
Abstract
Repair of bone and joint tissue to restore normal function is a unique endeavor that requires recreating tissue structure and the integrated healing of both organic and inorganic tissue components. Session 5 (Structural approaches to bone and joint repair) at the 36th annual Society of Toxicologic Pathology Annual Symposium included 2 talks covering methods, models, and regulatory considerations used to evaluate novel approaches for repairing bones and joints. Lyn Wancket provided a general overview of medical devices, with an emphasis on preclinical and clinical evaluations of bone and joint devices. Karen Manhart outlined regulatory review of medical devices by the Food and Drug Administration. This summary includes highlights from both talks.
This article represents a summary of content presented during 2 of the session 5 presentations of the 36th annual Society of Toxicologic Pathology Annual Symposium. Lyn Wancket presented “Evaluating Repair from the Ground (Substance) Up,” and Karen Manhart presented “Regulatory Considerations for Medical Devices.”
Defining Medical Devices
The U.S. Food and Drug Administration (FDA 2017) defines a medical device as “an instrument, apparatus, implement, machine, contrivance, implant, in vitro reagent, or other similar or related article, including a component part, or accessory which is recognized in the official National Formulary, or the United States Pharmacopoeia, or any supplement to them, intended for use in the diagnosis of disease or other conditions, or in the cure, mitigation, treatment, or prevention of disease, in man or other animals, or intended to affect the structure or any function of the body of man or other animals, and which does not achieve any of its primary intended purposes through chemical action within or on the body of man or other animals and which is not dependent upon being metabolized for the achievement of any of its primary intended purposes.”
Both speakers focused on the third category (affect the structure/function; primary intended purposes not achieved through chemical action or metabolism).
Bone and Joint Devices: Evaluating Repair from the Ground (Substance) Up (Lyn Wancket)
This portion of the session provided toxicologic pathologists with an overview of medical device preclinical studies. Emphasis was placed on the similarities and differences between evaluating medical devices and testing other classes of articles (e.g., small molecules, biologics). For all product types, the foremost concerns are evaluating safety and efficacy prior to exposing humans to the test article. Additionally, medical device studies are often performed in the same preclinical species used to evaluate other products. Extractables and leachables from medical devices are also tested for potential systemic effects, and these samples are dosed by the same routes as used for other articles (e.g., oral gavage, intravenous). Finally, all products have the potential for both local and systemic toxicity after article administration.
While there are many similarities between preclinical testing for medical devices and other products, there are also key differences to consider when evaluating studies with a medical device. The relative amount and timing of preclinical studies differ between types of articles (discussed in depth later). Additionally, since many devices are intended to be permanently implanted (e.g., total joint replacement), exposure is often more dependent on the length of time an individual will be exposed to the device than on a specific dose. Trauma at an implantation site is often unavoidable and requires toxicologic pathologists to differentiate tissue injury caused at the time of surgery from damage caused directly by the article. Finally, both material composition and shape/size play a role in how the body responds to a device.
Role of Composition and Shape/Size on the Host Response to Devices
Many medical devices have historically contained primarily or exclusively relatively inert materials such as stainless steel, titanium, and high-density polyethylene (commonly used as a negative control in preclinical device studies). For relatively inert materials with a smooth surface and solid structure, acute local tissue reactions are generally minimal, with a low cellular immune response and limited soft tissue reactions. Similarly, late stage healing is often limited to encapsulation by a thin fibrous tissue capsule and fat ingrowth with minimal to no active inflammation. Additionally, relatively inert materials are less likely to induce systemic effects because they are resistant to degradation or fragmentation (unless mechanically loaded).
In contrast to relatively inert materials, complex/nonuniform components such as xenogeneic materials (tissues or cells from a nonhost species), allografts, and absorbable articles are increasingly common and often have a higher potential for more widespread and severe host effects. At a local implantation site, acute foreign body responses are a common and expected response to xenogeneic and/or degradable materials (Anderson 2001). These factors lead to a higher potential for systemic effects if leachables or degradation fragments are released from the primary implant site.
In addition to material composition, the shape and size of medical devices influence the interaction with host tissue. The interface between the article and tissue can be smooth and regular (e.g., metals, plastics), which generally incites a minimal response (few immune cells and minimal soft tissue encapsulation). In contrast, irregular surface (e.g., collagen, braided suture) can incite tissue ingrowth and potentially a foreign body reaction. The surface area to volume ratio can also alter the host–device interaction, since an article with a large surface area will have more total tissue contact and may be more likely to incite a foreign body response (Anderson 2001). Finally, since larger articles can cause increased host tissue trauma at the time of implantation, a bigger device may increase the host reaction to the article independently of the underlying material composition.
