
Abstract
Osteoporosis increases fracture risk, a cause of crippling morbidity and mortality. The immunoskeletal interface (ISI) is a centralization of cell and cytokine effectors shared between skeletal and immune systems. Consequently, the immune system mediates powerful effects on bone turnover. Physiologically, B cells secrete osteoprotegerin (OPG), a potent anti-osteoclastogenic factor that preserves bone mass. However, activated T cells and B cells secrete pro-osteoclastogenic factors including receptor activator of Nuclear factor-kappaB (NF-kB) ligand (RANKL), Interleukin (IL)-17A, and tumor necrosis factor (TNF)-α promoting bone loss in inflammatory states such as rheumatoid arthritis. Recently, ISI disruption has been linked to osteoporosis in human immunodeficiency virus (HIV) infection/acquired immunodeficiency syndrome (AIDS), where elevated B cell RANKL and diminished OPG drive bone resorption. HIV-antiretroviral therapy paradoxically intensifies bone loss during disease reversal, as immune reconstitution produces osteoclastogenic cytokines. Interestingly, in estrogen deficiency, activated T cells secrete RANKL, TNF, and IL-17A that amplify bone resorption and contribute to postmenopausal osteoporosis. T cell–produced TNF and IL-17A further contribute to bone loss in hyperparathyroidism, while T cell production of the anabolic Wingless integration site (Wnt) ligand, Wnt10b, promotes bone formation in response to anabolic parathyroid hormone and the immunomodulatory costimulation inhibitor cytotoxic T lymphocyte–associated protein-4-IgG (abatacept). These findings provide a window into the workings of the ISI and suggest novel targets for future therapeutic interventions to reduce fracture risk.
Osteoporosis (Latin for porous bone) is a common malady of the skeleton and ensues when the rate of osteoclastic bone resorption outpaces the rate of osteoblastic bone formation, conditions that predispose to bone loss. From a clinical perspective, osteoporosis is most commonly defined as a bone mineral density (BMD) T score of less than or equal to −2.5 standard deviations from a young adult reference range, following a bone densitometry scan using dual-energy X-ray absorptiometry (DXA; Unnanuntana et al. 2010). From a biological standpoint and at the molecular level, osteoporotic bone is characterized by a loss of BMD and structural integrity, leading to a conformation with diminished load-bearing capacity and with an increased propensity to fracture.
The consequences of fragility fractures can be devastating and are commonly associated with crippling disability that necessitates intensive rehabilitation in as many as 75% of cases. In the case of hip fractures, mortality rates of 24% to 33% are common in the first year following a fracture (Bass et al. 2007; Johnell and Kanis 2006; Lewis et al. 2006). The associated loss of independence during and often beyond, the fracture rehabilitation period, can lead to a marked decline in quality of life especially in aged fracture victims (Kates, Kates, and Mendelson 2007; Johnell and Kanis 2006). It is estimated that almost 50% of women and 30% of men over the age of 50 will suffer a fragility fracture due to osteoporosis (Eisman et al. 2012), and the worldwide incidence of fracture is expected to rise to 6 million by 2050 (Cummings and Melton 2002) with costs projected to reach US$25 billion in the United States alone by 2025 (Burge et al. 2007).
An interesting and unexpected finding that has emerged in the field of bone biology is the repurposing by the skeleton of key immune elements including adaptive and innate immune cells and key immune-derived cytokines. As a result, physiological functions of the adaptive immune system beneficially regulate the skeleton. However, under states of pathologic immune dysfunction such as immunodeficiency or inflammatory responses to infection/disease, the skeleton is at the mercy of the immune response and serious collateral damage to bone may result leading to osteoporosis and an elevated risk for bone fracture (Weitzmann and Ofotokun 2016; Weitzmann and Pacifici 2006). Although bone loss is a natural consequence of aging, it is exacerbated by many common pathological conditions that afflict many people. This includes inflammatory diseases such as rheumatoid arthritis (RA), periodontal infection, and inflammatory bowel diseases including Crones disease. The immunodeficiency state resulting from HIV infection further promotes bone resorption. Another key protagonist of exacerbated skeletal decline is the result of estrogen decline following the menopause (postmenopausal osteoporosis). These bone loss states are all characterized by an increase in osteoclastic bone resorption relative to osteoblastic bone formation, leading to net bone loss (Weitzmann and Ofotokun 2016; Weitzmann and Pacifici 2006).
Although it has long been recognized that osteoclast precursors derive from the monocyte/macrophage lineage (Walker 1972, 1975; Buring 1975), cells of immune origin, in the past 2 decades, the new field of “osteoimmunology” has blossomed, revealing a deeply integrated immunoskeletal interface (ISI) in which immune cells direct physiological and pathological bone resorption and bone formation (Weitzmann and Ofotokun 2016; Weitzmann and Pacifici 2006).
Although many of the concepts described below have been uncovered using animal models, predominantly mice and rats, tantalizing evidence is beginning to emerge to support a role for the immune response in human bone diseases. It should be pointed out, however, that osteoimmunology does not replace traditional bone biology. In most cases, the ISI simply adds a new layer to the underlying principles already elucidated.
The following review outlines some of the past and ongoing investigations by our group and others into the role of the ISI in the regulation of the skeleton.
Osteoclasts and the Receptor Activator of Nuclear factor-kappaB (NF-kB) (RANK)/Receptor Activator of NF-kB Ligand (RANKL)/Osteoprotegerin (OPG) System
Although osteoclasts are the sole bone resorbing cells of the body and increased osteoclastic bone resorption underlies bone loss and osteoporosis development, it is only comparatively recently that the process underlying osteoclastogenesis has been unveiled. It has long been recognized that osteoclast precursors are significantly more frequent in inflammatory conditions in both humans and animals, particularly when inflammatory cytokines are elevated (Pacifici et al. 1991; Kitazawa et al. 1994; Kimble et al. 1996). However, adding inflammatory cytokines to osteoclast precursors is under most circumstances ineffective in promoting osteoclast formation suggesting an alternative mechanism.
In 1997 and 1998, a series of key discoveries unveiled a previously unknown pathway for stimulating osteoclast formation (Simonet et al. 1997). It is now evident that what differentiates an osteoclast precursor from a regular monocyte is expression on its surface of a specific molecule, RANK. RANK is the receptor for the key osteoclastogenic cytokine RANK ligand (RANKL; Simonet et al. 1997; Anderson et al. 1997; Wong et al. 1997), which upon binding to RANK in the presence of the trophic factor, macrophage colony-stimulating factor, causes the osteoclast precursor to differentiate into a preosteoclast (Lacey et al. 1998). These preosteoclasts further fuse together to form giant multinucleated bone resorbing mature osteoclasts. As with many biological pathways, there are checks and balances, and in this pathway, this takes the form of a soluble RANKL decoy receptor called osteoprotegerin (OPG; Lacey et al. 1998; Tsuda et al. 1997). OPG binds to RANKL and prevents it from associating with RANK, thus reducing the rate of osteoclast differentiation and bone resorption. As RANKL and OPG are the final downstream effectors of osteoclastogenesis, the ratio of RANKL to that of OPG in the bone marrow microenvironment is a key determinant of the rate of osteoclastic bone resorption occurring in humans and animals (Teitelbaum 2000). This pathway is summarized diagrammatically in Figure 1.
