
Abstract
Estrogen receptor alpha (ESR1) is 1 of the 2 intracellular receptors for estrogen and is expressed by hepatocytes in the liver. The role of ESR1 in the regulation of toxicant-induced liver injury and compensatory regeneration is not completely clear. We investigated the role of ESR1 in liver regeneration after carbon tetrachloride (CCl4)-induced liver injury using wild type (WT) and ESR1 knockout (ESR1-KO) rats. Adult female WT and ESR1-KO rats were treated with 1 mL/kg CCl4 and euthanized over a time course of 0 to 48 hours. Liver injury measured by serum alanine amino transaminase, and histopathological analysis showed significantly higher liver injury in ESR1-KO as compared to WT rats. Hematoxylin and eosin staining revealed 2-fold higher necrosis and significant inflammatory cell infiltration in ESR1-KO rats. Chloracetate esterase staining revealed higher neutrophil infiltration in ESR1-KO rat livers. Interestingly, proliferating cell nuclear antigen immunohistochemistry showed that in spite of 2-fold higher liver injury, the ESR1-KO rats had equal liver regeneration as compared to WT rats. Western blot analysis of cyclin D1 and phosphorylated Rb, proteins involved in the initiation of the cell cycle, was significantly higher at all time points in ESR1-KO rats. Further analysis revealed faster activation of canonical Wnt/β-catenin and NF-κB signaling in ESR1-KO rats characterized by higher activated β-catenin and phosphorylated p65 at 12 hours after CCl4 treatment. Taken together, these data indicate that ESR1-mediated signaling inhibits liver regeneration by downregulation of Wnt signaling resulting in lower cyclin D1 activation after chemical-induced liver injury.
Introduction
The liver has a remarkable ability to regenerate itself after injury or partial resection. Studies show that the ability of the liver to regenerate is the key determinant of final outcome after injury or tissue resection. 1 –5 The mechanisms of liver regeneration have been mainly studied using the surgical resection model of partial hepatectomy. 4,6 However, it is known that acute liver injury induced by overdose of drugs such as acetaminophen and experimental chemicals such as carbon tetrachloride (CCl4) is followed by compensatory increase in liver cell proliferation. 5,7 –9 In drug/chemical-induced injury models, there is extensive cell death followed by inflammatory response along with cell proliferation. 5 Carbon tetrachloride is a model toxicant used to study both liver injury and subsequent liver regeneration. Liver injury and recovery from CCl4 dosing follow a classic dose–response up to a threshold dose, past which there is no increase in regeneration and tissue repair. 10 Whereas it is known that liver regeneration is a critical determinant of the final outcome of toxicant exposure, the exact mechanisms of liver regeneration after toxic injury remain unclear.
Estrogen is the primary female steroid hormone that signals via binding to its cognate receptors called estrogen receptors, which exist in nuclear (intracellular) and membrane-bound forms. 11 The nuclear estrogen receptor exists in 2 isoforms, estrogen receptor alpha (ERα, also called ESR1) and ERβ (also called ESR2), both of which are members of the nuclear receptor subfamily 3 (NR3) of transcription factors. 11 Estrogen plays an important role in sexual development and reproduction, and disruption of ESR1 leads to infertility, small testes in males, and polycystic ovaries in females. 12 –14 In the liver, ESR1 is expressed exclusively by the hepatocytes, whereas biliary cells express both ESR1 and ESR2. 15 It has been previously shown that estrogen can act as a strong mitogen for hepatocytes and helps promote regeneration by increasing levels of cyclin D1 messenger RNA (mRNA) levels in hepatocytes. 16 Ovariectomized female rats given estrogen pellets showed a greater liver regeneration after portal branch ligation as compared to untreated ovariectomized female rats. 17 Similarly, the treatment of tamoxifen, an estrogen antagonist, to rats after partial hepatectomy (PHX) shows a decrease in hepatocyte proliferation. 18 Interestingly, estrogen-mediated signaling has been theorized to protect against the development of hepatocellular carcinoma (HCC), the primary hepatic malignancy. 19 –22 It is known that HCC incidence is significantly higher in males than females, and estrogen signaling has been hypothesized as the main protective force by inhibiting cellular proliferation. 20 Estrogen is also shown to be anti-inflammatory in the liver following injury. 19,23 This is important because inflammation is known to be a major driver of HCC pathogenesis. 19,24,25 A major component of HCC pathogenesis is tissue injury and subsequent compensatory regeneration, which is missing in the PHX model. 4 It is possible that estrogen and ESR1-mediated signaling plays a different role in situations involving injury and regeneration. The exact role of estrogen and ESR1 signaling in regulation of hepatocyte proliferation in general and specifically after toxicant-induced liver injury remains unclear.
