
Abstract
We previously reported the contribution of constitutive androstane receptor (CAR) in cytotoxicity-related hepatocarcinogenesis induced by oxadiazon (OX) or acifluorfen (ACI), two pesticides categorized as protoporphyrinogen oxidase (PROTOX) inhibitors. The molecular characteristics of preneoplastic and neoplastic lesions induced by OX and ACI were immunohistochemically compared to those by phenobarbital (PB), a typical CAR activator, in wild-type (WT) and CAR knockout (CARKO) mice after diethylnitrosamine initiation. We focused on changes in β-catenin and its transcriptional product glutamine synthetase (GS). In PB-promoted foci and adenomas, nuclear accumulation of mutated β-catenin was increased with high frequency. PB treatment also increased the multiplicity and area of GS-positive foci and adenomas in WT mice. No foci and adenomas showed nuclear accumulation of β-catenin and expression of GS in CARKO mice, similar to both genotypes of mice treated with OX and ACI. Interestingly, hepatocellular carcinoma induced in ACI-treated WT mice showed nuclear accumulation of β-catenin and was positive for GS. Our results indicated that β-catenin mutations were not involved in early-stage hepatocarcinogenesis induced by PROTOX inhibitors in mice, although activation of β-catenin and CAR is important in PB-induced tumorigenesis. The significant differences in molecular profiles suggested involvements of multiple mode of actions for hepatocarcinogenesis induced by PROTOX inhibitors.
Protoporphyrinogen oxidase (PROTOX) is an enzyme that oxidizes protoporphyrinogen to protoporphyrin IX in the chlorophyll and heme synthetic pathway in plants and animals. PROTOX inhibitors are herbicides that induce accumulation of protoporphyrin IX and photo-oxygenation in the presence of light (Matringe et al. 1989).
In rodents, PROTOX inhibitors have been reported to induce hepatocellular carcinoma (HCC) in long-term oral studies (Von Burg 1994; Quest et al. 1989). Our recent studies demonstrated that the PROTOX inhibitors oxadiazon (OX) and acifluorfen (ACI) have a complex mode of action (MoA) in the hepatocarcinogenicity process, including porphyrin accumulation, cytotoxicity, and activation of the constitutive androstane receptor (CAR) and peroxisome proliferator-activated receptor alpha (PPARα) in the mouse liver (Kuwata et al. 2016a, 2016b). We also reported that CAR partially affects the multiplicity of proliferative lesions induced by OX (Kuwata et al. 2016b). In contrast to OX, CAR plays a major role in ACI-induced cytotoxicity and subsequent tumor development in the liver, although the specific mechanisms of CAR-mediated cytotoxicity induced by ACI remain unknown (Kuwata et al. 2016a). The results of these two reports have suggested that CAR and cytotoxicity play important roles in tumor development induced by OX and ACI.
CAR is a nuclear receptor involved in hepatocarcinogenicity in rodents. Yamamoto et al. (2004) demonstrated that liver tumor induction by phenobarbital (PB) is mediated by CAR. Although all of the molecular/cellular events in CAR-mediated liver tumor development have not yet been clarified, CAR activation is known to trigger cell cycle-, apoptosis-, and Wnt-related signaling pathways (Marx-Stoelting et al. 2009). In particular, the strong interaction between CAR and β-catenin has been shown to be related to the suppressive effects of PB-tumor promotion in liver-specific β-catenin knockout (KO) mice (Singh et al. 2014).
Mutations in β-catenin have been found in more than 80% of macroscopically visible liver tumors promoted by PB treatment after diethylnitrosamine (DEN) initiation in mice (Loeppen et al. 2002). Mutation of β-catenin causes stabilization and accumulation of β-catenin protein in the nucleus, resulting in upregulation of β-catenin target genes, such as glutamine synthetase (GS) and axin2 in hepatocytes (Nejak-Bowen and Monga 2011). β-catenin mutations, as demonstrated by GS expression, have been reported in both adenomas and HCCs in PB-treated mice (Braeuning, Gavrilov, et al. 2014; Delgado et al. 2015).
In contrast to tumors promoted by PB, DEN-induced tumors contain mutations in Ha-ras (about 50%) or B-raf (about 20%), with few β-catenin mutations (Aydinlik et al. 2001; Jaworski et al. 2005). β-catenin mutations are not present in adenomas but are frequently observed in HCCs induced by DEN in mice (Ogawa et al. 1999). The molecular events involved in hepatocarcinogenesis are known to be altered according to the specific insult. Tumors harboring ras or raf mutations have been reported to be positive for phosphorylated extracellular signal-regulated kinase 1/2 (ERK1/2), which functions downstream of mitogen-activated protein kinase (Braeuning, Bucher, et al. 2014).
Several studies have shown that CAR is involved in hepatocarcinogenesis; however, the MoAs of both compounds are predicted to be significantly different from those of the typical CAR activator PB. In this study, we aimed to clarify the characteristics of protein and gene expression profiles of tumor development induced by OX and ACI and compare these characteristics with those of PB-induced tumors. We investigated changes from early to late stages of carcinogenesis, that is, altered cell foci, adenomas, and carcinomas. In addition, we focused on alterations in β-catenin-mutated lesions, which are promoted specifically by PB.
Materials and Methods
Collection of Samples
Liver tissues were obtained from mice used in two previous studies (Kuwata et al. 2016a, 2016b). Male C3H/HeNCrl (WT) mice and CARKO mice in the C3H/HeNCrl background were administered a single intraperitoneal injection of 90 mg/kg DEN (Tokyo Chemical Industry, Tokyo, Japan) as a liver tumor initiator at 6 weeks of age. Two weeks after DEN initiation, mice were fed a basal diet, 1,000 ppm OX (Wako, Osaka, Japan), 2.500 ppm ACI (Wako), or 500 ppm PB (Wako) for 26 weeks. At the necropsy, all animals were euthanized by exsanguination. All experimental animal protocols in the previous studies were approved by the Animal Care and Utilization Committee of National Institute of Health Sciences (NIHS) and were carried out following NIHS guidelines for the care and use of laboratory animals (Kuwata et al. 2016a, 2016b).
Histopathological Examination
The numbers of samples examined in this study are shown in Table 1. Twelve samples per group were randomly selected from our previous studies. However, in the ACI-treated group, 12 samples from WT mice were purposely selected to examine a certain number of HCCs.
Numbers of Samples Examined in the Present Study.
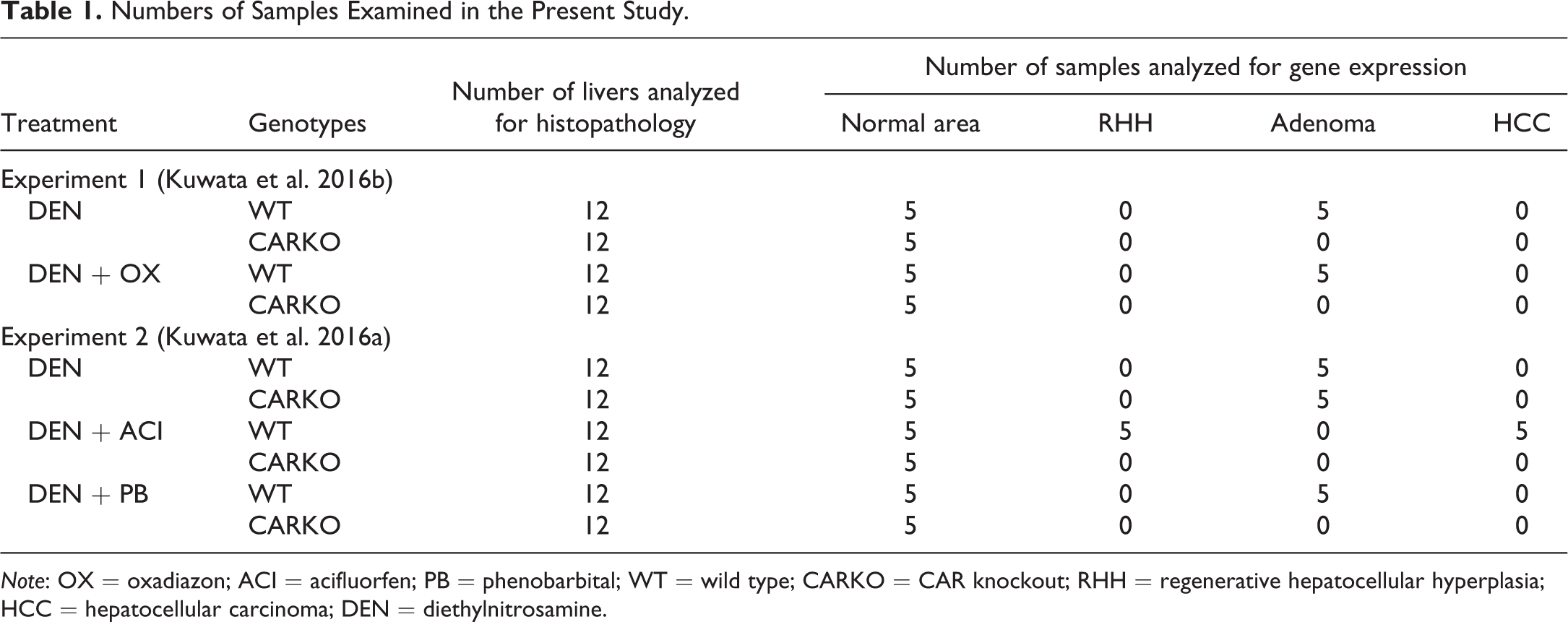
Note: OX = oxadiazon; ACI = acifluorfen; PB = phenobarbital; WT = wild type; CARKO = CAR knockout; RHH = regenerative hepatocellular hyperplasia; HCC = hepatocellular carcinoma; DEN = diethylnitrosamine.