Specific Considerations for Bone and Joint Devices
Bone and joint devices are often primarily designed to restore movement (e.g., joint replacements), provide structural support (either temporarily or permanently), or aid in tissue reconstruction (e.g., craniofacial repair after a traumatic injury). A device designed to provide structural support and/or restore movement often requires the formation of a host/device interface. The interaction between device and host tissue is often assessed with bone-specific evaluations of the microarchitecture. Potential interactions include osteoinduction (article stimulates proliferation/differentiation of stem cells to osteoprogenitor cells), osteoconduction (bone ingrowth along surfaces of an implant, e.g., scaffold effect), and osteointegration (direct structural–functional connection between bone and load-bearing implant).
A variety of methods can be used to either reduce or enhance the strength of a bone/article interface. If an article is intended to be removed at a later date (e.g., intramedullary pin, temporary bone pate), reduced host/device integration can be achieved by using inert materials (e.g., metals, plastics) or specific surface coatings to limit tissue ingrowth. In contrast, if strong binding between tissue and article is required (e.g., joint replacement), tissue/device interactions can be enhanced by changing surface chemistry or increasing the surface porosity of the device.
Considerations for Designing, Performing, and Evaluating Bone/Joint Device Preclinical Studies
There are physical, biological, and practical factors to consider when selecting a preclinical species for bone/joint device studies (reviewed in Wancket’s [2015] study). Physical factors include identifying anatomic sites in the model with similar composition (cortical vs. cancellous bone) to the intended site in humans. Species have different potentials for remodeling bone (e.g., very limited to absent in rodents). Additionally, there can be wide variability in macroscopic shape and microarchitecture (illustrated in Muschler et al.’s [2010] study). Biological factors to consider when selecting a model include age (which influences the growth plate) and the potential to overestimate device performance (when using a young animal with rapid bone growth to model a clinical condition in elderly humans). Practical considerations include selecting modes that are both suitable and cost effective (profit potential for devices is low compared with other treatments such as biologics) and availability/ease of handling (e.g., challenges associated with preventing fractures in large animals during initial healing postsurgery).
Once a species is selected, different types of studies are used to evaluate specific attributes of a device. Three general classes of studies include (1) assessing de novo bone formation (osteoinduction assays), (2) evaluating local and systemic tissue injury (biocompatibility; the ability of a material to perform with an acceptable host response in a specific application), and (3) testing device performance (functional studies that determine whether a device performs as intended in a model that simulates the human clinical condition). In some cases, one model may be used to assess multiple attributes of an individual device.
Osteoinduction studies are used to determine whether a device has intrinsic qualities that induce new host bone. Material is typically implanted in heterotopic sites (e.g., skeletal muscle) to ensure that all new bone formed is due to the presence of the device. Since many articles tested for osteoinduction are xenogeneic (e.g., human cadaver bone), studies are typically performed in athymic rodents.
Biocompatibility studies evaluate the potential for a device to induce injury locally or systemically. Local reactions are assessed by implanting the device in the site of intended clinical use. Sites that are commonly used include the femoral diaphysis (cortical bone), femoral/tibia epiphysis (cancellous bone), or skull and vertebral column (mixed/atypical bone). If a device is expected to degrade or may produce wear debris when loaded, draining lymph nodes may also be assessed. Adjacent tissues that may be compressed by bone growth or remodeling (e.g., bone marrow and central nervous system tissues) should also be examined. Local implant studies are performed in species that have bones large enough to support human-sized implants (rabbits, dogs, small ruminants, and pigs). Systemic reactions are often assessed after subcutaneous (rats, rabbits) or osseous (rabbits) implantation.
Functional assays are used to test whether a device performs as intended. If the amount of article implanted can be scaled (e.g., bone void filler), multiple species can be used. For devices that are sized for adult humans (e.g., hip replacement), species selection is generally limited to dogs, pigs, and small ruminants. Clinical conditions that are commonly modeled include fracture (long bone, vertebral, and craniofacial), tendon repair, infection, wear debris, defect filling, cartilage degeneration, vertebral fusion, and critical size defects.
Auxiliary Evaluation Methods and Clinical Complications
While describing all available auxiliary methods to evaluate medical devices was outside the scope of the session, a brief overview of techniques that complement histologic evaluations was provided. Methods include macroscopic observations, advanced imaging, mechanical testing, and histomorphometry. Additionally, device composition and size can limit the number of options available for histological processing, as can evaluations that require bone samples to remain mineralized (e.g., some fluorescent markers that bind to mineral). Finally, common clinical complications associated with bone/joint devices were summarized, including infection, mechanical failure, wear debris, granulomatous inflammation (Anderson 2001), and the potential for neoplasia (solid state carcinogenesis).