We investigated the role of estrogen and ESR1 signaling in liver injury and regeneration after CCl4-induced injury model using the newly developed ESR1 knockout (ESR1-KO) rats. Carbon tetrachloride-induced liver injury is not only a well-studied model of chemical-induced liver injury and regeneration but also exhibits several components of HCC pathogenesis including liver injury, inflammation, stellate cell activation, and compensatory proliferation. Because of the comprehensive nature of CCl4 model, it can provide insights into both regeneration and HCC pathogenesis mechanisms and connections between them.
Materials and Methods
Animals and Tissue Preparation
All animal studies were approved by and performed in accordance with the Institutional Animal Care and Use Committee (IACUC) at the University of Kansas Medical Center. The generation, genotyping, and characterization of the ESR1-KO rats have been described in detail previously. 12 The rats used in this study were provided by Dr Soares laboratory. Two to three-month-old female wild type (WT) or ESR1-KO rats (n = 3-4 per time point) were treated intraperitoneally with CCl4 (1 mL/kg; Sigma, St Louis, Missouri) in a 1:1 mixture with corn oil. Rats were euthanized over a time course of 0 to 48 hours. Liver and blood were collected and processed as previously described. 26 Liver and serum samples were used to determine markers of injury and regeneration as described before. 27
Protein Isolation and Western Blot
Proteins were isolated from liver samples for Western blot using methods previously described. 26 All Western blot antibodies were purchased from Cell Signaling Technology (Danvers, Massachusetts) except for active β-Catenin (Millipore, Billerica, Massachusetts).
Histology and Immunohistochemistry
Hematoxylin and eosin (H&E)-stained paraffin sections of livers from CCl4-treated WT and ESR1-KO rats were used for percent necrosis scoring as described before. 27 Two sections per slide per rat and 3 to 4 rats per time point per genotype were used for necrosis scoring. Proliferating cell nuclear antigen (PCNA) and H&E staining were performed using 4-µm thick paraffin-embedded liver sections as described previously. 26 Proliferating cell nuclear antigen positive cells were quantified in 10 high-power (400×) fields of liver sections from at least 3 individual rats.
Neutrophil Staining
Neutrophils were monitored using a chloroacetate esterase stain (Sigma) according to the manufacturer’s suggested methods. Slides were counterstained with hematoxylin for 2 minutes. Neutrophils were counted in ten 400× fields for quantification.
Statistical Analysis
All bar graphs depict the mean (standard deviation). To determine statistical significance, 1-way analysis of variance (ANOVA) on the time course within a group was used. For nonparametric analysis, the Mann-Whitney U test was used. Finally, the Dunn post hoc test was used to compare samples versus control for nonparametric and Student-Newman-Keuls for normally distributed data.
Results
Significantly Higher Liver Injury and Increased Inflammation in ESR1-KO
There is no difference in baseline histology of the liver between the WT and ESR1-KO rats (data not shown). Liver injury after CCl4 was measured by serum alanine amino transaminase (ALT) activity and H&E staining of paraffin-embedded liver sections (Figure 1). Hepatocyte balloon degeneration and necrosis were evident between 6 to 48 hours in both groups (Figure 1A). Both groups had a peak ALT levels at 6 hours and a subsequent decrease from 12 to 48 hours. Serum ALT increased in both groups at 6 hours after CCl4 treatment where it was moderately higher in ESR1-KO rats but was not statistically significant (Figure 1B). Alanine amino transaminase levels decreased in both groups at 12 to 48 hours but were significantly higher in ESR1-KO rats. Necrosis scoring demonstrated a 2-fold higher liver injury and cell death in ESR1-KO livers at 6, 12, and 24 hours (Figure 1C). Increased inflammatory cell foci appeared in both WT and ESR1-KO livers after 12 hours after CCl4 treatment and persisted throughout the time course.