After fixation in 10% phosphate-buffered formalin, left lobes of livers were dissected into 4 to 5 sections and routinely processed. Serial liver sections were stained with hematoxylin–eosin (HE) or immunohistochemically stained for β-catenin, GS, and phosphorylated ERK1/2 (p-ERK) using the following diluted antibodies: rabbit anti-β-catenin (Abcam, Cambridge, United Kingdom; 1:200 dilution), rabbit anti-GS (Abcam; 1:10,000 dilution), and rabbit anti-p-ERK (Cell Signaling Technology, Danvers, MA; 1:200 dilution). After incubation with primary antibodies at 4°C overnight, the sections were reacted with secondary antibodies conjugated to peroxidase-labeled dextran polymers (Histofine Simple Stain Mouse MAX PO; Nichirei, Tokyo, Japan) and then visualized by 3-3′-diaminobenzidine reactions (Dojindo Laboratories, Kumamoto, Japan).
The classification of proliferating lesions was in accordance with the criteria of the International Harmonization of Nomenclature and Diagnostic Criteria of Lesions in Rats and Mice (Thoolen et al. 2010). The multiplicity (average number per animal) of proliferative lesions was analyzed and expressed per total area of each liver section. Each focus and adenoma was subclassified into three types: eosinophilic, basophilic, and others (containing many vacuoles). Regenerative hepatocellular hyperplasia (RHH), which represents a regenerative response to continuous hepatocellular damage, was also observed. The nodular lesion usually maintained a lobular architecture containing a central vein and bile duct or oval cell proliferation (Thoolen et al. 2010).
The positive percentages of β-catenin, GS, and p-ERK were counted in each lesion. In addition, the numbers and areas of β-catenin-, GS-, or p-ERK-positive foci and adenomas per total area of each liver section were calculated. The areas of β-catenin-positive foci and adenomas were not measured because nuclear accumulation of β-catenin was counted as positive, and its expression area was limited. The areas of GS- and p-ERK-positive foci and adenomas and total areas of liver sections were measured using Image J 1.48 (NIH, Bethesda, MD).
Quantitative Real-time Reverse Transcription Polymerase Chain Reaction (RT-PCR) Analysis
At necropsy in our previous studies, liver tissues from left lobes (normal areas) and large nodules (>4 mm in diameter) were sampled. Half parts of large nodules were fixed and routinely stained with HE for histopathological examination, and the other half of nodules were applied for RT-PCR analysis. The numbers of liver samples examined in this study are shown in Table 1. Five normal areas and adenomas from WT mice and normal areas from CARKO mice in each group were used for gene expression analysis. In the ACI-treated group, 5 normal areas, RHH, and HCC samples from WT mice and normal areas from CARKO mice were analyzed. In the ACI-treated group of WT mice and in all groups of CARKO mice, sufficient numbers of adenomas could not be sampled. Samples were randomly selected from our previous studies.
Total RNA was extracted from frozen samples using ISOGEN (Nippon Gene Co. Ltd., Tokyo, Japan). After reverse transcription using 1 µg of total RNA with a High-capacity Reverse Transcription Kit (Applied Biosystems, Foster City, CA), real-time PCR was performed on a 7900HT Fast Real-time PCR System (Applied Biosystems) using TaqMan Fast Universal PCR Master Mix (Applied Biosystems) and TaqMan Gene Expression Assays (Applied Biosystems) following the manufacturer’s protocols. The primer-probe sets used in the present study were for the following genes: Gs (Mm00725701_s1), Axin2 (Mm00443610_m1), α fetoprotein (Afp, Mm00431715_m1), Car (Mm01283978_m1), and Cyp2b10 (Mm01972453_s1). The expression level of each gene was calculated using the relative standard curve method and normalized to endogenous Gapdh (TaqMan Rodent GAPDH Control Reagent; Applied Biosystems).
Statistical Analyses
Quantitative data are expressed as group means and standard deviations. In histopathological examinations, the values from the treated groups were compared with those from the control group for the same genotype, and the values from CARKO mice were compared with those from the corresponding group of WT mice. In RT-PCR analysis, the values from adenomas, RHHs, and HCCs in WT mice and normal areas from CARKO mice were compared with those from normal areas in WT mice in the same treatment group. To compare two groups of data, Student’s or Welch’s t tests (two-sided) were performed for homogenous or heterogeneous variances, respectively, after evaluating the homogeneity of variance with F tests. To compare multiple groups of data, Dunnett’s or Steel’s tests (two-sided) were performed for homogenous or heterogeneous variances, respectively, after evaluating the homogeneity of variance with Bartlett’s test. Fisher’s exact tests were performed for incidences of proliferative lesions. Results for these tests were considered to be significantly different when the p value was less than .05.
Results
Proliferative and Nonproliferative Lesions in HE Slides
The incidences and multiplicities of proliferative lesions used for the current analysis are shown in Tables 2 and 3. OX treatment increased the incidences and multiplicities of eosinophilic foci, adenomas, and basophilic adenomas in both genotypes of mice. OX treatment increased the incidences and multiplicities of basophilic foci in WT mice, but not in CARKO mice. ACI treatment resulted in increased incidences and multiplicities of eosinophilic and basophilic foci and adenomas in WT mice. However, the incidences and multiplicities of these proliferative lesions in the DEN + ACI group of CARKO mice were comparable to those in the DEN group of CARKO mice. RHHs and HCCs were observed only in the DEN + ACI group of WT mice. PB treatment increased the incidences and multiplicities of eosinophilic foci and adenomas in WT mice, but not in CARKO mice compared with that in the DEN groups of corresponding mice.
Incidences of Proliferative Lesions in the Livers of WT and CARKO Mice Treated with OX, ACI, or PB for 26 Weeks.
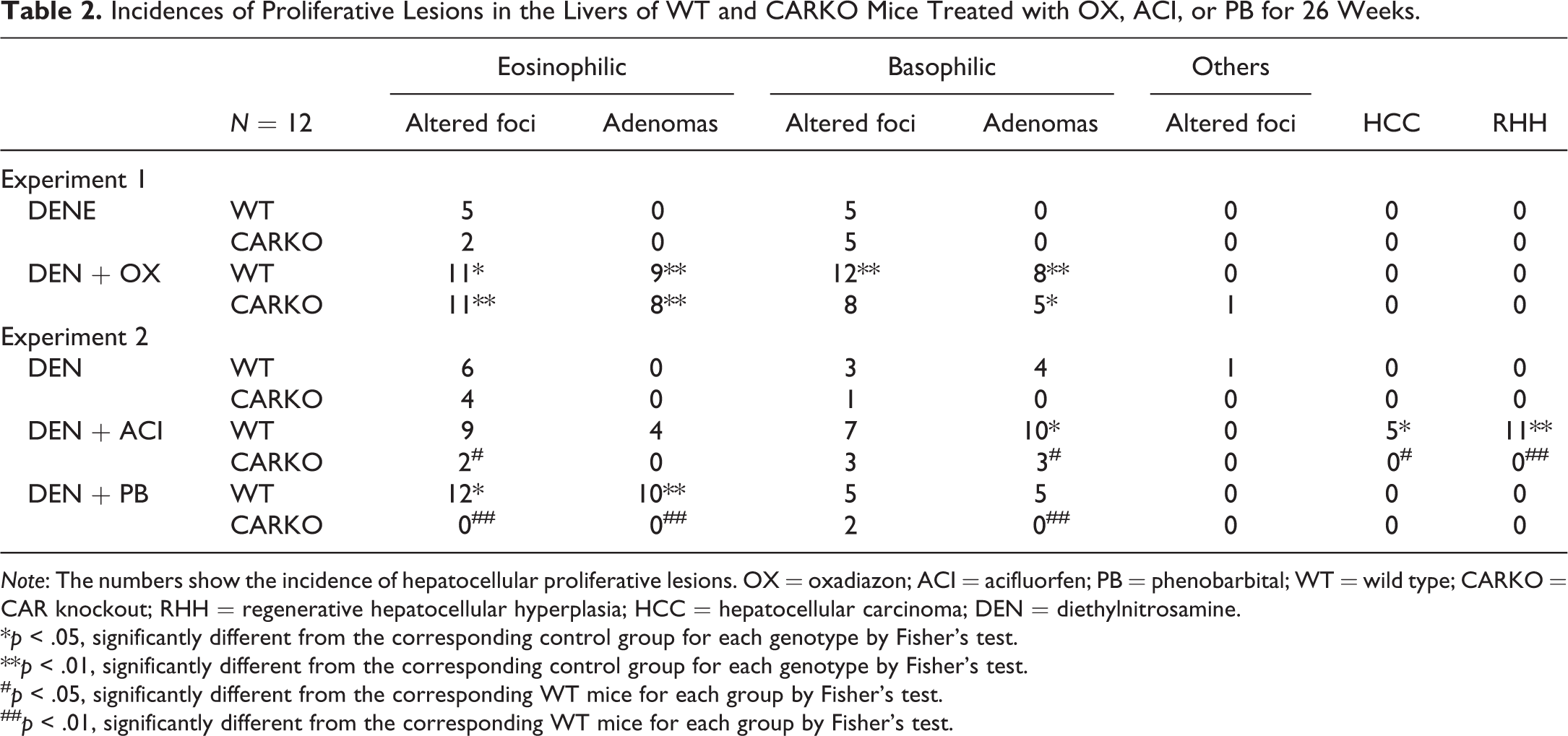
Note: The numbers show the incidence of hepatocellular proliferative lesions. OX = oxadiazon; ACI = acifluorfen; PB = phenobarbital; WT = wild type; CARKO = CAR knockout; RHH = regenerative hepatocellular hyperplasia; HCC = hepatocellular carcinoma; DEN = diethylnitrosamine.