Regulatory Considerations for Medical Devices (Karen Manhart)
This summary was prepared by Lyn Wancket from the material presented at the symposium. While the speaker is an FDA employee, the author has no affiliation with the FDA, and this summary is not an official statement from the FDA.
FDA Organization
As reviewed in the session by Karen Manhart, the FDA’s core mission is to protect and promote public health by regulating foods, veterinary medicine, consumer products, drugs, biologics, medical devices, and tobacco. The Center for Devices and Radiologic Health (CDRH) is one of the 6 FDA branches and focuses on approval and/or clearance of medical devices. At the time of the presentation, the CDRH consisted of 8 offices including 2 that perform premarket review (Office of Device Evaluation, Office of In Vitro Diagnostics and Radiological Health). Divisions within the Office of Device Evaluation are grouped by clinical specialty or system (e.g., general surgery, respiratory, and orthopedics). Each division is subdivided into branches that review specific device types with teams of interdisciplinary specialists. Each team member reviews the data in the applications related to their area of expertise. Team members provide their reviews and recommendations to the lead reviewer who works with management to coordinate communication with the sponsors.
While products that exert their effects strictly though a physical mode of action are reviewed entirely by CDRH, the continuing growth of combination product applications can require involvement by multiple FDA centers (Table 1). Combination products are evaluated by a primary center with appropriate cross center consultations. To determine which FDA center will conduct the primary review and be responsible for ongoing regulation, both the mode of action and the intended use of a device are considered. Products that include drugs (mode of action is based on chemical interactions) are reviewed by the Center for Drug Evaluation and Research. Products of a biological origin (e.g., tissue, cells, genetic material) include review by the Center for Biologics Evaluation and Research. While all centers focus on assessing safety, effectiveness, and device ability to appropriately deliver the product to the targeted site, there are different application types and data requirements for each center. Therefore, the assignment of the lead center will impact the review process.
Summary of Combination Devices and Ways That Components May Be Combined in Combination Devices.

Device Classes
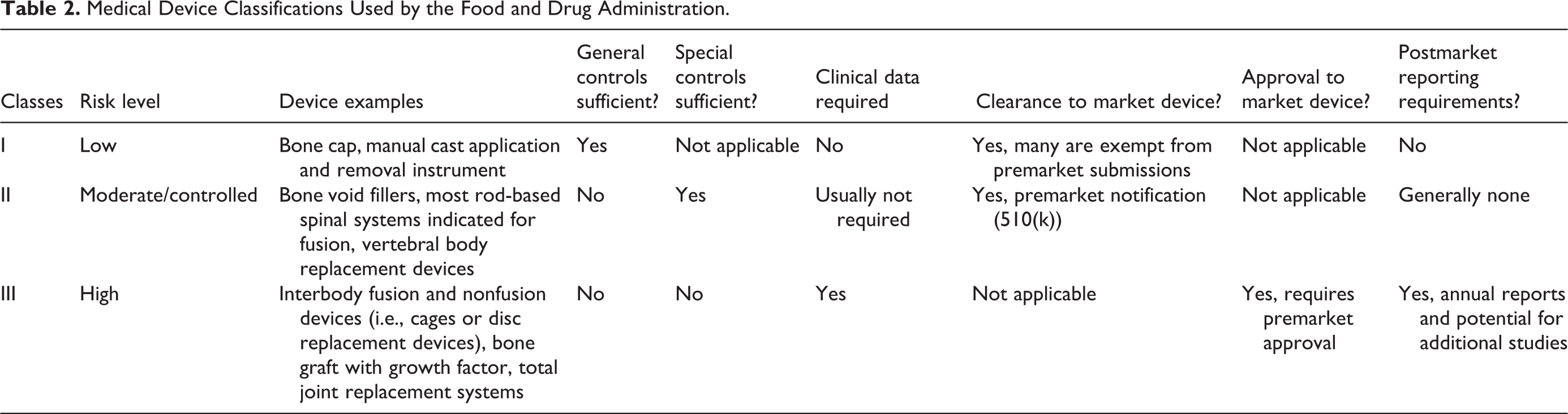
The Medical Device Amendments of 1976 to the Federal Food Drug and Cosmetic Act established the current FDA policies regarding medical device approval. This framework uses a 3-tiered risk-based classification system. Classifications for approximately 1,700 different generic types of devices exist, which are grouped into 16 medical specialties referred to as panels. Each of these generic types of devices is assigned to one of 3 regulatory classes based on the level of control necessary to assure the safety and effectiveness of the device. Class I is considered as low risk, class II as moderate to high, and class III as the highest risk. All novel devices are automatically considered to be class III (highest level of risk). If controls are established to provide a reasonable assurance of safety and effectiveness based on risk, devices can be reassigned to class I or class II. Details about each class are summarized below and in Table 2.