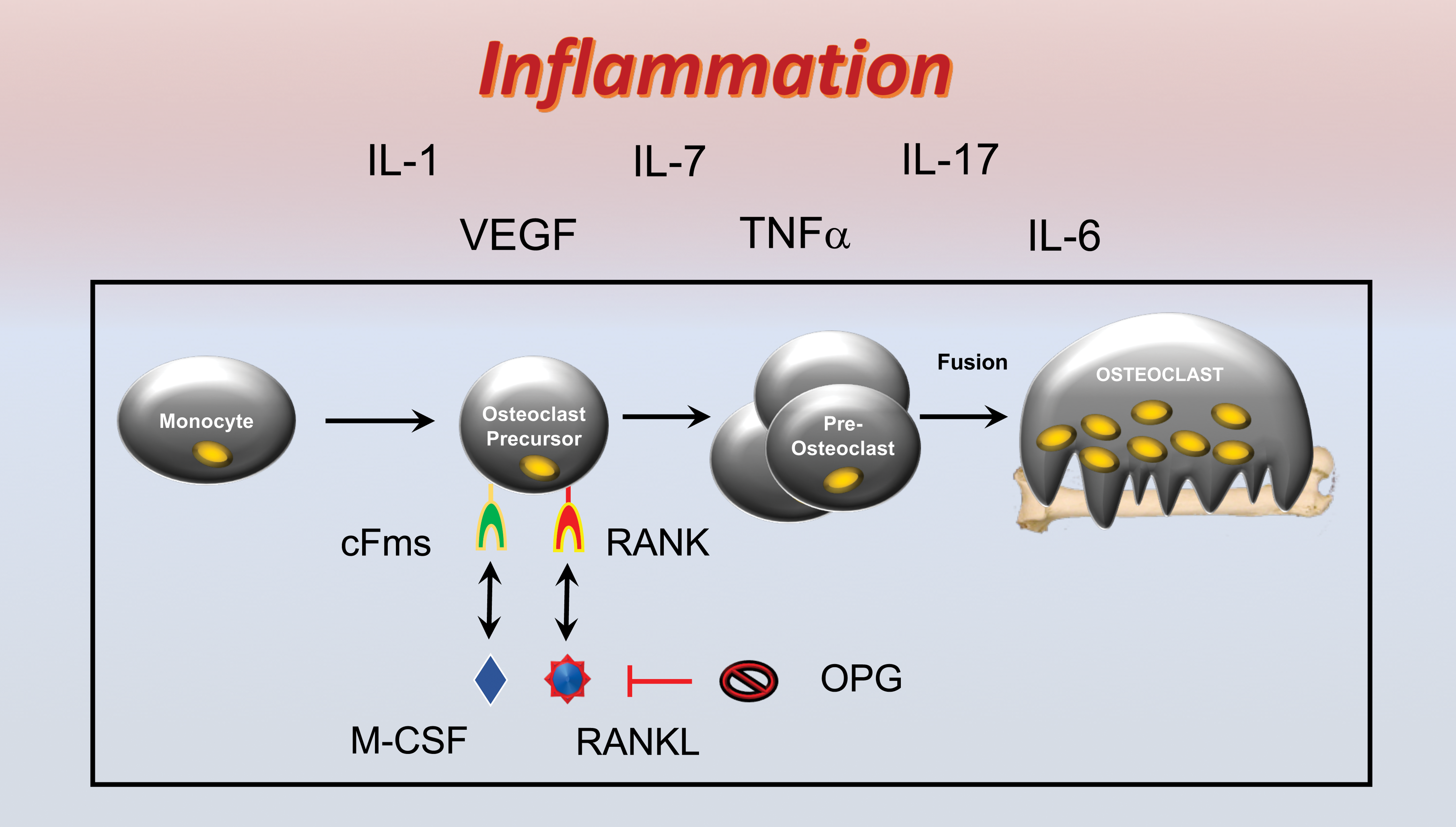
Osteoclastogenesis and the receptor activator of NF-kB (RANK)/receptor activator of NF-kB ligand (RANKL)/osteoprotegerin (OPG) axis. Osteoclast precursors derive from the monocyte lineage and express the RANK that is a receptor for the key osteoclastogenic cytokine RANKL. RANKL, in the presence of permissive concentrations of the tropic factor, macrophage colony-stimulating factor (M-CSF), causes differentiation of osteoclast precursors into preosteoclasts that fuse together to form mature multinucleated bone resorbing osteoclasts. RANKL activity is moderated by a soluble decoy receptor OPG. Inflammatory cytokines promote osteoclastogenesis and bone resorption by stimulating RANK and/or increasing production of RANKL and/or suppressing expression of OPG.
It is further recognized that the role of inflammatory cytokines in promoting osteoclastogenesis is indirect and mediated through the regulation of the RANK/RANKL/OPG axis. For example, tumor necrosis factor alpha (TNF), a potent inflammatory and indeed osteoclastogenic cytokine, drives up bone resorption by promoting RANK expression on monocytes thus converting them into osteoclast precursors, upregulates RANKL expression by osteoblast lineage cells (Hofbauer et al. 1999), and down modulates osteoblastic production of OPG. Although other inflammatory cytokines such as IL-1, IL-6, IL-7, and Interleukin (IL)-17A also upregulate osteoclastogenesis by regulating RANK, RANKL, and/or OPG production, TNF mediates 1 other unique function making it an extremely potent osteoclastogenic factor. TNF synergizes with RANKL at the signal transduction level to further intensify osteoclastogenesis and bone resorption (Cenci et al. 2000; Lam et al. 2000; Zhang et al. 2001; Fuller et al. 2002).
The ISI
Although long recognized that osteoclasts derive from cells of the monocyte lineage (immune cells), an unexpected finding was the existence of a deeply rooted nexus between the immune and skeletal systems, the ISI. This is the result of a centralization of multiple cells of the adaptive immune response and their cytokine effectors, which serve dual functions in the regulation of the skeleton. One key aspect of this involves activated lymphocytes, both T- and B cells, which secrete RANKL and TNF under inflammatory conditions, driving up osteoclast formation and bone resorption in inflammatory states including the autoimmune disease RA, inflammatory bowel diseases including Crohn’s disease, and periodontal infection. Consequently, these conditions are all associated with bone loss (Weitzmann and Ofotokun 2016).
In contrast to these inflammatory states, under physiological conditions, B cells are a significant source of the osteoclast inhibitor OPG. Early studies recognized that human tonsil B cells secrete OPG and that in vitro activation of cluster of differentiation (CD) 40 costimulatory pathway on B cells further upregulates OPG production (Yun et al. 1998). We later ratified these data in the murine system where we demonstrated that in mouse bone marrow, the B cell lineage contributed 64% of the total OPG production (Y. Li, Toraldo, et al. 2007). Consistent with these data, B cell knockout (KO) mice were found to have a bone marrow deficit in OPG, an enhanced rate of osteoclastic bone resorption, and significantly diminished BMD and bone mass. Reconstitution of B cells into young B cell null mice rescued these alterations to the ISI and prevented bone loss (Y. Li, Toraldo, et al. 2007).
As CD40 on B cells functions as a receptor for CD40 ligand (CD40L) expressed predominantly by activated T cells, we further investigated a role for T cell CD40/CD40L costimulation and found that deletion of CD40 or CD40L or of T cells also led to diminished B cell OPG production and bone loss (Y. Li, Toraldo, et al. 2007).
Taken together, these data demonstrate that under inflammatory conditions B cells and T cells damage the skeleton though copious secretion of RANKL and inflammatory cytokines such as TNF. By contrast, under basal conditions, B cells, regulated by costimulatory actions through T cells, protect the skeleton by promoting production of OPG. This pathway is summarized diagrammatically in Figure 2.
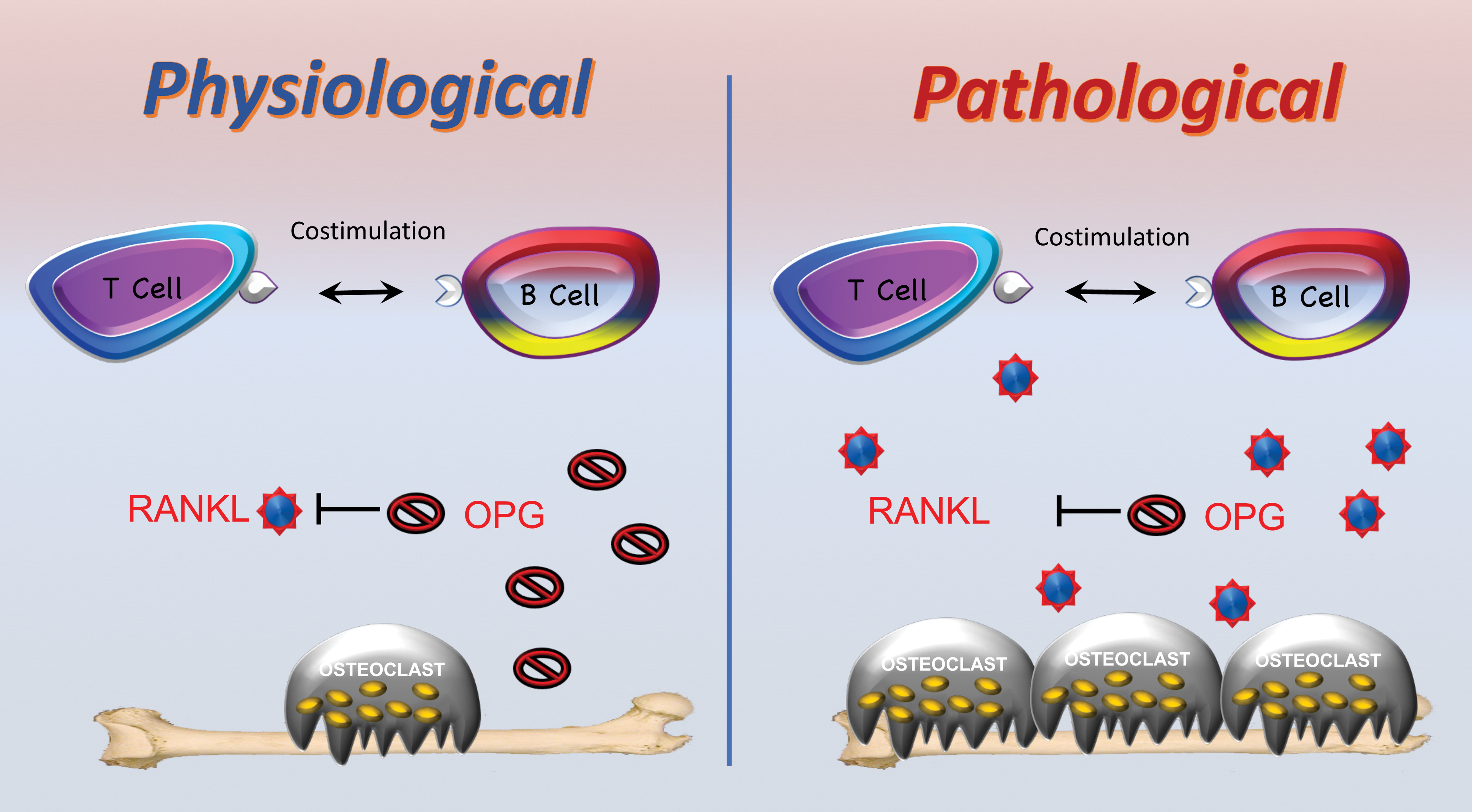
The immunoskeletal interface (ISI). The ISI is a nexus between immune and skeletal systems with key immune components repurposed for skeletal functions. Under basal physiological conditions, B cell, under the regulation of T cells, secretes copious concentrations of the receptor activator of NF-kB ligand (RANKL) decoy receptor osteoprotegerin (OPG), moderating osteoclastogenesis and maintaining homeostatic bone turnover. Under pathological conditions such as inflammation, activated B cells and T cells secrete large concentrations of RANKL, driving up osteoclastogenesis, disrupting basal bone homeostasis, and leading to bone loss.