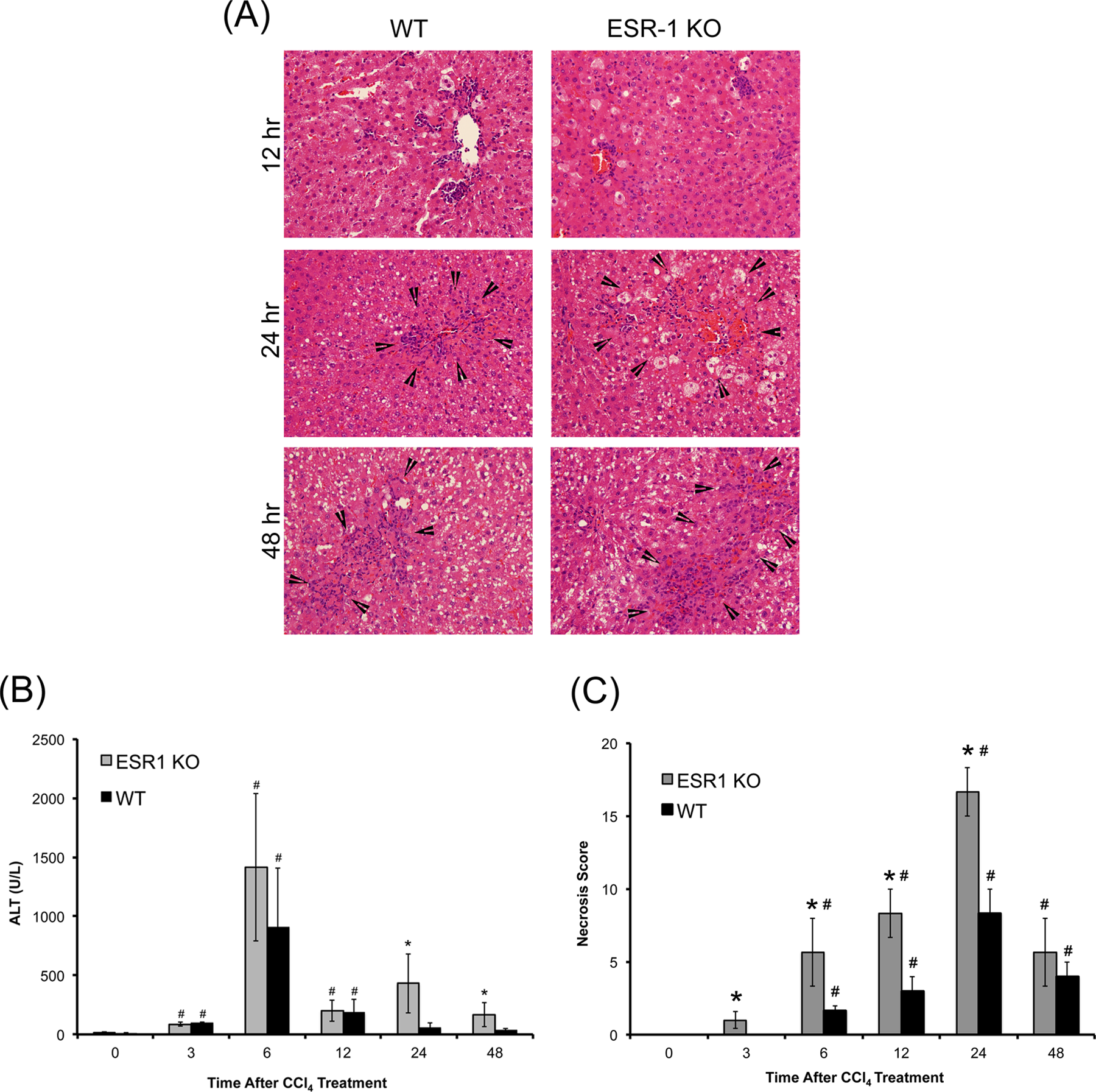
Carbon tetrachloride (CCl4)-induced liver injury in wild type (WT) and estrogen receptor alpha knockout (ESR1-KO) rats. A, Representative photomicrographs (400×) of Hematoxylin and eosin (H&E)-stained liver sections of WT and ESR1-KO rat liver at 12, 24 and 48 hours after CCl4 treatment. Black arrowheads point to centrilobular necroinflammatory foci. Bar graphs showing serum alanine amino transaminase (ALT; B) and (C) percent necrosis in WT and ESR1-KO rats after CCl4 treatment. *Significant difference at P
Higher Neutrophil Infiltration in ESR1-KO Rats
To further evaluate neutrophil infiltration in the liver after CCl4 treatment, we stained liver sections from WT and ESR1-KO rats using chloracetate esterase histochemistry (Figure 2A) and counted the number of neutrophils, evident from their lobulated nuclei (Figure 2B). These data indicate that both WT and ESR1-KO rats had significant neutrophil infiltration in the liver after CCl4 treatment. At 12 hours after CCl4, both groups had similar number of neutrophils. However, at 24 and 48 hours, ESR1-KO rats had significantly higher neutrophils congregated in and around the necrotic foci.
