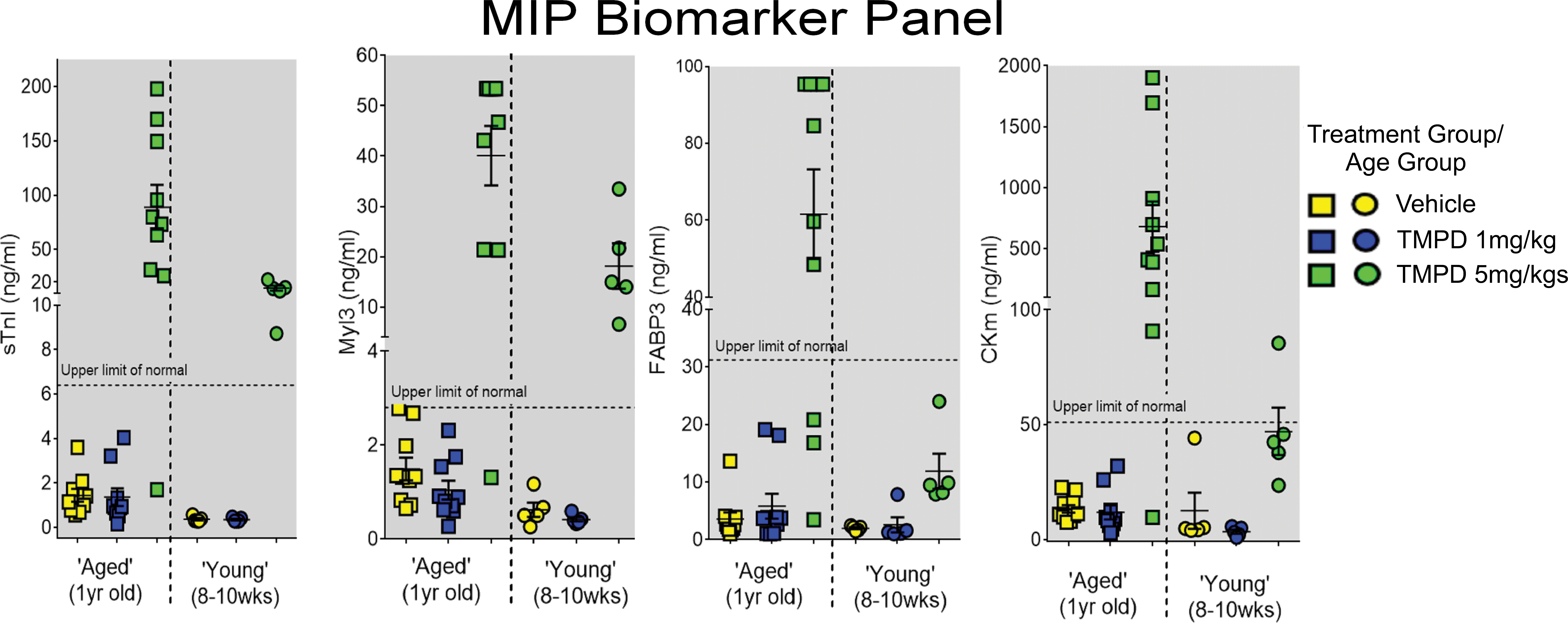
The impact of animal age on the response to skeletal muscle toxicant tetramethyl p-phenylenediamine (TMPD) is demonstrated by the muscle injury biomarker panel (MIP) biomarkers measured on study day 2. No elevations of the MIP biomarkers concentrations were observed in either the “young” or “aged” 1 mg/kg TMPD-treated rats. The 5 mg/kg TMPD “aged” rats treated had greatly elevated MIP biomarker concentrations while the 5 mg/kg TMPD “young” rats had no or minimal increases over the analytes upper limit of normal reference range values.
These MIP biomarker results demonstrate the potential utility of these assays for in-life monitoring for SKM injury. Added drug safety information may be gained by including the MIP biomarkers in projects where the potential for SKM injury exists and the project includes subjects with a range of ages and/or metabolic status.
The correlation between MIP biomarker response, microscopic SKM findings, and the TMPD dosing levels also suggests potential utility of the MIP biomarkers to support dose-escalation studies that could help define no-observed-adverse-effect-level concentrations.