The Bone Loss of HIV-1 Infection
Given the existence of the ISI and the protective role of immune cells on basal bone turnover, one might predict that conditions that disrupt adaptive immunity would lead to an imbalance in bone turnover promoting osteoclast formation, increased bone resorption and bone loss. A test of this prediction involves infection by the HIV-1 virus that devastates adaptive immunity leading to AIDS. HIV dramatically depletes CD4+ T cells, the lynchpin of adaptive immunity, leading to a decline in number and function of other adaptive immune components including B cells and CD8+ T cells.
In fact, bone loss is widespread in human subjects infected by HIV and a meta-analysis has concluded that overall there is a 50% to 70% incidence of osteopenia and 15% incidence of osteoporosis in this population (Brown and Qaqish 2006). Importantly, this loss of BMD has been demonstrated in several large population-based studies to translate into a significantly elevated prevalence of bone fracture (Prior et al. 2007; Triant et al. 2008; Young et al. 2011; Womack et al. 2011; Guerri-Fernandez et al. 2013; Prieto-Alhambra et al. 2014; Sharma et al. 2015). In fact, both men and women are affected and with fracture prevalence affected over a wide age range, such that in the context of HIV, even relatively younger men are at increased risk of a fragility fracture (Triant et al. 2008).
The mechanisms underlying bone loss and increased fracture incidence have been difficult to assess in humans, given a significant number of coexisting osteoporosis risk factors in this demographic. These include metabolic complications of AIDS such as hypogonadism and renal disease and lifestyle factors including high rates of smoking and alcohol and recreational drug use, which can all impact the skeleton. Finally, complex invasive mechanistic studies in humans are severely limited in scope, ultimately necessitating the use of an animal model. The animal model we chose for these studies was the HIV transgenic (Tg) rat, a small animal model of HIV infection that develops multiple clinical manifestations of human AIDS (Reid et al. 2001). To interrogate changes in the skeleton, we performed bone densitometry to quantify BMD using DXA. Tg rats displayed significantly diminished BMD at lumbar vertebrae and femurs and in both trabecular and cortical compartments as assessed by microcomputed tomography (μCT; Vikulina et al. 2010). Biochemical markers of in vivo bone resorption further identified a significant increase in the bone resorption marker C-terminal telopeptide of type I collagen (CTx), a sensitive and specific marker of bone resorption, while osteocalcin, a marker of bone formation, was unchanged. Histological and histomorphometric analysis of tartrate resistant acid phosphatase positive multinucleated cells (osteoclasts) confirmed a significant increase in osteoclast number and the area of bone surface covered by osteoclasts in HIV Tg rats. To understand the mechanistic basis for increased osteoclasts, we next examined the expression of RANKL and OPG in the bone marrow by real-time reverse transcriptase (RT)-polymerase chain reaction (PCR) analysis of whole bone marrow and observed a significant elevation in expression of RANKL and a simultaneous significant decline in OPG. These data suggest an imbalance in the RANKL/OPG ratio may underlie the osteoclastic bone loss in HIV Tg rats. As many cell types have the capacity to secrete both OPG and RANKL, we next investigated production of these factors by specific cell populations. Immunomagnetic isolation of B cells followed by RT-PCR confirmed that the changes in RANKL and OPG observed were exclusively a consequence of altered B cell function (Vikulina et al. 2010).
To determine whether these changes in B cell production of RANKL and OPG are representative of human HIV, we performed a translational clinical study in which we recruited 58 HIV-negative control subjects and 62 HIV-positive patients and collected peripheral blood mononuclear cells and plasma/serum (Titanji et al. 2014). As expected, we identified a significant elevation in bone resorption marker (CTx) in HIV-infected subjects compared to uninfected controls. Flow cytometric analysis of peripheral blood B cells further ratified a significant 20% decline in the percentage of OPG producing B cells and a significant 60% increase in the percentage of B cells expressing RANKL in conditions of HIV infection. Although cells in the peripheral circulation may not accurately represent conditions in the bone microenvironment and may only provide muted evidence of changes at other sites, overall the data strongly support an altered B cell RANKL/OPG ratio in HIV-infected patients. Importantly, we further identified a significant inverse correlation between BMD at the femoral neck and total hip with the B cell RANKL/OPG ratio, suggesting a pathophysiological effect of B cells on BMD (Titanji et al. 2014).
Although bone loss in HIV-infected subjects is likely multifactorial and exacerbated by AIDS sequelae and lifestyle factors, we propose that 1 major underlying cause is an immunoskeletal defect in B cell RANKL and OPG production. HIV infection likely results in significant alterations in CD4+ T cell costimulation, a consequence of severe CD4 depletion as well as functional changes. Irrespective of the underlying disturbance, B cell function changes from that of OPG production to that of RANKL production (an activated condition). The increase in free bioactive RANKL thus increases osteoclast differentiation, driving up bone resorption and leading to bone loss. This model is presented diagrammatically in Figure 3.