Medical Device Classifications Used by the Food and Drug Administration.
Class I
General controls (Good Manufacturing Practices, reporting, labeling, registration, and listing) are deemed sufficient to assure the safety and effectiveness of class I devices. These devices are exempt from premarket review and are highly unlikely to present a safety risk.
Class II
General controls alone are not sufficient to provide reasonable assurance of the safety and effectiveness of class II devices. However, there is sufficient information to establish special controls to assure safety and effectiveness. Manufacturers are typically required to demonstrate basic similarities between a new device and a predicate device and typically provide descriptive information (e.g., comparison of specifications, materials, and technology). In contrast, FDA generally evaluates differences between the new device and the predicate device to determine their effect on safety and effectiveness (where the FDA has previously determined that the approved predicate device has reasonable safety and effectiveness).
Class II devices require a premarket notification submission (510(k) application). The FDA has 90 calendar days to complete the review (after the application is officially accepted into the data center). If deficiencies are identified within the first 60 days of review and those deficiencies require additional information and/or data, the file is put on administrative hold and an additional information deficiency letter is issued. The sponsor then has 180 calendar days to adequately address the deficiencies and provide a response (which can include data from additional preclinical studies). Once the sponsor response is received, the FDA has an additional 30 calendar days to complete the review and assign either a substantially equivalent or not substantially equivalent status. If a decision of substantial equivalency is made, the device is cleared and the sponsor can legally market the device in the United States. A new 510(k) application is required each time a device design change is made or there is a proposed change in the indications for use.
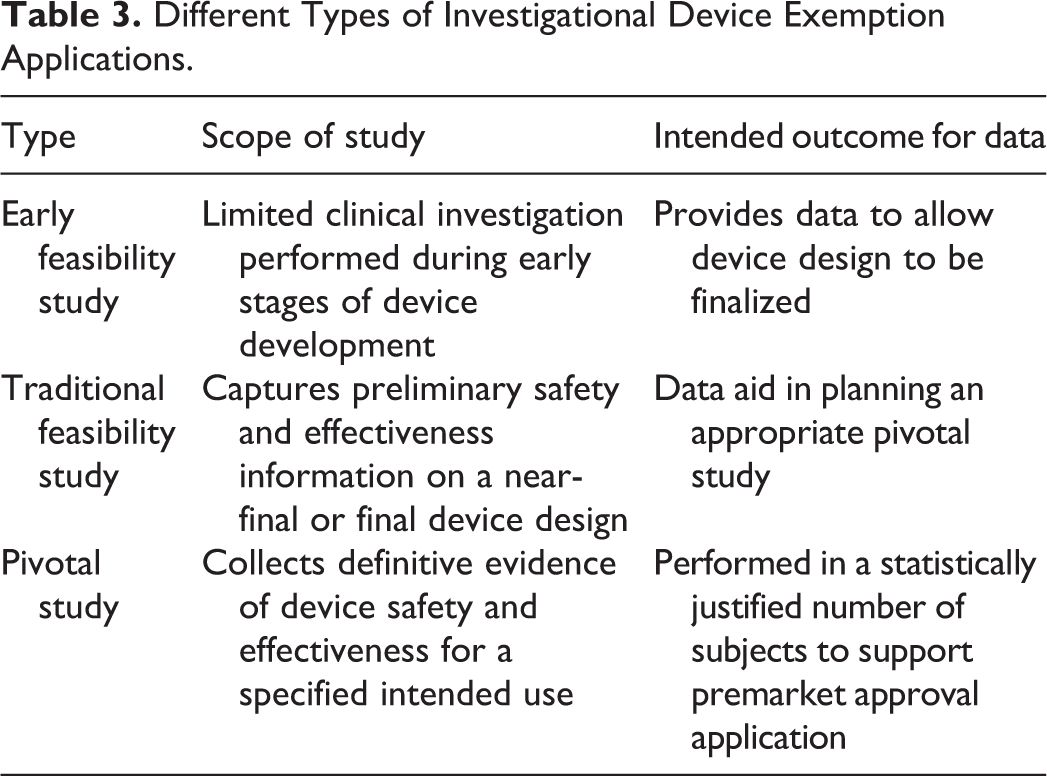
Investigational Device Exemption (IDE) Application
For a subset of class II and III devices, clinical trial data are required for device approval. An IDE application is required to perform a clinical trial in the United States (different types outlined in Table 3). The data included in an IDE are primarily focused on evidence that subject safety will be protected. Preclinical data often include engineering, biocompatibility, Shelf Life packaging, electrical/MRI, and labeling. Preclinical Good Laboratory Practice (GLP) Animal Studies (both acute and chronic) can also play a key role in supporting an IDE application. The clinical trial component includes a detailed investigational plan with list of investigators and institutional review boards (IRBs) as well as informed consent and the statistical plan. FDA has 30 calendar days to review an IDE and provide an approval, conditional approval, or disapproval decision.