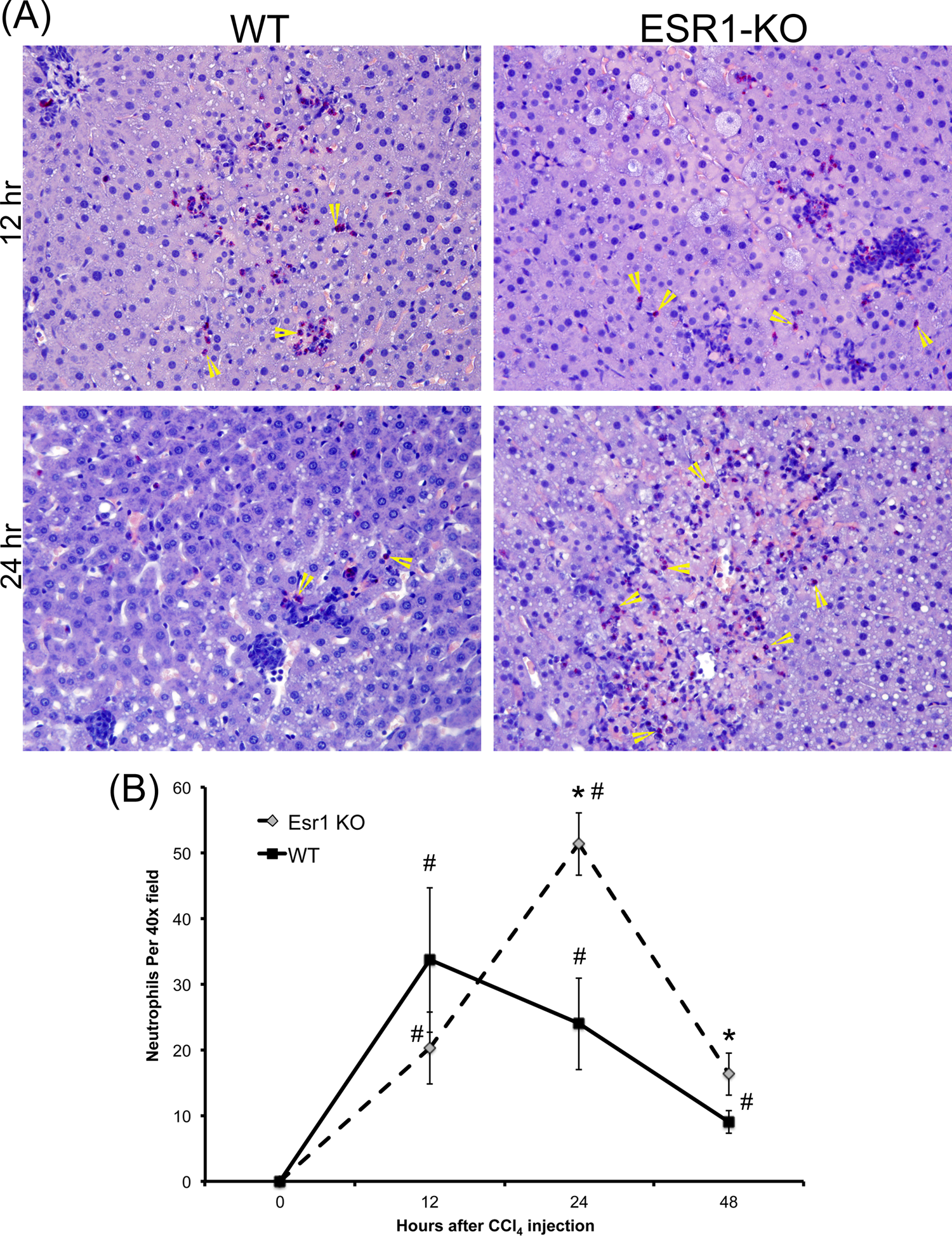
Increased neutrophil infiltration in estrogen receptor alpha knockout (ESR1-KO) rat livers after carbon tetrachloride (CCl4) treatment. A, Representative photomicrographs (400×) of chloracetate esterase (CAE)-stained liver sections of wild type (WT) and ESR1-KO rat liver at 12 and 24 hours after CCl4 treatment. Yellow arrowheads point to neutrophils. B, Line graph showing neutrophil count of CAE-stained liver sections of WT and ESR1 rat livers at various time points. * Significant difference at P
Stimulated Liver Regeneration in ESR1-KO
Compensatory liver regeneration was studied using Western blot analysis of critical cell cycle proteins such as cyclin D1, cyclin E, cyclin-dependent kinase 4 (CDK4), and phosphorylated retinoblastoma (Rb) protein. Western blot analysis showed that cyclin D1 protein expression is greater in ESR1-KO rats as compared to WT rats at all time points studied, with peak expression at 24 hours post-CCl4 treatment (Figure 3A and B). Cyclin E expression increased in both WT and ESR1-KO livers from 12 to 24 hours and was undetectable at 48 hours. There was no significant difference in cyclin E expression between WT and ESR1-KO groups at any time point. Protein expression of CDK4 was unchanged in the ESR1-KO rats when compared