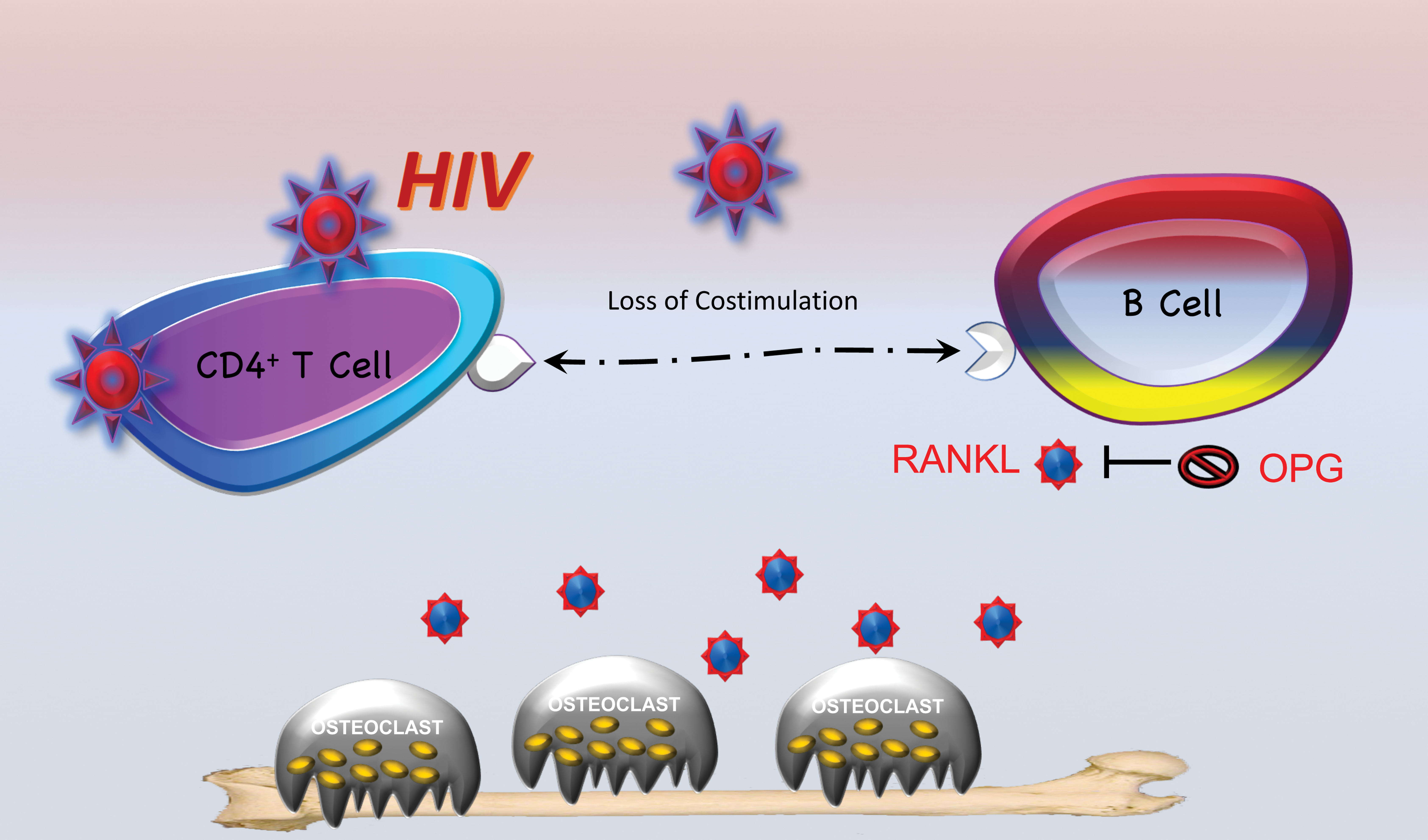
The bone loss of human immunodeficiency virus (HIV) infection. HIV infection leads to a disruption of the immunoskeletal interface. Damage to adaptive immune components, especially CD4+ T cells, leads to a loss of costimualtion and changes in cytokine production, causing a significant decline in B cell osteoprotegerin (OPG) production and a significant upswing in production of receptor activator of NF-kB ligand (RANKL). This RANKL/OPG imbalance drives up osteoclastogenesis and bone resorption, leading to bone loss.
The Bone Loss of HIV Antiretroviral Therapy (ART)
Years (often decades) of undiagnosed HIV infection drive a significant deterioration of the skeleton. Once diagnosed, however, the majority of patients begin ART also known as highly active antiretroviral therapy and combinatorial antiretroviral therapy. In most cases, ART comprises a cocktail of multiple agents from different drug classes. Modern ART drug combinations are extremely effective at reducing HIV viral load and reversing many of the classic manifestations of AIDS.
Paradoxically, the skeleton does not improve with ART and in fact, in most cases, actually undergoes a further deterioration with an additional average loss of up to 6% in BMD within the first 1 to 2 years of therapy initiation (McComsey et al. 2010). Although specific ART classes and indeed specific drugs within those classes appear to mediate more dramatic bone loss than others (particularly tenofovir disoproxil fumarate containing regimens; Wohl et al. 2016), it is now recognized that virtually all ART mediate some degree of bone loss, and recent evidence supports the contention that much, if not most, of the bone loss is independent of the specific antiviral agents used (Brown et al. 2009; Piso et al. 2011; Bruera et al. 2003).
How ART affects the skeleton is unclear and has traditionally been considered to be a direct toxic effect on bone cells such as osteoblasts increasing RANKL production. However, studies of antiretroviral drugs in culture systems (Gibellini et al. 2010; Grigsby, Pham, Gopalakrishnan, et al. 2010; Grigsby, Pham, Mansky, et al. 2010) or in vivo in mice (Wang et al. 2004) generally fail to recapitulate the effects on bone turnover that are observed when administered to humans with HIV infection. Furthermore, recent studies in which ART is used to treat human hepatitis B virus infection cause little bone loss compared to HIV-infected subjects (Chan et al. 2016). Taken together, the data do not support a direct toxic action of ART on bone cells and suggest a mechanism unique to HIV infection.
What all HIV agents have in common is that they suppress viral replication and reduce viral load. As a consequence, adaptive immune function undergoes, in part, reconstitution and reactivation. The recovery of CD4+ T cells involves homeostatic reconstitution, a process involving T cell proliferation and expansion to fill available immunological space (Ernst et al. 1999; Surh and Sprent 2000). Similar processes are involved in CD8+ T cell and B cell recovery and are driven in part through cytokine-mediated processes. Through costimulatory interactions and cytokine production, CD4+ T cell subsets further regulate other adaptive immune components including humoral immunity (B cells) and antigen presenting cells including macrophages, dendritic cells, and B cells. The regeneration and rekindling of adaptive immunity thus has the potential to produce inflammatory events that may have the capacity to drive osteoclastogenesis and bone loss, as is characteristic of other inflammatory states (Kotake et al. 2001; P. Li and Schwarz 2003).
To test this hypothesis, we mimicked the process of T cell repopulation following ART in humans by reconstituting T cells by syngeneic adoptive transfer into T cell deficient TCRβ KO mice and quantified bone turnover and structure over 3 months (Ofotokun et al. 2015). Using prospective DXA to quantify BMD, we found a dramatic loss of bone mass in reconstituted mice, as T cells homeostatically expanded and engaged adaptive immunity. μCT further revealed significant deterioration of both cortical and trabecular bone compartments. A marker of bone resorption (CTx) revealed significantly increased bone resorption and serum levels of RANKL and TNF were found to be significantly elevated, similarly to human HIV patients initiating ART (Ofotokun et al. 2016). Using flow cytometry of whole bone marrow and/or spleen, we further demonstrated that both CD4+ and CD8+ T cells produced significantly increased levels of RANKL, while multiple adaptive immune components including CD4+ and CD8+ T cells, B cells, and macrophages all produced significant levels of TNF. Reconstitution of T cells from RANKL KO mice led to significant protection from bone loss, suggesting a key role of T cell RANKL while transplantation of TNF KO T cells only partly diminished bone loss, consistent with TNF production by not only T cells but also other adaptive immune cells (Ofotokun et al. 2015).
Taken together, the data confirm that homeostatic repopulation of T cells leads to an inflammatory environment capable of driving significant bone resorption and loss of BMD and mass. This model is presented diagrammatically in Figure 4.
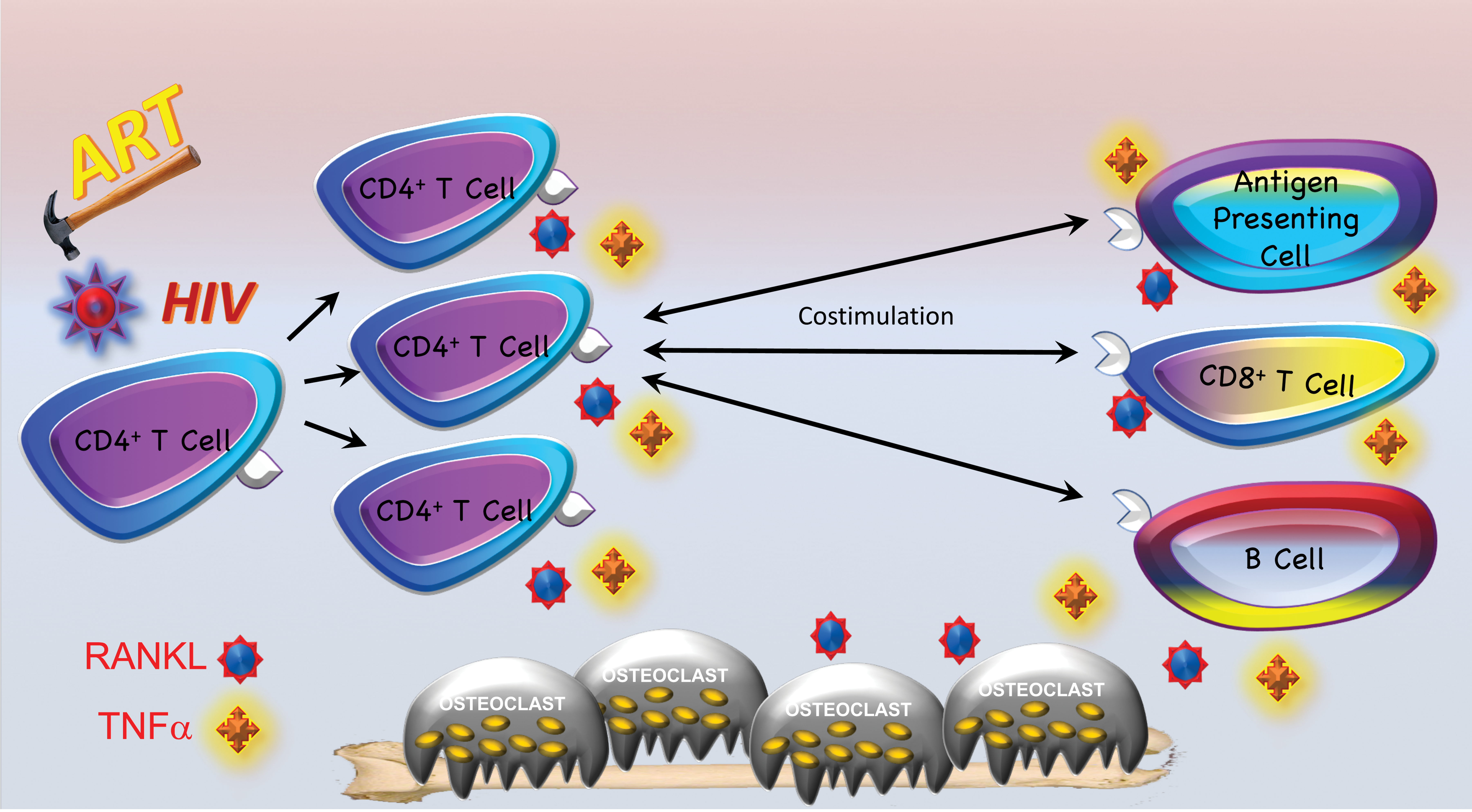
The bone loss of human immunodeficiency virus (HIV) antiretroviral therapy (ART). HIV ART significantly diminishes viral load allowing a partial CD4+ T cell recovery. CD4+ T cells undergo homeostatic expansion and repopulation of immunological niches. This process is inflammatory and leads to the release of receptor activator of NF-kB ligand (RANKL) and tumor necrosis factor (TNF) driving up osteoclastogenesis. In addition, reactivation of adaptive immune function causes additional RANKL and/or TNF production by APC, CD8+ T cells, and B cells. These high levels of osteoclastogenic cytokines promote elevated osteoclastogenesis and stimulate bone resorption, leading to additional bone loss over and above that causes by HIV infection itself.