Different Types of Investigational Device Exemption Applications.
Class III
General and special controls alone are insufficient to assure the safety and effectiveness of class III devices. Since a class III device “supports or sustains human life or is of substantial importance in preventing impairment of human health or presents a potential, unreasonable risk of illness or injury,” the FDA requires scientific evidence independent of comparisons to a similar or predicate device. To obtain marketing clearance, class III devices require a premarket approval (PMA) application under section 515 of the Federal Food, Drug, and Cosmetic Act. A PMA must show an independent demonstration of the new device’s safety and effectiveness and requires the highest level of data including a clinical trial. The clinical trial provides clinical data to provide safety and effectiveness data for a PMA marketing application. The FDA has 180 days to review a PMA from the day of filing. The data included are primarily focused on marketing and use of the device (preclinical animal data are less common unless a change was made in the device design). New technology or a device with safety or efficacy concerns may go to a panel of outside experts to weigh in on approval, with the FDA having the final decision. PMAs are either approved or deemed not approvable. PMA includes the requirement for annual reports that contain postmarket data for as long as the device is marketed and may require postmarketing studies.
Other Approval Routes
The FDA also provides mechanisms for designating devices for compassionate of humanitarian uses. Investigational devices that are not yet proven to be safe and effective can be designated for compassionate use if they are intended to treat life-threatening or serious disease that have no alternative treatment, and there is no time to obtain FDA approval. The FDA has 10 days to review a request, and the sponsor must provide follow-up information within 45 days of the implantation procedure.
A device designated for Humanitarian Use Device (HUD) is intended to treat or diagnose a disease or condition that affects/is manifested in fewer than 8,000 individuals in the United States annually. A Humanitarian Device Exemption (HDE) application must contain sufficient information that the device does not pose an unreasonable/significant risk of illness or injury and that the probable benefits outweigh the risk of injury or illness from its use. The applicant must also demonstrate that no comparable devices are available to treat/diagnose the disease/condition and cannot charge in excess of the costs of research, development, fabrication, and distribution. HDE approval authorizes marketing of an HUD but limits its use to facilities with an established local IRB that has approved the use of the device to treat/diagnose the disease and is able to provide appropriate clinical oversight.
CDRH also has a presubmission program that allows an applicant to obtain agency feedback prior to their regulatory submission. This can include review of animal study protocols by a pathologist or other scientists prior to the initiation of the study to make sure that the types and amount of data that are collected will be adequate to address regulatory requirements. FDA’s goal for providing nonbinding feedback is 75 to 90 days after receipt and is based on the information available at that time (feedback may be modified, if important new issues materially relevant to a determination of safety or effectiveness emerge). Presubmissions can also discuss reasons for disapproval and strategies that can be used to address deficiencies.
Additional Considerations for Preclinical Animal Studies for Bone/Joint Devices
Multiple FDA guidance documents are available by searching for device-specific, file type–specific, GLP related, or animal study related. In addition to FDA regulations, a wide variety of guidances are published that provide suggestions on designing bone and joint studies. One useful resource is the 10993-6 guidance published by the International Organization for Standardization (2016) that provides specific study designs and factors to consider when evaluating local tissue effects after implantation of a medical device. While the provided test methods are detailed, individual protocols should be tailored to the specific device. As always, it is important to consult the federal registry or to contact an FDA employee to determine whether specific guidances are recognized by the agency.
Footnotes
Acknowledgments
The author would like to thank Karen Manhart for presenting.
Author Contributions
The author (LW) contributed to conception or design, data acquisition, analysis, or interpretation; drafting the manuscript; and critically revising the manuscript. All authors gave final approval and agreed to be accountable for all aspects of work in ensuring that questions relating to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.