*p < .05, significantly different from the corresponding control group for each genotype by Fisher’s test.
**p < .01, significantly different from the corresponding control group for each genotype by Fisher’s test.
# p < .05, significantly different from the corresponding WT mice for each group by Fisher’s test.
## p < .01, significantly different from the corresponding WT mice for each group by Fisher’s test.
Multiplicities of Proliferative Lesions in the Livers of WT and CARKO Mice Treated with OX, ACI, or PB for 26 Weeks.
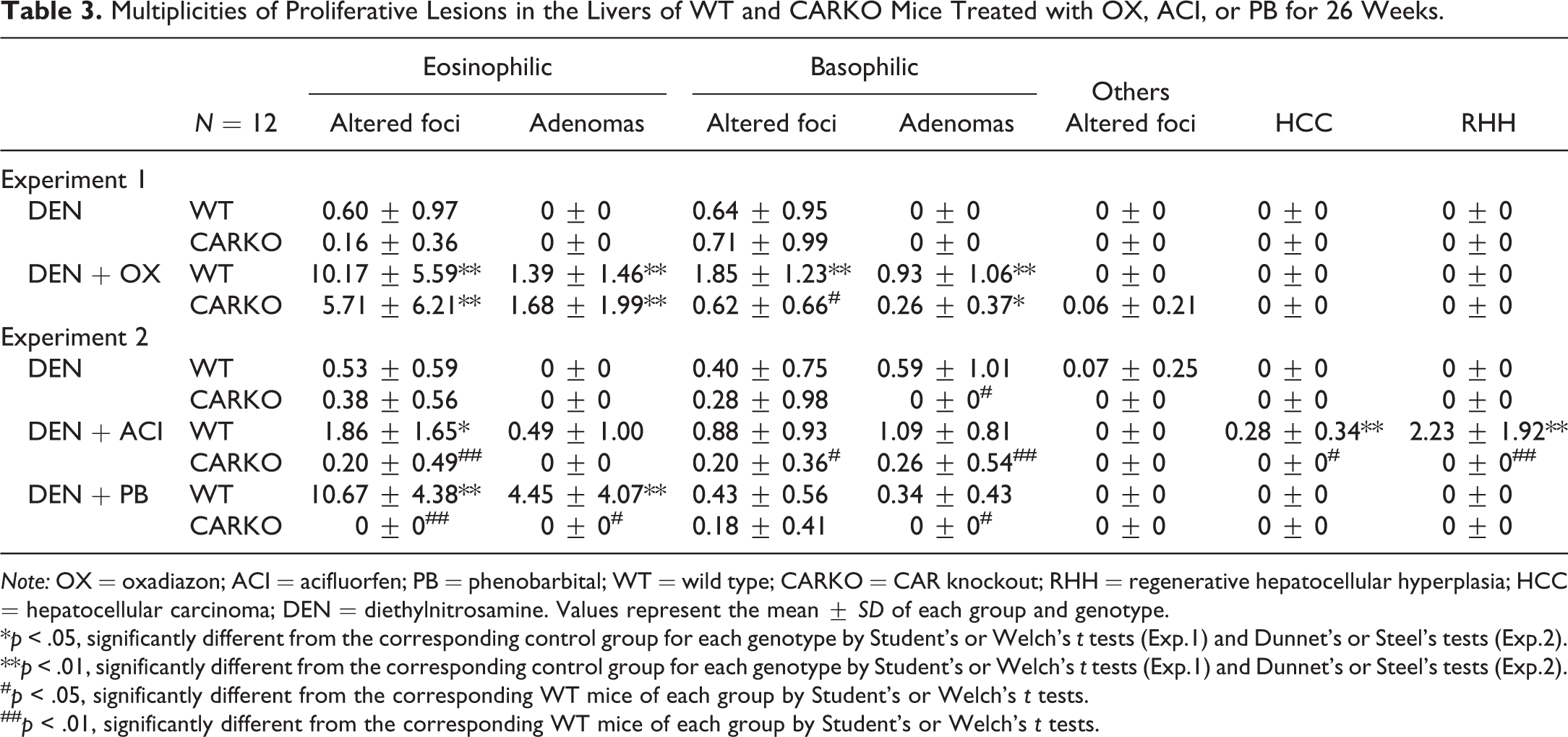
Note: OX = oxadiazon; ACI = acifluorfen; PB = phenobarbital; WT = wild type; CARKO = CAR knockout; RHH = regenerative hepatocellular hyperplasia; HCC = hepatocellular carcinoma; DEN = diethylnitrosamine. Values represent the mean ± SD of each group and genotype.
*p < .05, significantly different from the corresponding control group for each genotype by Student’s or Welch’s t tests (Exp.1) and Dunnet’s or Steel’s tests (Exp.2).
**p < .01, significantly different from the corresponding control group for each genotype by Student’s or Welch’s t tests (Exp.1) and Dunnet’s or Steel’s tests (Exp.2).
# p < .05, significantly different from the corresponding WT mice of each group by Student’s or Welch’s t tests.
## p < .01, significantly different from the corresponding WT mice of each group by Student’s or Welch’s t tests.
As for non-proliferative lesions, cytotoxic changes including single-cell necrosis of hepatocytes and diffuse infiltration of mononuclear cells were observed in OX- and ACI-treated mice of both genotypes (Kuwata et al. 2016a, 2016b). Those cytotoxic changes induced by ACI were attenuated in CARKO mice than in WT mice. Cytotoxic changes were not apparent in PB-treated mice in both genotypes.
Immunohistochemical Characteristics of Proliferative Lesions
Total numbers of β-catenin-, GS-, and p-ERK-positive lesions are summarized in Tables 4 and 5. Representative histological and immunostaining images are shown in Figures 1–4. Positive nuclear staining for β-catenin indicated its mutation. GS- and p-ERK-positive staining were observed in both the nuclei and cytoplasm. In nonproliferative lesions, a thin membranous β-catenin signal delineated the normal hepatocytes and did not display nuclear accumulation of β-catenin. GS expression was observed around the central vein, and there was no specific staining for p-ERK. In the DEN groups of both genotypes, all foci and adenomas were negative for β-catenin and GS, and several foci and adenomas were positive for p-ERK in both studies. Basophilic adenomas were observed in the DEN groups of WT mice in Exp. 1. In the DEN + OX groups of both genotypes, all foci and adenomas were negative for β-catenin and GS, and several foci and adenomas were positive for p-ERK. The number of foci and adenomas positive for p-ERK was higher in basophilic foci and adenomas than in eosinophilic foci and adenomas, respectively. There were no significant differences between the incidences of foci and adenomas which were positive for p-ERK. In the DEN + ACI group of both genotypes, all foci and adenomas were negative for β-catenin and most of them were negative for GS. Moreover, several foci and adenomas were positive for p-ERK. Basophilic foci and adenomas showed higher incidences of p-ERK staining. There were no significant differences between the incidences of foci and adenomas which were positive for p-ERK. All RHHs were negative for β-catenin and GS, and half of RHHs in ACI-treated WT mice were positive for p-ERK. All HCCs were positive for β-catenin and GS. One of the 5 HCCs was positive for p-ERK. In the DEN + PB group of WT mice, more than 50% of eosinophilic types of foci and adenomas were positive for β-catenin and GS. However, basophilic types of foci and adenomas were negative for β-catenin and GS, and some of them were positive for p-ERK. Expressions of β-catenin and GS in eosinophilic foci and adenomas and expression of p-ERK in basophilic foci and adenomas were clearly observed. In CARKO mice, 2 of the 2 basophilic foci were positive for p-ERK.