Whether this mechanism is a significant contributor to bone loss in humans initiating ART remains to be determined, although in support of this mechanism, recent clinical data by us (Ofotokun et al. 2016) and others (Grant et al. 2013) reveal that subjects with low starting CD4+ T cell number and those undergoing the most robust T cell reconstitution (Ofotokun et al. 2016) undergo to most aggressive bone loss.
A translational clinical study to validate homeostatic reconstitution and immune activation as a key mechanism underlying ART bone loss is presently in progress.
Estrogen Deficiency Bone Loss and the Role of the ISI
Postmenopausal osteoporosis is the archetypal bone disease of women and is the result of an imbalance in bone turnover as a consequence of estrogen deficiency following the menopause (Weitzmann and Pacifici 2006). As in other osteoporotic conditions, estrogen deficiency leads to an enhanced rate of bone resorption relative to bone formation, leading to net bone loss. Ovariectomy in mice or rats is a traditional animal model of postmenopausal osteoporosis, as removal of the ovaries causes a rapid drop in estrogen levels that leads to increased bone resorption and significant cortical and trabecular bone loss within a period of just 2 to 4 weeks (Weitzmann et al. 2002).
Estrogen is established to mediate potent anti-inflammatory effects in the body, and loss of estrogen has been shown to cause significant expansion of lymphocytes, both T cells (Cenci et al. 2003) and B cells (Miyaura et al. 1997). To test whether T cell expansion following estrogen decline promotes bone loss as in other inflammatory conditions, we performed ovariectomy in T cell deficient nude mice (Cenci et al. 2000). While control Wild Type (WT) mice underwent significant loss of BMD due to increased bone resorption, nude mice were protected from osteoclastic bone loss. Although activated T cells are a significant source of RANKL, this cytokine is not typically observed to be increased in T cells under conditions of estrogen deficiency in mice. By contrast, TNF is significantly increased in the bone marrow, a consequence of T cells (Cenci et al. 2000). Although TNF production per cell was not increased, the expansion of T cells in bone marrow and other peripheral sites leads to an overall increase in TNF. TNF has indeed been shown to be significantly elevated in both human peripheral blood cells in surgically ovariectomized women (Pacifici et al. 1991) and in mice (Cenci et al. 2000). Attesting to the importance of TNF in ovariectomy bone loss, TNF and TNF receptor I (p55) KO mice fail to undergo bone loss in response to ovariectomy (Cenci et al. 2000). These studies led to a model whereby estrogen decline leads to an expansion in T cells secreting TNF, and this TNF amplifies RANKL-induced osteoclastic bone resorption causing bone loss (Weitzmann and Pacifici 2006). Although this general model still appears to hold true, the regulation of the signaling cascades that lead to T cell expansion has turned out to be exceedingly complex. Among the key upstream events, driving T cell expansion is an increase in IL-7 production by multiple tissues within the body (Ryan et al. 2005). IL-7 is a potent lymphopoietic cytokine that controls multiple steps in T cell biology. IL-7 increases the sensitivity of T cells to otherwise tolerogenic antigens decreasing the threshold for antigen-dependent T cell activation (Weitzmann and Pacifici 2006). Differentiation of T cells into different T-helper subsets including Th1 cells leads to production of TNF but also of Interferon (IFN)γ, a cytokine that upregulates the transcription factor transactivator (CIITA) in macrophages causing upregulation of major histocompatibility complex II (MHCII) and increased antigen presentation to T cells that further amplifies T cell activation (Cenci et al. 2003). Another helper subset induced by this process is Th17, characterized by production of IL-17A, a potent osteoclastogenic cytokine that induces production by osteoblast lineage cells of RANKL, the final osteoclastogenic effector cytokine. IL-17 production is elevated in ovariectomy conditions (Tyagi et al. 2012) and anti-IL-17 antibodies (Tyagi et al. 2014), and IL-17 genetic deletion (DeSelm et al. 2012) ameliorates bone loss in ovariectomized mice. This process is further sustained by downregulation of Transforming growth factor (TGF)β, an estrogen-regulated cytokine, which mediates immunosuppressive effects in part, through induction of regulatory T cells (Tregs) that down modulate T cell activation. This model is presented diagrammatically in Figure 5.