to ESR1-WT rats as shown by Western blot. Cyclin-dependent kinase 4 phosphorylates its downstream target, the Rb protein. Western blot analysis showed that phosphorylation of Rb protein was significantly increased at 12 and 24 hours in ESR1-KO rats when compared to ESR1-WT rats (Figure 3C). Cell proliferation was further studied using PCNA immunohistochemistry (Figure 3D and E). No PCNA positive cells were observed at 12 hours after CCl4 treatment in either group. Equal numbers of PCNA positive cells were observed in WT and ESR1-KO rat livers at 24 and 48 hours. These data indicate that ESR1-KO rats exhibit equal stimulation of compensatory liver regeneration as compared to WT mice despite 2-fold higher liver injury.
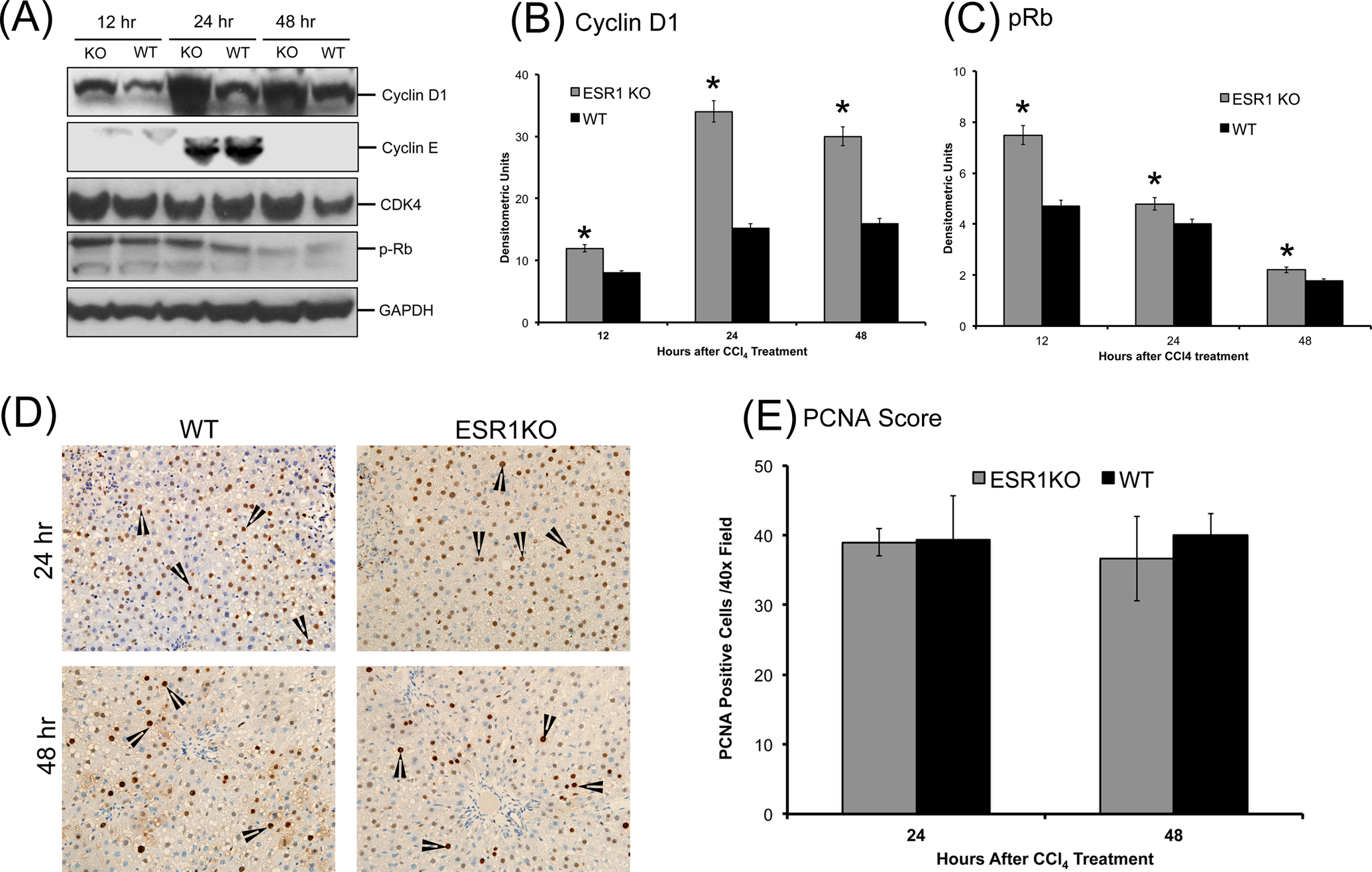
Accelerated compensatory cell proliferation in estrogen receptor alpha knockout (ESR1-KO) rats after carbon tetrachloride (CCl4) treatment. A, Western blot analysis of cyclin D1, cyclin E, cyclin-dependent kinase 4 (CDK4), and phosphorylated retinoblastoma (Rb) proteins at 12, 24, and 48 hours after CCl4 treatment performed using total liver cell extracts of WT and ESR1-KO rat livers. B and C, Densitometric analysis of cyclin D1 and pRb blots. D, Representative photomicrographs of proliferating cell nuclear antigen (PCNA) immunohistochemistry (400×) performed on WT and ESR1-KO rat livers at 24 and 48 hours after CCl4 treatment. Black arrowheads point to cells in S-phase of cell cycle (E) bar graph showing the number of PCNA positive cells. *Significant difference at P
Increase in Wnt/β-Catenin and p65 Signaling in ESR1-KO Rats
To determine the mechanism behind the stimulated liver regeneration in ESR1-KO rats despite higher injury, we investigated several pathways known to induce cyclin D1 and cell proliferation. Our investigation revealed higher activation of canonical Wnt signaling pathway in ESR1-KO rats. Western blot analysis showed an increase in activated (nonphosphorylated) β-catenin expression at 12 hours after CCl4 administration (Figure 4). Further, phosphorylation of GSK3β, the upstream regulator of β-catenin was also moderately higher in the ESR1-KO rats at 12 hours. Both active β-catenin and p-GSK3β increased in WT rat liver at 24 hours, coinciding with increased cyclin D1 expression and cell proliferation. We also studied expression and activation of p65, which homodimerizes to form active NF-κB signaling molecule. There was no difference in total p65 protein (data not shown), but the phosphorylated active form of p65 was significantly higher in the ESR1-KO mice at 12 hours after CCl4 treatment (Figure 4A).