Incidences of β-catenin-, GS-, and p-ERK-positive Lesions in the Livers of WT and CARKO Mice in Exp. 1.
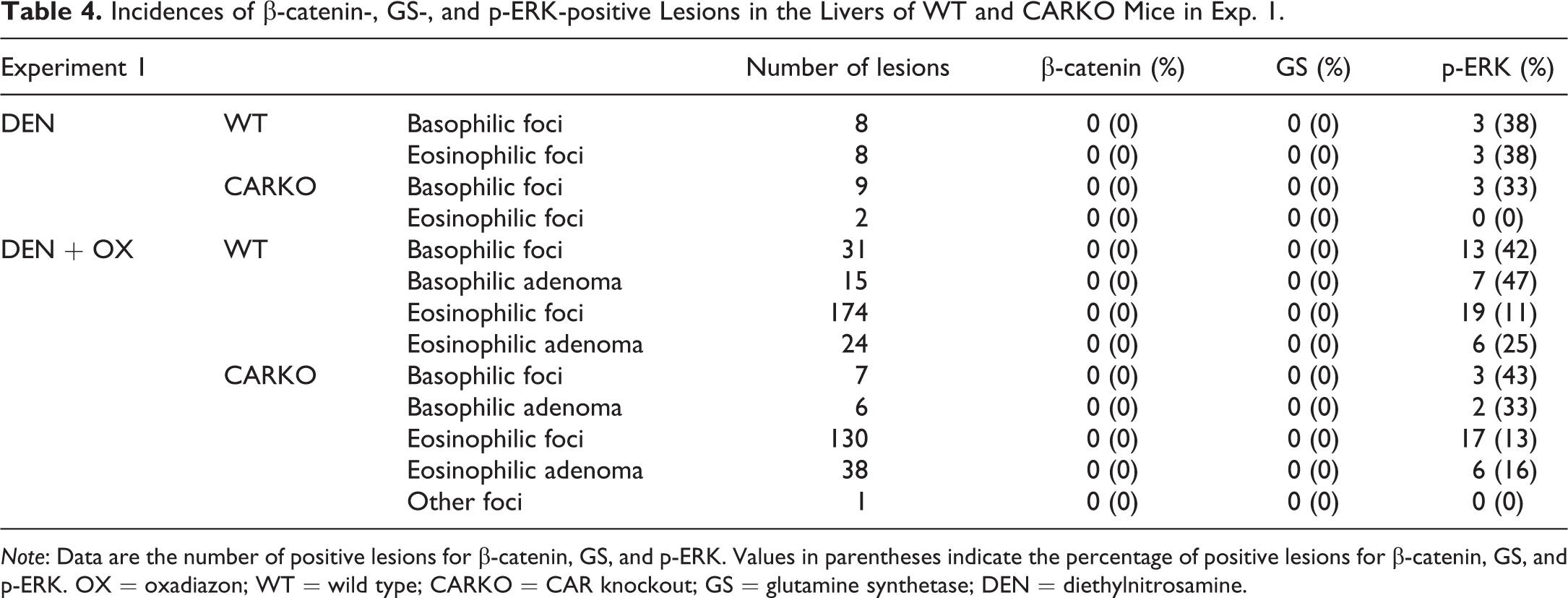
Note: Data are the number of positive lesions for β-catenin, GS, and p-ERK. Values in parentheses indicate the percentage of positive lesions for β-catenin, GS, and p-ERK. OX = oxadiazon; WT = wild type; CARKO = CAR knockout; GS = glutamine synthetase; DEN = diethylnitrosamine.
Incidences of β-catenin-, GS-, and p-ERK-positive Lesions in the Livers of WT and CARKO Mice in Exp. 2.
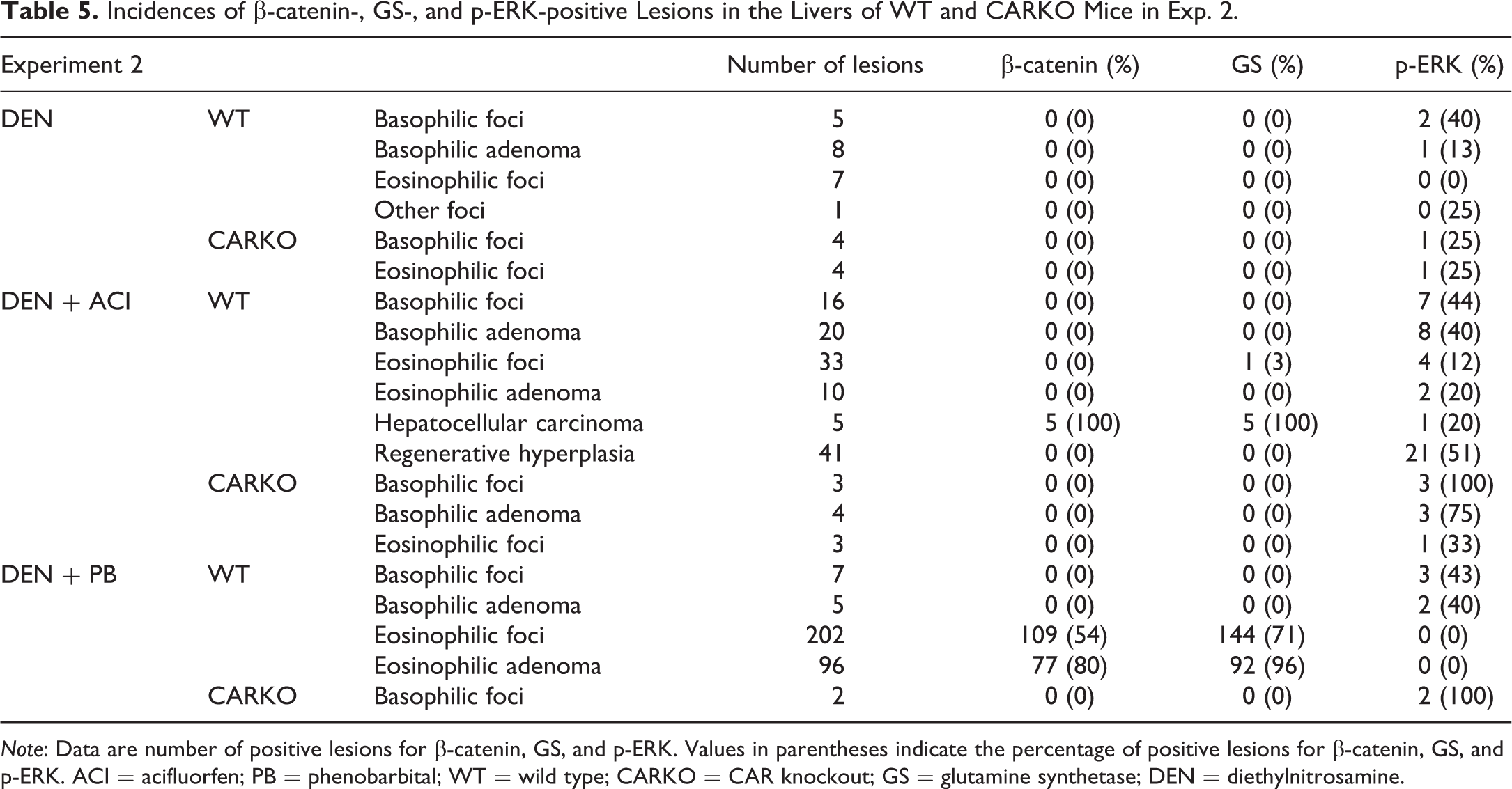
Note: Data are number of positive lesions for β-catenin, GS, and p-ERK. Values in parentheses indicate the percentage of positive lesions for β-catenin, GS, and p-ERK. ACI = acifluorfen; PB = phenobarbital; WT = wild type; CARKO = CAR knockout; GS = glutamine synthetase; DEN = diethylnitrosamine.