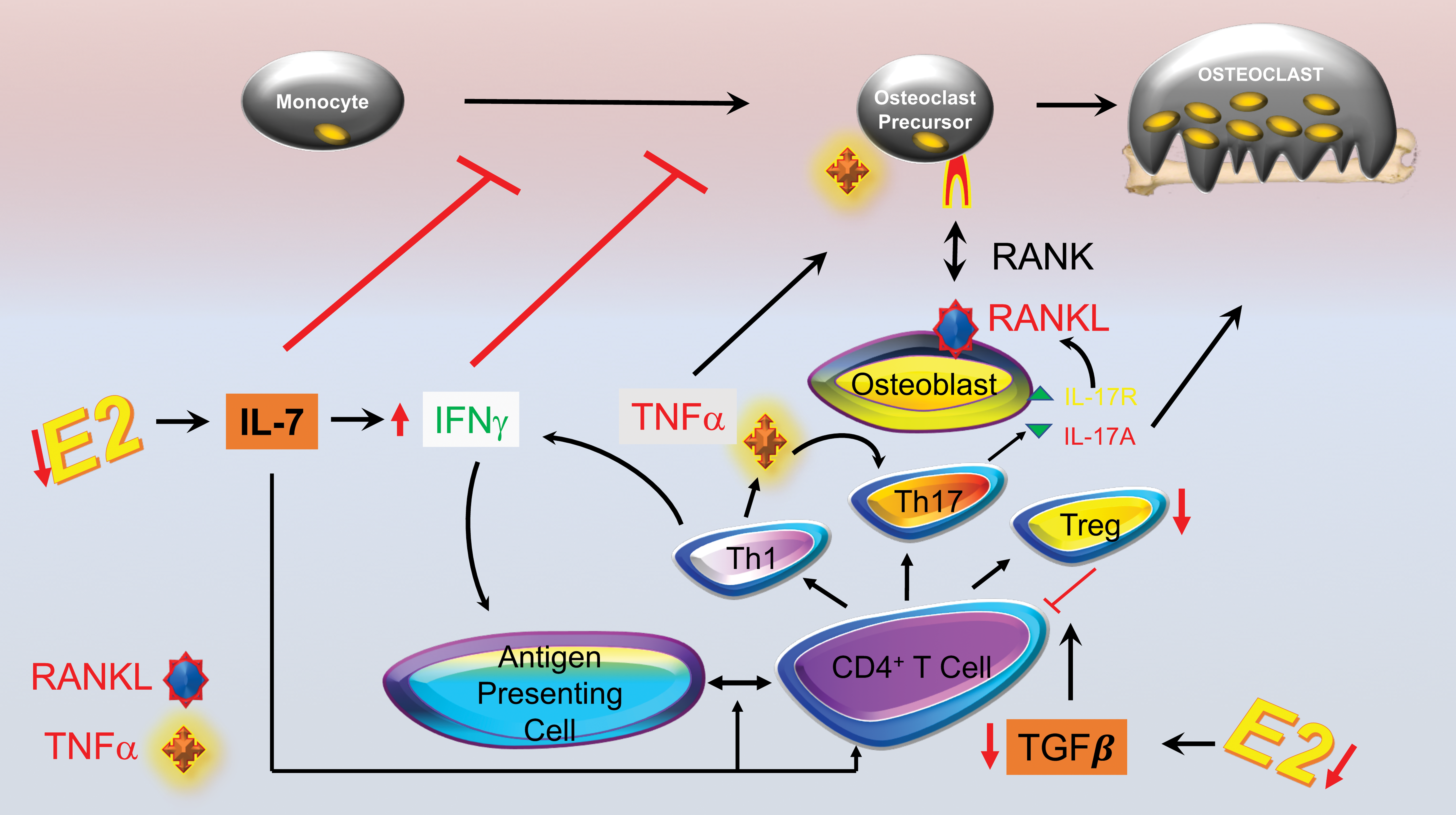
The bone loss of estrogen deficiency. Estrogen (E2) deficiency leads to bone loss though a complex cascade of interacting pathways involving the immunoskeletal interface. Among the upstream events involved is an upswing in IL-7 production driving CD4+ T cell proliferation and activation. These events are amplified by production of IFNγ that upregulates antigen presentation further stimulating T cell activation. Th1 effector T cells secrete tumor necrosis factor (TNF) which is key to the inflammatory response in estrogen deficiency leading to bone loss. TNF amplifies receptor activator of NF-kB ligand (RANKL)-induced osteoclastogenesis and stimulates RANKL production through formation of Th17T cells which secrete IL-17A, a cytokine that promotes RANKL production by osteoblasts. Finally, estrogen deficiency leads to downregulation of the anti-inflammatory cytokine TGFβ, leading to a decline in formation of regulatory T cells (Treg). Decline in Tregs further leads to increased immune activation sustaining the inflammatory cascade.
A perplexing aspect of ovariectomy-induced bone loss is the apparent antigen-dependent nature of T cell activation in this response. To validate a need for antigens, we performed ovariectomy in mice with silenced antigen presentation due to a transgenic T cell receptor in all T cells, responsive only to ovalbumin, a protein not endogenously present in mice (Cenci et al. 2003). In the absence of antigen presentation, these mice were fully protected from ovariectomy-induced bone loss. However, after exogenous administration of antigen (ovalbumin), robust bone loss was again elicited by estrogen deficiency. These data confirmed the requirement for antigen presentation in this response but also suggested that no specific type of antigen was needed and that any antigen capable of activating a T cell might suffice. Although a low baseline of antigen presentation is always occurring even in healthy humans and animals, recently, the likely nature of the antigens that are permissive for ovariectomy bone loss have been uncovered. Evidence now suggests that these antigens are gut derived (J. Y. Li et al. 2016). The gut of humans and animals is a vast receptor for a myriad of microorganisms collectively referred to as the gut microbiota, which have the capacity to control bone mass, as germ-free mice have increased BMD (Sjogren et al. 2012). The efflux of bacterial antigens into the circulation is controlled by tight junctions in the cells of the gut wall. Interestingly, these adhesion molecules are regulated by estrogen, and their expression is down modulated by estrogen deficiency (J. Y. Li et al. 2016). The net effect is a leaky gut lining that allows bacterial and other antigens to enter the circulation. Attesting to the importance of the gut microbiota in estrogen deficiency bone loss, germ-free mice do not loss bone mass after estrogen deprivation but again undergo bone resorption and loss following recolonization of the gut microbiota (J. Y. Li et al. 2016). Other studies have shown that treatment with probiotics can prevent ovariectomy-induced bone loss (Britton et al. 2014; Ohlsson et al. 2014).
Addition of other factors to this complex network driving estrogen deficiency bone loss continues relentlessly with identification of additional participants including insulin-like growth factor (IGF)-1 (Lindberg et al. 2006) and reactive oxygen species to the scheme (Grassi et al. 2007; Almeida et al. 2007).
Although early studies using B cell KO mice did not identify a role for B cells in ovariectomy-induced bone loss in mice (Y. Li, Li, et al. 2007), recent studies using sophisticated RANKL conditional KO in the B and T lineage have revealed a partial contribution of B cells to trabecular (but not cortical) bone loss in ovariectomy (Onal et al. 2012). As in our previous studies of murine ovariectomy, this study found no role for RANKL production by T cells in ovariectomy-induced bone loss (Onal et al. 2012).
Although studies validating these responses in the human context remain few and far between, at least one examination of human bone marrow cells has revealed that both T cells and B cells produce significantly elevated concentrations of RANKL, in postmenopausal women compared to premenopausal controls (Eghbali-Fatourechi et al. 2003). Treatment of postmenopausal women with estrogen replacement therapy significantly diminished T cell and B cell RANKL production. Additional evidence in humans has also been provided by a clinical study examining peripheral blood T cells which found that RANKL and TNF production was significantly increased in postmenopausal women with osteoporosis but not postmenopausal women without osteoporosis or in premenopausal women (D’Amelio et al. 2008).
The role of the ISI in postmenopausal bone loss remains controversial, as not all studies have validated significant effects of T cells in the response and some mouse and rat models of T and/or B cell deficiency have produced variable outcomes (Lee, Kadono, et al. 2006; Lee, Kalinowski, et al. 2006; Sass et al. 1997).
Studies in animal and human systems continue, however, and new findings are constantly being added to the literature. If validated, an interesting conclusion may be that estrogen deficiency bone loss has significant hallmarks of an inflammatory state (Weitzmann and Pacifici 2006).
Bone Formation and the ISI
An interesting finding is that the ISI not only plays key roles in regulation of bone resorption but also mediates potent effects on bone formation. In recent year’s potent, immunomodulatory agents have found therapeutic application in RA (Kuek, Hazleman, and Ostor 2007), an autoimmune inflammatory disease characterized by focal destruction of bone and cartilage in joints and by systemic osteoporosis resulting from release of inflammatory cytokines into the circulation (Feldmann, Brennan, and Maini 1996). Much of these cytokines emanate from activated immune cells, and animal studies have demonstrated that conditional loss of RANKL production in the adjuvant-induced osteoporosis animal model of RA protects mice from bone loss (Kong et al. 1999).
Given the role of inflammation in RA etiology, a T cell costimulation inhibitor cytotoxic T lymphocyte-associated protein-4 (CTLA-4) immunoglobulin (Ig), also referred to as abatacept, is now Food and Drug Administration (FDA) approved for intractable cases of RA. CTLA4-Ig blunts T cell activation and diminished inflammation and bone loss downstream of the inflammatory cascades (McCoy and Le Gros 1999).
Because of the role of lymphocytes in the regulation of basal osteoclastogenesis, an interesting question is whether long-term therapy with CTLA4-Ig would be detrimental to the skeleton by causing a RANKL/OPG imbalance and hence causing collateral damage to the skeleton as was observed in HIV-induced immunodeficiency. To test this theory, we injected abatacept into normal C57BL6 mice to examine its long-term effects (6 month) on basal bone turnover (Roser-Page et al. 2014). After 6 months of therapy, BMD was significantly elevated in CTLA4-Ig-treated mice compared to mice receiving Ig control. Surprisingly, metabolic turnover markers did not reveal any changes in baseline bone resorption; however, markers of bone formation were significantly increased. These data were confirmed by quantitative bone histomorphometry, the gold standard for assessment of dynamic changes in bone formation in vivo (Roser-Page et al. 2014).
The mechanism of CTLA4-Ig action involves suppression of CD28 signaling in the T cell. T cell activation occurs through a process involving 2 separate steps (the dual signal hypothesis; Sayegh 1999). The first signal involves the presentation of an antigen as part of an major histocompatibility complex (MHC) complex by an Antigen presenting cell (APC) to the T cell receptor complex. This is not an activation signal but in fact renders the T cell anergic (dormant) unless a second costimulatory signal is received. The second signal is transmitted by the APCs CD80 or CD86 costimulatory ligands to the CD28 receptor on the T cell. Activation of CD28 alleviates the anergic block allowing T cell activation, differentiation into T helper cells, and effector function (Sayegh 1999). CTLA4 is a physiological molecule produced by activated T cells and Tregs that acts as a decoy receptor for CD28, blocking CD80 and CD86, and preventing CD28 activation. CTLA4 thus terminates immune responses following resolution of infection, a necessary requirement to pacify inflammation and prevent a runaway immune response. CTLA4-Ig is a pharmacological derivative of the natural CTLA4 molecule and functions similarly to resolve inflammation by rendering T cells anergic (McCoy and Le Gros 1999).