In summary, results from this project using single-dose treatments of TMPD or APAP in young and aged rats demonstrate that the MIP biomarker panel analytes and the AST activity assay display similar performance in the ability to detect SKM injury. The MIP biomarkers demonstrated significantly larger fold-changes or a wider dynamic range over baseline concentrations than AST in subjects with histologically confirmed SKM injury. This observation may translate into improved sensitivity of the MIP biomarkers to detect SKM injury. The MIP biomarkers demonstrated improved specificity over AST by detecting SKM injury in only the TMPD-treated rats with histologically confirmed SKM injury, while AST concentrations were elevated in both the TMPD-treated rats with SKM injury and the APAP-induced liver injury animals.
MIP Biomarkers: Building the Foundation for Clinical Translation
A goal of biomarker qualification is the identification of tools that can be used to support drug safety and efficacy assessments. The FDA biomarker Letter of Support is a result of demonstrated preclinical utility of the MIP biomarkers as drug development tools. In their biomarker letter of support, the FDA encouraged the use of these biomarkers as exploratory translational tools to support clinical projects.
Consistent with the FDA recommendations, we designed projects to evaluate the ability to translate the MIP biomarkers to clinical drug development tools. A translational MIP biomarker project was designed that included serial serum samples collected from mdx mice, a model of Duchene muscular dystrophy (DMD) from 6 months to 14 months of age, and serum samples from DMD subjects ranging in age from 2 to 26 years. The MIP analyte data demonstrated similar trends in the mdx mice and DMD subjects with a negative correlation of decreasing biomarker concentrations associated with increasing subject age and disease progression (see Figure 7). Similar data were observed for sTnI and CKm (data not shown).
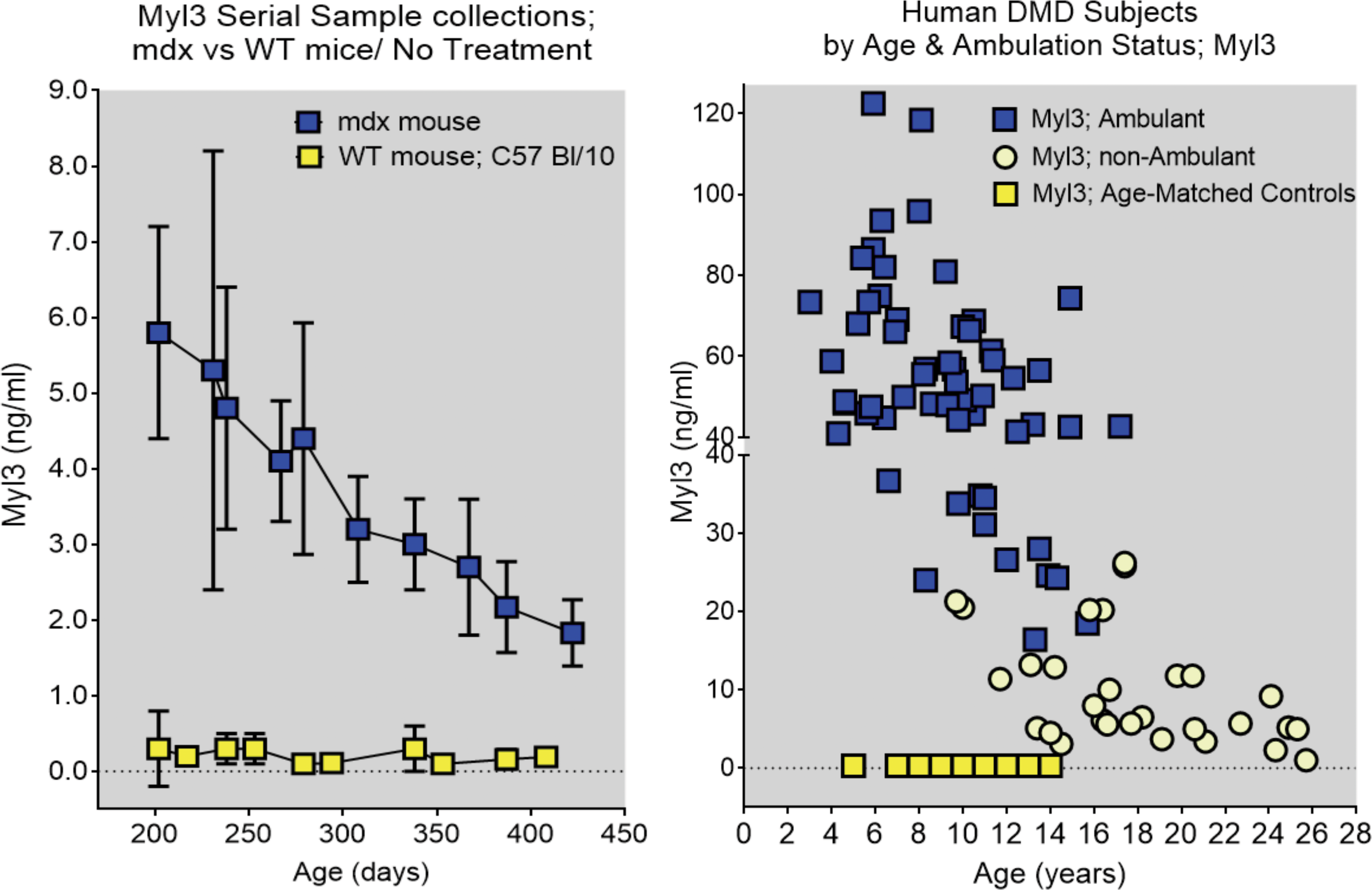
Muscle injury biomarker panel biomarker Myosin Light Chain 3 in Duchene muscular dystrophy subjects and mdx mice show similar decreases in serum biomarker concentrations with increasing age and ambulation status demonstrating utility as a translatable skeletal muscle biomarker. Similar data were observed for Skeletal Troponin I and creatine kinase measured by a mass assay (data not shown).
MIP biomarkers were measured in a project utilizing serum samples collected from healthy human volunteers and subjects with Duchene, Becker, or Limb-Girdle 2B muscular dystrophy. The MIP biomarker concentrations in the muscular dystrophy subjects showed strong negative correlations with subject age, having decreasing biomarker concentrations associated with increased subject age and disease progression. The MIP biomarkers had a positive correlation with the functional parameters of disease progression of ambulation, respiratory forced vital capacity, and cardiomyopathy status, with decreased biomarker concentrations being associated with decreased functional performance. (Burch et al. 2015).