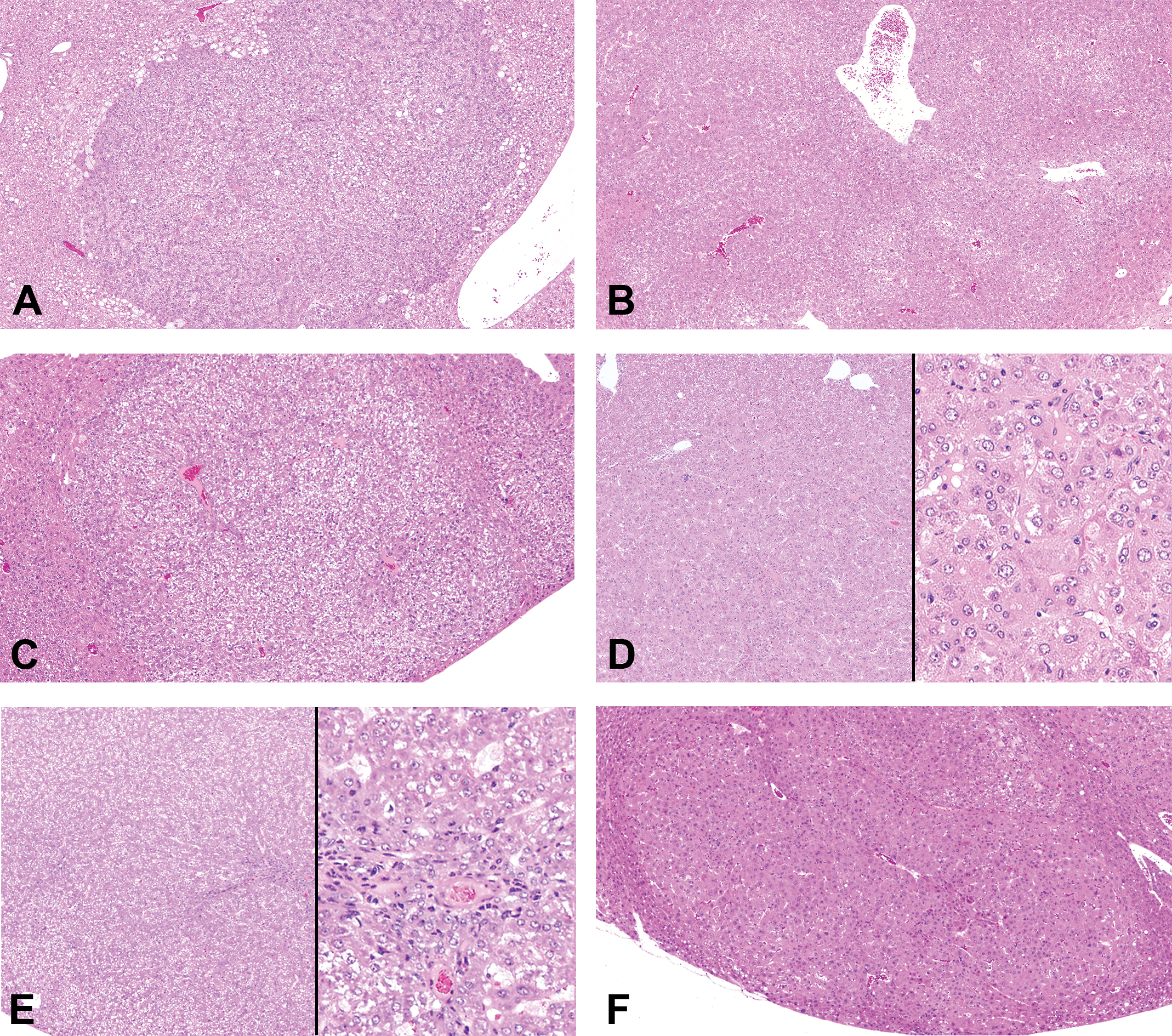
Representative histological (hematoxylin and eosin staining) images of adenomas, hepatocellular carcinomas (HCCs), and regenerative hepatocellular hyperplasias (RHHs) in wild-type mice in each group. Adenomas in diethylnitrosamine (DEN, A), DEN + oxadiazon (B), and DEN + acifluorfen (ACI) groups (C). A HCC in DEN + ACI group (D). A RHH in DEN + ACI group (E). RHHs contained bile ducts or oval cell components. Adenomas in DEN + phenobarbital group (F).
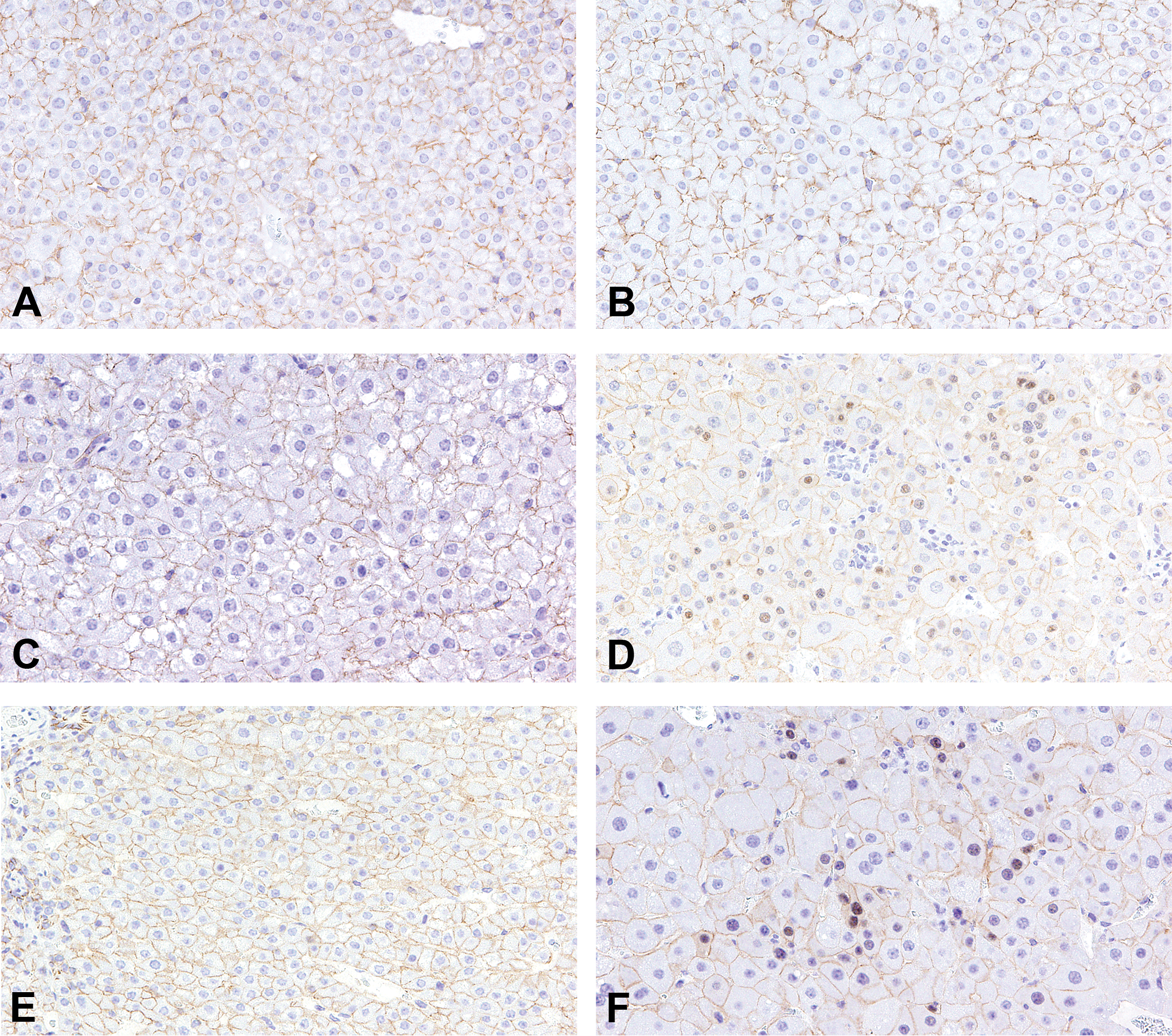
Representative immunohistochemical images of β-catenin in adenomas, hepatocellular carcinomas (HCCs), and regenerative hepatocellular hyperplasias (RHHs) in wild-type mice in each group. Nuclear accumulation of β-catenin was observed in HCCs in diethylnitrosamine (DEN) + acifluorfen (ACI) group (D) and adenomas in DEN + phenobarbital group (F). The accumulation was not observed in adenomas in DEN (A), DEN + oxadiazon (B), and DEN + ACI groups (C), and RHHs in DEN + ACI group (E). In all groups, β-catenin was expressed in the membranes of hepatocytes.