Interestingly, it has been reported that anergic T cells secrete high levels of a Wingless integration site (Wnt) pathway agonist called Wnt10b (Zha et al. 2006). The Wnt pathway is potently bone anabolic and canonical Wnt signaling modulates many aspects of osteoblast physiology including proliferation, differentiation, bone matrix formation/mineralization, and apoptosis (Bodine and Komm 2006). Consequently, a logical hypothesis is that anergic T cells promote bone anabolic responses by secretion of Wnt10b. To test this model, we quantified Wnt10b production by real-time RT-PCR in an in vitro APC assay in which transgenic T cells (bearing a T cell receptor specific for ovalbumin) are cocultured with ovalbumin (antigen)-loaded APC (dendritic cells). While T cells alone or APC alone did not express Wnt10b, T cell engagement of antigen on dendritic cells led to a small induction of Wnt10b expression. Addition of CTLA4-Ig, breaking costimulation and rendering the activating T cells anergic, potently upregulated Wnt10b expression by more than an order of magnitude (Roser-Page et al. 2014).
To further validate this hypothesis in vivo, we treated mice with CTLA4-Ig and then immuno-magnetically purified T cells. T cells were cultured overnight, and secretion of Wnt10b into the medium was quantified by ELISA. Secretion of Wnt10b protein was significantly elevated by in vivo treatment of mice with CTLA4-Ig (Roser-Page et al. 2014).
Taken together, the data show a surprising T cell mediated capacity to not only promote inflammatory bone loss but also a capacity to potentially repair collateral damage to bone following resolution of inflammation through bone anabolic actions involving T cell secretion of Wnt10b. This model is presented diagrammatically in Figure 6.
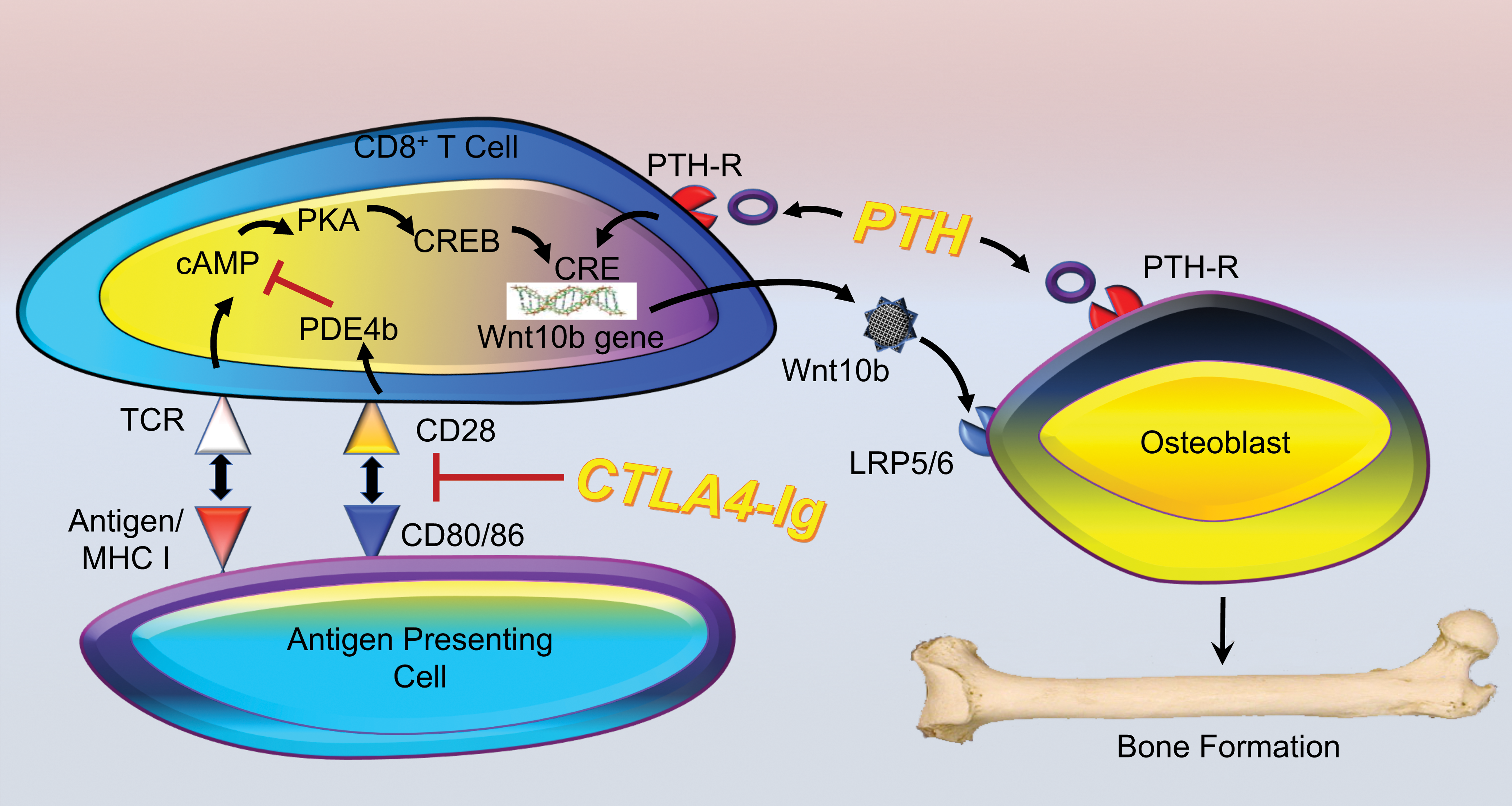
Bone formation by cytotoxic T lymphocyte-associated protein-4 immunoglobulin (CTLA4-Ig) and parathyroid hormone (PTH). CTLA4-Ig targets and disrupts the CD28 costimulatory interaction with CD80/86. This signal is necessary to activate phosphodiesterase 4b (PDE4b), which prevents accumulation of Cyclic adenosine monophosphate (cAMP) initiated by T cell receptor (TCR) binding to an antigen bearing major histocompatibility complex (MHC) I complex on an antigen presenting cell. Sustained cAMP signaling renders the T cell anergic (dormant) through Protein kinase A (PKA)-induced activation of cAMP responsive element-binding protein (CREB), anergic T cells also secrete the bone anabolic ligand Wnt10b, following binding of CREB to CREB responsive elements (CRE) in its promoter. Wnt10b binds to its receptors (LDL receptor related protein (LRP)5/6) on the osteoblasts leading to bone formation. PTH, when administered in an intermittent fashion, is also able to stimulate Wnt10b production by CD8+ T cells following binding to its receptor (PTH-R), although the intracellular signaling pathways have yet to be defined. PTH action is also dependent on direct effects mediated on osteoblasts that promote proliferation, differentiation, and increase longevity of the bone-forming cells.
Role of T Cells in Parathyroid Hormone (PTH)-Induced Bone Formation and Bone Loss
PTH is a complex and paradoxical factor. As an important regulator of calcium metabolism in the body, it defends against hypocalcemia by stimulating the release of calcium from the skeleton by promoting bone resorption (Weitzmann and Pacifici 2017). However, primary or secondary hyperparathyroidism causes sustained overproduction of PTH driving severe osteoclastic bone resorption and skeletal deterioration (Pacifici 2010). PTH-induced bone loss may be modeled in mice by the chronic administration of PTH through mini-osmotic pumps leading to a balance in bone turnover in which bone resorption outpaces that of bone formation leading to bone loss. By contrast, when administered in an intermittent (pulsatile) manner paradoxically, PTH leads to a weak bone resorptive response that is overshadowed by a robust increase in bone formation leading to a significant net bone gain. In fact, teriparatide, a fragment of human PTH injected daily, is an FDA approved bone anabolic modality for treatment of osteoporosis through stimulation of bone anabolism in humans (Pacifici 2013).