In a separate project, the MIP biomarkers plus myostatin, a highly conserved negative regulator of SKM mass, were measured in a sample set from subjects having 7 different forms of muscular dystrophy. The MIP and myostatin biomarkers concentrations were similarly negatively correlated with subject age and had a positive correlation with functional performance parameters in each muscle disease group (Burch et al. 2017).
The MIP biomarkers are now being included as exploratory biomarkers in several clinical trial projects. The utility of this MIP biomarker data as SKM safety or efficacy end points to these therapeutic interventions remains to be determined at this time.
Summary
There is a clear need to identify and qualify translatable, sensitive, and specific SKM injury biomarkers to aid in the detection of drug-induced injury and the evaluation of therapeutics for muscular dystrophies and other muscle disorders. The FDA and EMA Letters of Support confirmed that the SKM injury biomarkers evaluated (sTnI, Myl3, FABP3, and CKm) outperform or add value to the conventional SKM injury biomarkers CK and AST in preclinical species.
A demonstrated clinical “COU” remains to be defined for the MIP biomarker panel analytes. Inclusion of the MIP biomarkers in preclinical and clinical projects should help to define their utility to assess SKM safety during drug development and the potential therapeutic effectiveness of treatments for inherited muscle diseases.
Footnotes
Author’s Note
All procedures performed on the animals were in accordance with regulations and established guidelines and were reviewed and approved by Pfizer Institutional Animal Care and Use Committee. Human studies were approved by the appropriate local ethics committees. All persons gave their informed consent prior to inclusion in the study, and all information about the participants was provided as anonymized data.
Acknowledgment
The author would like to thank Peter Burch (Pfizer, Summit Therapeutics) and Warren Glaab (Merck) for support in the generation and use of material presented in this article and would also like to acknowledge and thank the members of Predictive Safety Testing Consortium for their scientific, financial, and in-kind contributions that supported these research activities, as well as the input from Food and Drug Administration and European Medicines Agency scientists who serve as advisors.
Author’s Contribution
The author (RG) contributed to conception or design; data acquisition, analysis, or interpretation; drafting the manuscript; and critically revising the manuscript. The author gave final approval, and agreed to be accountable for all aspects of work in ensuring that questions relating to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This material is based upon work supported by Critical Path Institute’s Predictive Safety Testing Consortium.