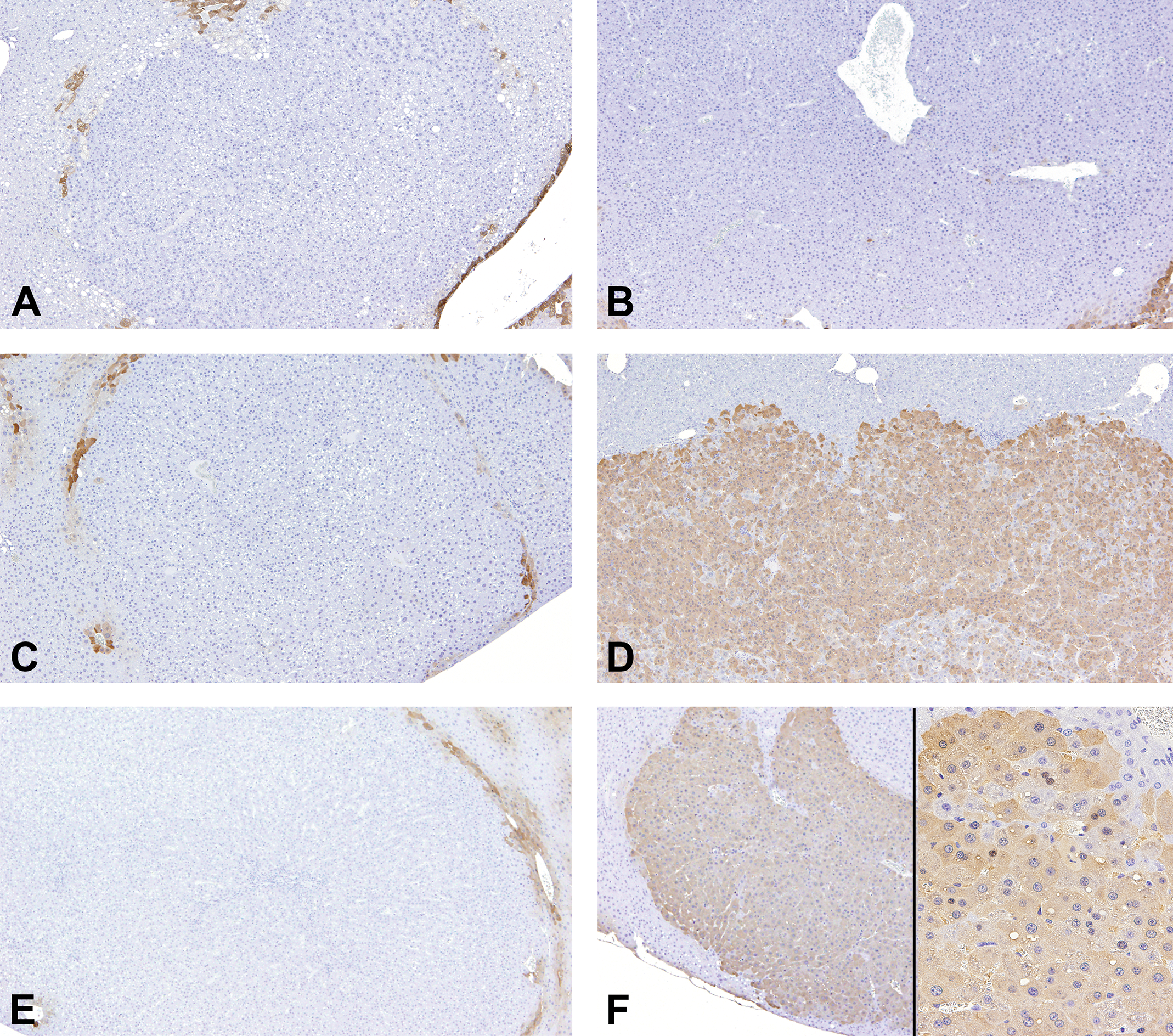
Representative immunohistochemical images of glutamine synthetase (GS) in adenomas, hepatocellular carcinomas (HCCs), and regenerative hepatocellular hyperplasias (RHHs) in wild-type mice in each group. HCCs in diethylnitrosamine (DEN) + acifluorfen (ACI) group (D), and adenomas in DEN + phenobarbital group (F) were positive for GS. GS expression was observed in both the nuclei and cytoplasm. Adenomas in DEN (A), DEN + oxadiazon (B), and DEN + ACI groups (C), and RHHs in DEN + ACI group (E) were negative for GS.
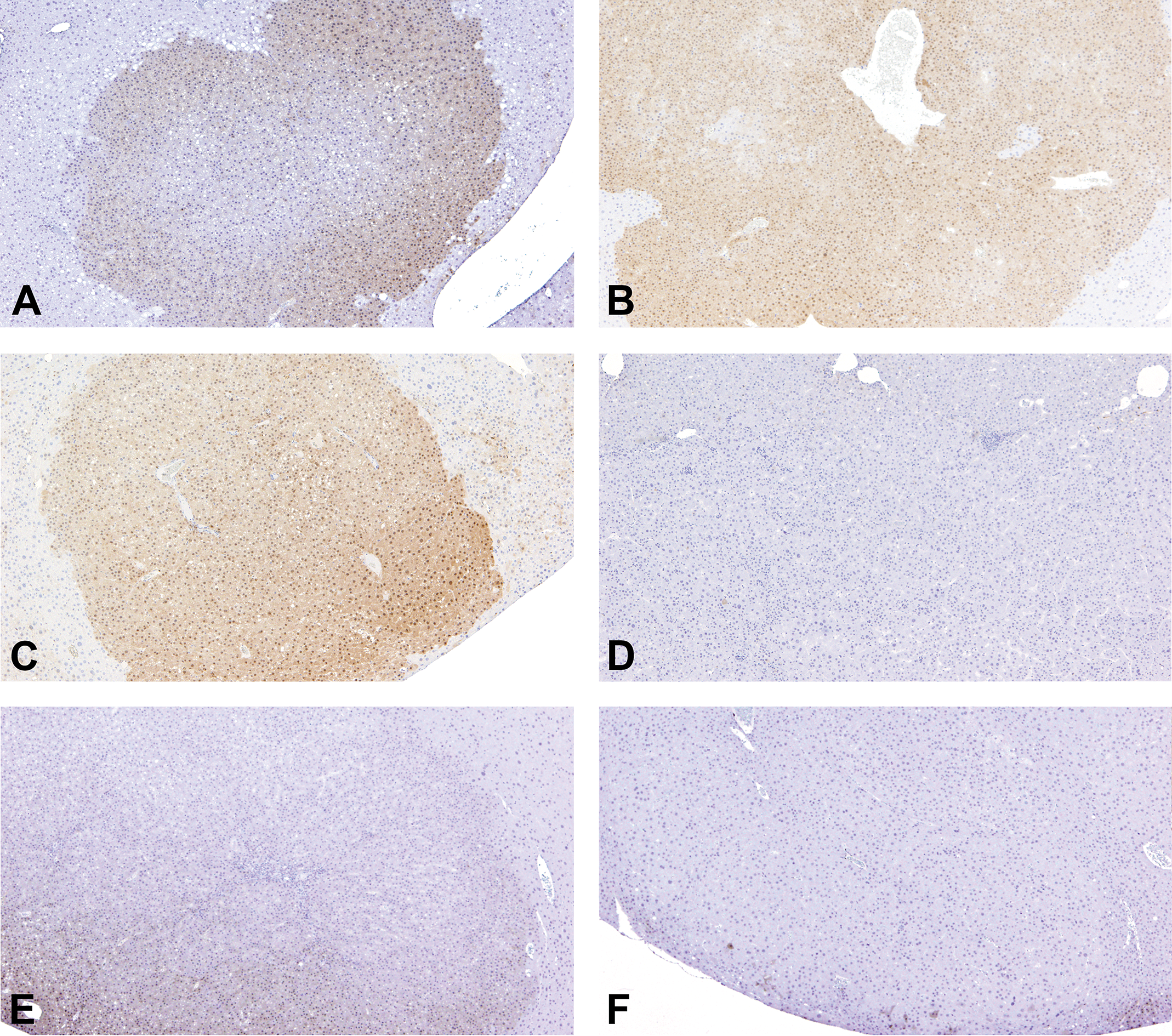
Representative immunohistochemical images of p-ERK in adenomas, hepatocellular carcinomas (HCCs), and regenerative hepatocellular hyperplasias (RHHs) in wild-type mice in each group. Adenomas in diethylnitrosamine (DEN, A), DEN + oxadiazon (B), and DEN + acifluorfen (ACI) groups (C), and RHHs in DEN + ACI group (E) were positive for p-ERK. Expression of p-ERK was observed in both the nuclei and cytoplasm. HCCs in DEN + ACI group (D) and adenomas in DEN + phenobarbital group (F) were negative for p-ERK.
The multiplicities of foci and adenomas positive for β-catenin, GS, and p-ERK and the areas of foci and adenomas positive for GS and p-ERK are shown in Figure 5. The multiplicities of β-catenin- and GS-positive foci and adenomas and the areas of GS-positive foci and adenomas were significantly increased in PB-treated WT mice. There were no significant increases in foci and adenomas positive for GS in the other groups. In the DEN + OX group, the multiplicities and areas of p-ERK-positive foci in both genotypes and adenomas in WT mice were significantly increased. In the DEN + ACI, the multiplicities and areas of p-ERK-positive foci and adenomas were increased in WT mice, although not all values achieved statistical significance. There were no significant increases in foci and adenomas positive for p-ERK in the DEN + PB group.