Anabolic Actions of PTH and the ISI
Interestingly, anabolic actions of PTH involve the ISI. A traditional view of anabolic PTH is that PTH induces direct osteoblast proliferation and differentiation, activation of quiescent lining osteoblasts, increasing osteoblast life span by suppressing apoptosis, and down modulation of the Wnt receptor antagonist sclerostin in osteocytes (Pacifici 2013). Although the Wnt pathway has been recognized as a key target of PTH in bone anabolic effects, the source of the Wnt ligands driving bone formation in this context is poorly characterized and largely considered to be secreted in a cell autonomous fashion by osteoblast-lineage cells themselves. However, recent data suggest that a critical source of Wnt ligand is in fact the T cell. As with CTLA4-Ig, PTH induces significant production of Wnt10b from T cells. Attesting to the importance of T cell–produced Wnt10b, reconstitution of WT T cells into TCRβ KO mice restores the anabolic activity of PTH, while adoptive transfer of T cells from Wnt10b KO mice fails to rescue the anabolic activity of PTH (Terauchi et al. 2009). This model is presented diagrammatically in Figure 6.
Catabolic Actions of PTH and the ISI
The traditional view of PTH-induced bone resorption is that PTH targets cells of the osteoblast lineage to increase the production of RANKL and decrease the production of OPG. This rebalances the RANKL/OPG ratio in favor of increased osteoclastogenesis and bone resorption (Pacifici 2013).
Evidence for a role of T cells in PTH-induced bone resorption came from studies almost 2 decades ago reporting that transplantation of human parathyroid glands from patients with hyperparathyroidism into T cell deficient nude mice failed to induce bone loss, suggesting a role for T cells in the catabolic activity of PTH (Hory et al. 2000). Follow-up studies by our group almost a decade later confirmed that administration of PTH in TCRβ KO T cell deficient mice does not induce a catabolic effect on the skeleton. However, adoptive transfer of WT T cells into TCRβ KO mice rescued the catabolic action (Gao et al. 2008). Evidence that PTH acts directly on T cells came from additional studies in which the PTH receptor was conditionally deleted on T cells leading to a failure of PTH to induce bone loss (Bedi et al. 2012). Mechanistically, PTH promotes secretion of TNF from T cells, which in addition to its amplificatory role in RANKL signaling and suppression of OPG feeds back on T cells to simulate their differentiation to Th17 subsets that secrete IL-17A leading to additional RANKL production by osteoblast lineage cells. Finally, TNF upregulates CD40, a costimulatory receptor, on bone marrow stromal cells. Binding of CD40 with its ligand CD40L on T cells imparts proliferative and survival cues to osteoblast precursors further contributing to the catabolic action of PTH (Gao et al. 2008; Bedi et al. 2010; Tawfeek et al. 2010). This model is presented diagrammatically in Figure 7.
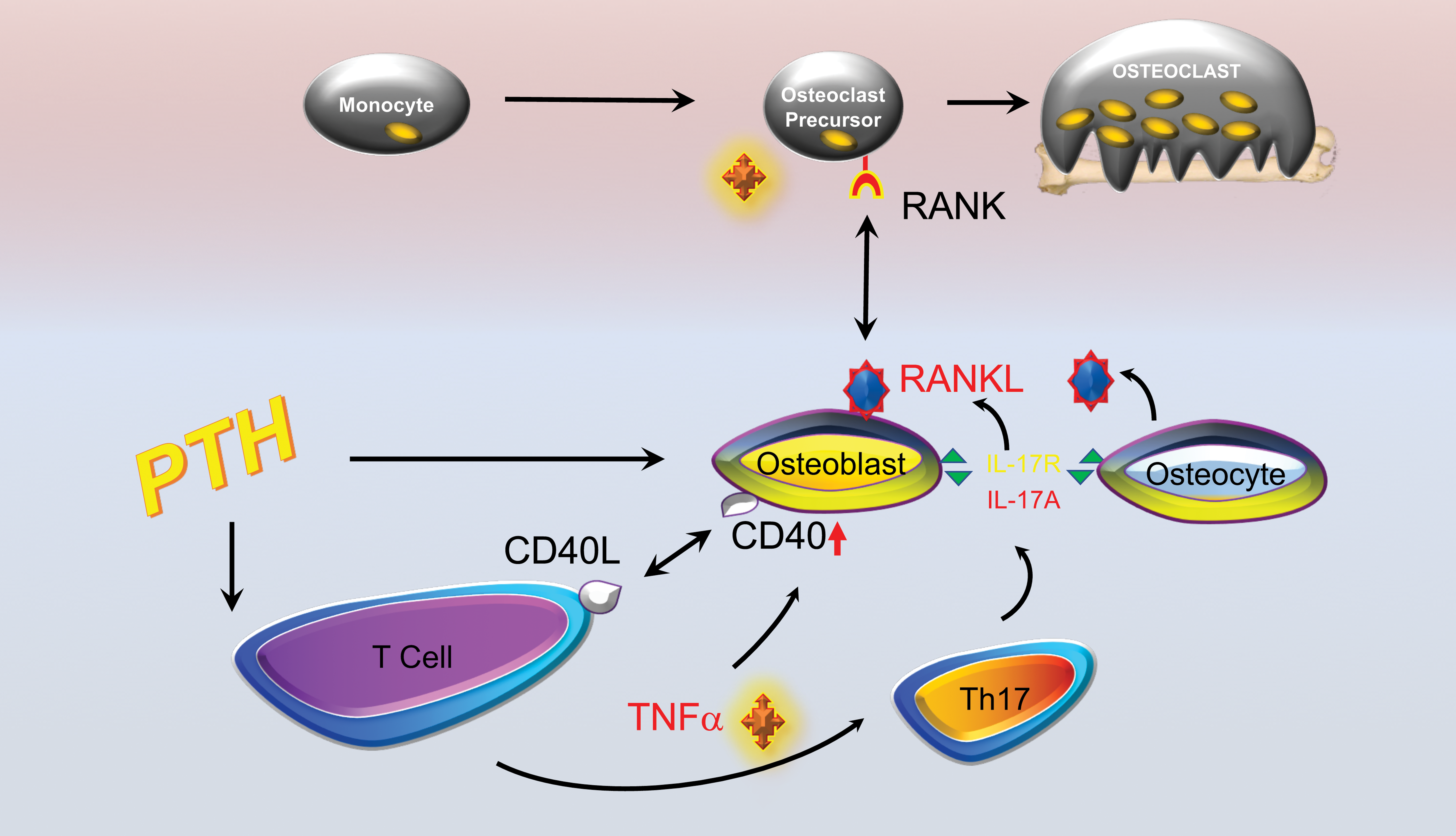
Parathyroid hormone (PTH) and bone loss. The bone catabolic effects of chronic PTH result from production of tumor necrosis factor (TNF) by T cells that feeds back though cell autonomous effects to promote Th17 effector cell formation. Th17T cells secrete IL-17A, a cytokine that binds to receptors (IL-17R) on osteoblasts and osteocytes stimulating receptor activator of NF-kB ligand (RANKL) production that promotes osteoclastogenesis and bone resorption leading to bone loss. Finally, TNF upregulates CD40 receptor on bone marrow stromal cells. Binding of CD40 with its ligand CD40L on T cells imparts proliferative and survival cues to osteoblasts.
Conclusions
Our understanding of the ISI and the field of osteoimmunology continues to evolve rapidly with new discoveries attesting to the considerable depth of the ISI and the influence of the immune system on bone. The immune system mediates protective actions on physiological bone turnover under basal conditions but causes pathological destruction when activated (or compromised as in immunosuppressive states) and causes increased fracture incidence in a number of disease states. The immune response further regulates bone resorption and bone formation depending on context. The latter discovery may have important implications for the future development of bone anabolic agents to combat osteoporosis. Teriparatide (anabolic PTH) is already an FDA approved modality for increasing bone formation. Our recent work is further focusing on the use of anergic T cells, as bone anabolic agents and immunosuppressive drugs such as abatacept to further promote bone anabolism alone or to amplify the effects of teriparatide. With new understanding of the integration between immune and skeletal functions, yet additional immune targets are likely to emerge for amelioration of bone loss in the future.
Footnotes
Author’s Note
The contents of this manuscript do not represent the views of the Department of Veterans Affairs, the National Institutes of Health, or the United States Government.
Acknowledgments
The author gratefully acknowledges research support from the Biomedical Laboratory Research and Development Service of the VA Office of Research and Development (5I01BX000105) and from the National Institutes of Health (the National Institute of Arthritis and Musculoskeletal and Skin Diseases [grant numbers AR056090, AR059364, AR068157, and AR070091] and the National Institute on Aging [grant number AG040013]).
Author Contribution
All authors (MW) contributed to conception or design; data acquisition, analysis, or interpretation; drafting the manuscript; and critically revising the manuscript. All authors gave final approval and agreed to be accountable for all aspects of work in ensuring that questions relating to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Biomedical Laboratory Research and Development Service of the VA Office of Research and Development (5I01BX000105) and from the National Institutes of Health (the National Institute of Arthritis and Musculoskeletal and Skin Diseases [grant numbers AR056090, AR059364, AR068157, and AR070091] and the National Institute on Aging [grant number AG040013]).