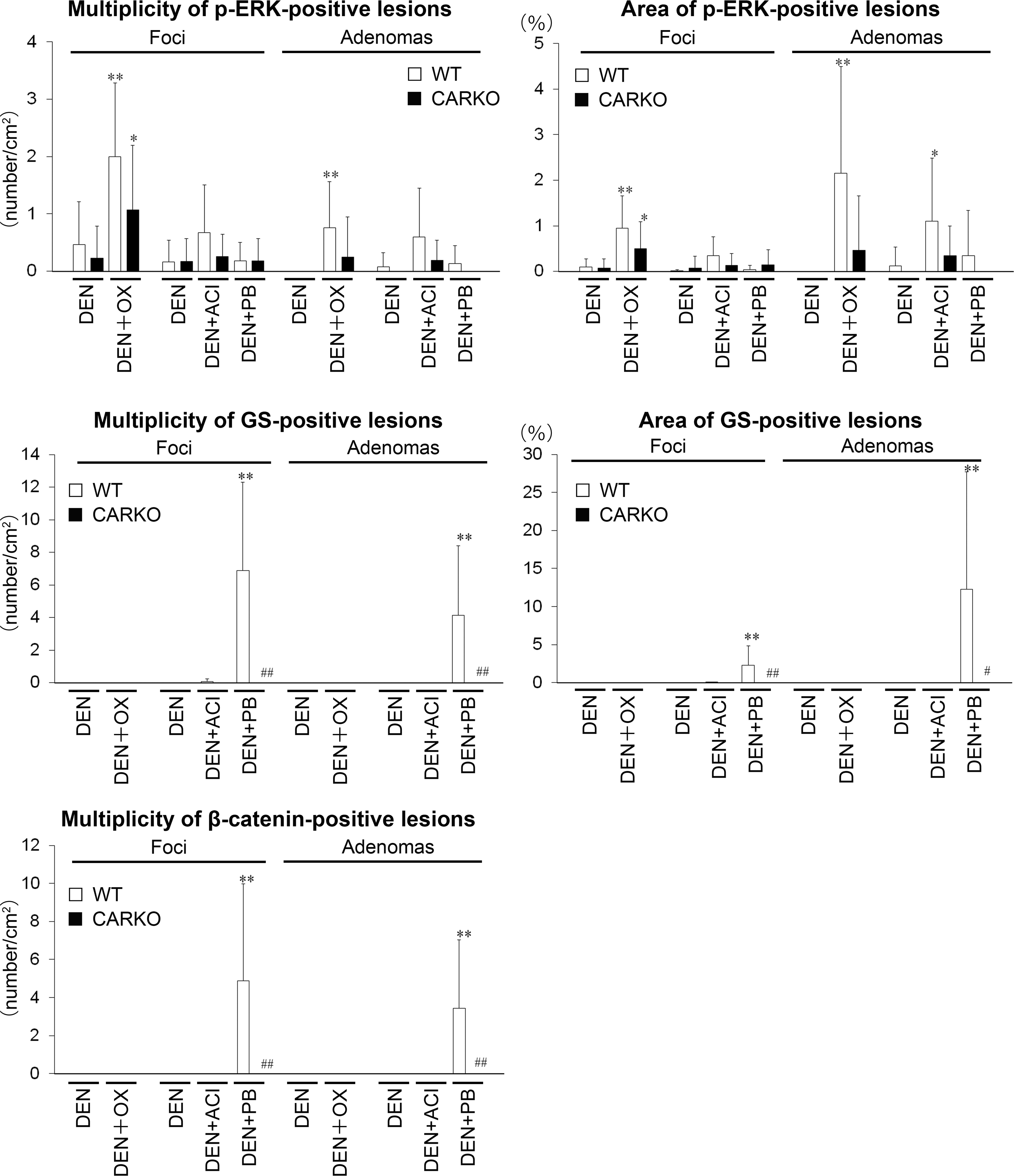
The multiplicities of foci and adenomas positive for β-catenin, glutamine synthetase (GS), and p-ERK and areas of foci and adenomas positive for GS and p-ERK. Values represent the mean ± SD of each group and genotype. *p < .05 and **p < .01, significantly different from the corresponding control group for each genotype by Student’s t test, Welch’s t test, Dennett’s test, or Steel’s test. # p < .05 and ## p < .01, significantly different from the corresponding wild-type mice of each group by Student’s or Welch’s t tests.
Gene Expression
Gene expression levels are shown in Figure 6. The expression level of Afp, an HCC marker, was significantly increased in HCCs in ACI-treated WT mice. The expression levels of GS and Axin2, target genes of β-catenin, tended to decrease in adenomas in the DEN and DEN + OX groups compared with those in corresponding normal areas of WT mice. In HCCs of ACI-treated WT mice, the expression levels of the two genes were increased, although the GS level did not reach statistical significance. There were no significant differences in the expression levels of GS and Axin2 between normal areas and adenomas in PB-treated WT mice. The expression level of CAR tended to decrease in adenomas and HCCs compared with those in normal areas of corresponding WT mice. In correlation with the decreased expression of CAR, the expression level of Cyp2b10, a prototypical CAR target gene, also tended to decrease in adenomas and HCCs compared with those in normal areas of corresponding WT mice.
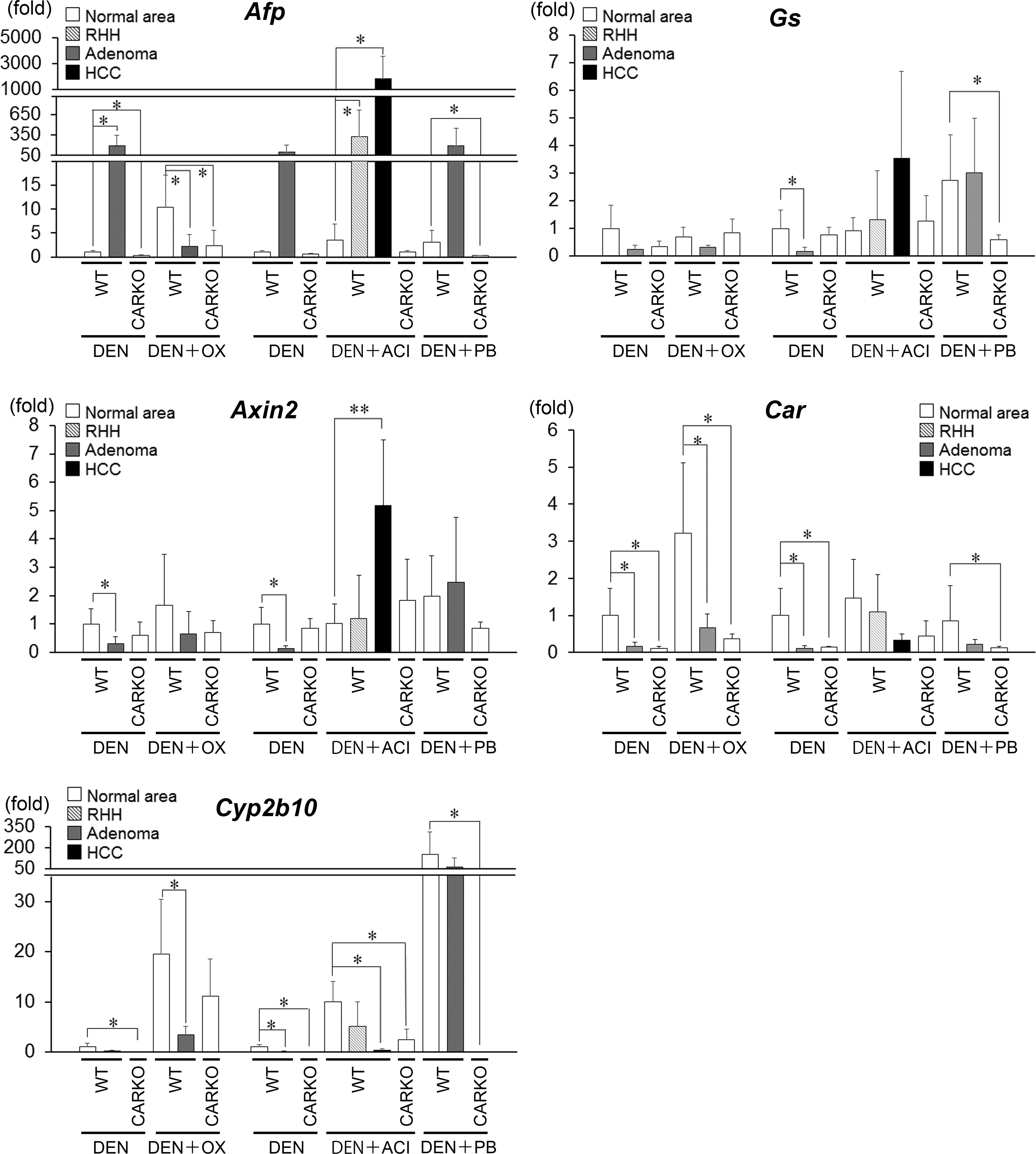
Relative expression levels of mRNAs. The average fold change for each gene is shown based on the basal expression level in control wild-type (WT) mice. *p < .05 and **p < .01, significantly different from the normal areas of corresponding WT mice by Student’s t test, Welch’s t test, Dennett’s test, or Steel’s test.
Discussion
In this study, we found significant differences in molecular profiles of hepatocellular foci/adenomas between PROTOX inhibitors and PB. Immunohistochemical profiles of major types of foci/adenoma in each group are summarized in Table 6. PB treatment after DEN initiation in WT mice increased the multiplicities of eosinophilic foci and adenomas, which showed nuclear accumulation of β-catenin and diffuse expression of GS, a target of β-catenin. Both changes are accepted as markers of β-catenin mutations (Nhieu et al. 1999). These data suggested that PB treatment after DEN initiation promoted β-catenin mutated foci/adenomas, consistent with a previous study in PB-treated mice (Loeppen et al. 2002). The lack of β-catenin- and GS-positive foci and adenomas in CARKO mice with DEN + PB supported a previous report demonstrating that CAR plays an important role in initiation of tumor development in collaboration with β-catenin (Dong et al. 2015).
Immunohistochemical Profiles of Major Types of Foci/Adenomas Induced by OX, ACI, or PB in Mice.
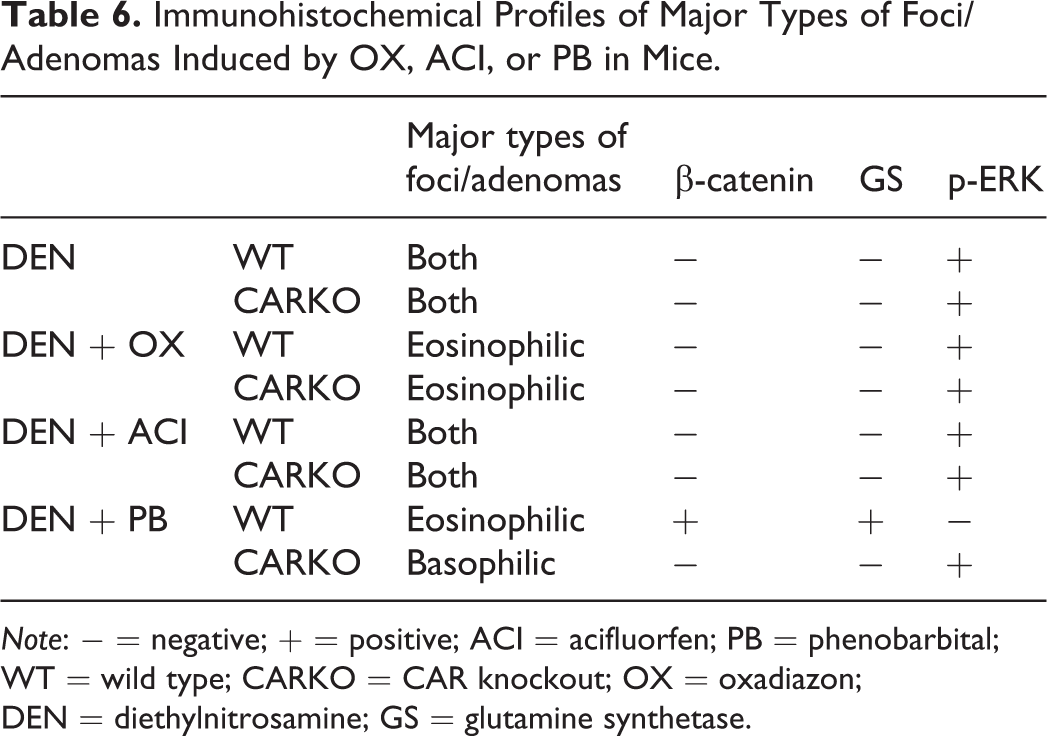
Note: − = negative; + = positive; ACI = acifluorfen; PB = phenobarbital; WT = wild type; CARKO = CAR knockout; OX = oxadiazon; DEN = diethylnitrosamine; GS = glutamine synthetase.
The present study demonstrated that almost all foci and adenomas induced by both PROTOX inhibitors were negative for β-catenin and GS, whereas CAR was important for one of the MoAs of tumor development in our previous studies (Kuwata et al. 2016a, 2016b). As described above, PB treatment after DEN initiation promoted β-catenin-mutated foci and adenomas. Liver tumors promoted by PB and the CAR agonist 1,4-Bis-[2-(3,5-dichloropyridyloxy)] benzene show positive expression of β-catenin signaling-related genes (Dong et al. 2015; Giera et al. 2010). Therefore, the lack of contribution of β-catenin signaling in foci and adenomas, both of which are involved in early-stage liver tumor development, indicated that the MoAs of liver tumor induction by OX and ACI are different from that of PB. Additionally, our results indicated that rodent CAR had complex roles in liver tumor development depending on the compound used for cancer induction.
Interestingly, in ACI-treated WT mice, HCCs were positive for β-catenin and GS, although β-catenin signaling was not involved in early stages of liver tumor development. Both the immunohistochemical characteristics and the increased expression of β-catenin target genes GS and Axin2 in HCCs supported mutational activation of β-catenin in HCC. Thus, these findings suggested that β-catenin plays an important role in ACI-induced tumor progression from adenoma to carcinoma. β-catenin mutations have been reported to be important factors contributing to tumor progression in rodent and human HCC (Inagawa et al. 2002; Ogawa et al. 1999). DEN, which does not activate CAR, induces HCCs harboring β-catenin mutations in mice but does not induce foci and adenomas harboring β-catenin mutations (Ogawa et al. 1999). Additional studies are needed to confirm these findings; however, the present study suggested that key molecular events may be shifted during hepatocarcinogenesis induced by ACI treatment in mice.
ERK phosphorylation was mainly observed in basophilic foci and adenomas in all groups of both genotypes. DEN initiates liver tumors containing Ha-ras (∼50%) or B-raf (∼20%) mutations, leading to phosphorylation of ERK (Buchmann et al. 2008; Jaworski et al. 2005). Thirty percent of spontaneous liver tumors in B6C3F1 mice contain the Ha-Ras proto-oncogene (Maronpot et al. 1995). Therefore, ERK phosphorylation in foci and adenomas may be caused by DEN-induced or spontaneous mutations in Ha-ras or B-raf. In our study, OX and ACI increased the multiplicities and areas of p-ERK-positive foci and adenomas in WT mice, whereas PB did not. This difference may be related to differences in the promoting effects of CAR on the development of p-ERK-positive foci and adenomas. Importantly, ERK phosphorylation is important for cellular proliferation and tumor development (Bobrovnikova-Marjon et al. 2010). However, the specific mechanism through which p-ERK contributes to tumor development induced by OX and ACI was unclear because the frequency of p-ERK positivity in foci and adenomas was limited.
ERK phosphorylation was also observed in RHHs in ACI-treated WT mice. RHHs have been accepted as a regenerative response to continuous cytotoxicity, and the pathogenesis of RHHs is different from that of neoplastic lesions, including adenomas (Thoolen et al. 2010). In adult rodent hepatocytes and the regenerating liver, p-ERK plays a key role in proliferation (Talarmin et al. 1999). However, in the present study, we could not detect the differences of molecular profiles between foci, adenomas, and RHHs. Further studies are needed to elucidate the mechanisms of ERK phosphorylation and its contribution to tumor development or RHH.
PB-induced foci and adenomas showed clear differences in immunohistochemical characteristics between eosinophilic and basophilic types based on positive expressions of β-catenin and GS in eosinophilic lesions and of p-ERK in basophilic lesions, respectively. In contrast, their characteristics induced by OX and ACI were not clear because both eosinophilic and basophilic types of lesions expressed p-ERK. In the case of promoted tumors, investigation of the molecular profiles rather than classic histopathological classification of foci and adenomas in the liver may be useful for investigation of the MoAs of tumor development.
Conclusion
We found that β-catenin mutations were not involved in early-stage hepatocarcinogenesis induced by PROTOX inhibitors in mice, although combined activation of β-catenin and CAR is known to be important in PB-induced tumorigenesis. The significant differences in molecular profiles suggested that multiple MoAs are involved for hepatocarcinogenesis induced by PROTOX inhibitors.
Footnotes
Authors’ Note
Kazunori Kuwata is an employee of Mitsubishi Tanabe Pharma Corporation, Saitama, Japan.
Acknowledgments
We thank Ms. Yoshimi Komatsu and Ms. Ayako Saikawa for their expert technical assistance in performing the processing histological materials.
Author Contribution
Authors contributed to conception or design (KK, KI, and MY); data acquisition, analysis, or interpretation (KK, KI, RI, MT, YK, MS, and MY); drafting the manuscript (KK); and critically revising the manuscript (KI, RI, MT, YK, MS, and MY). All authors gave final approval and agreed to be accountable for all aspects of work in ensuring that questions relating to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was commissioned under a grant for the 2013 Cabinet Office Research for Assessment of the Effect of Food on Human Health, Japan (Topic No. 1303).