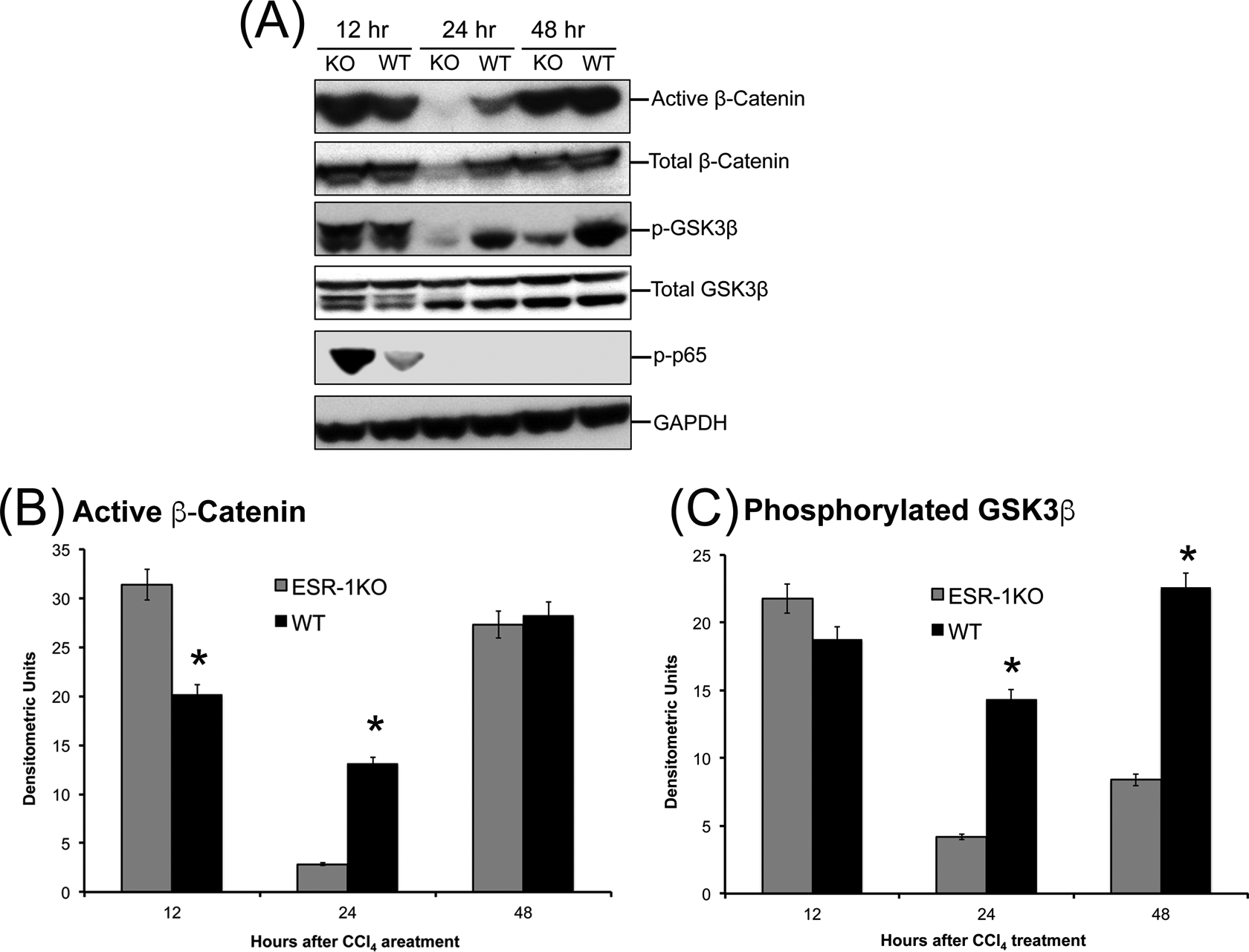
Increased β-catenin activation in estrogen receptor alpha knockout (ESR1-KO) livers after carbon tetrachloride (CCl4) treatment. A, Western blot analysis of total and activated β-catenin, phosphorylated nuclear p65, and total and phosphorylated GSK3β proteins at 12, 24, and 48 hours after CCl4 treatment performed using total liver cell extracts of wild type (WT) and ESR1-KO rat livers. B, Bar graphs showing densitometric analysis of the active β-catenin Western blot. C, Bar graphs showing densitometric analysis of the phosphorylated GSK3β Western blot. *Significant difference at P
Discussion
Estrogen and estrogen receptor signaling play a central role not only in reproduction but also cell injury and repair. Here, we demonstrate that the disruption of ESR1 leads to significantly higher liver injury after CCl4 treatment. However, in spite of higher liver injury, the ESR1-KO rats exhibit rapid increase in compensatory regeneration after liver injury by CCl4. Previous studies on the role of estrogen-induced signaling have revealed contradictory roles in liver cell proliferation. Estrogen and estrogen receptor signaling seems to promote liver regeneration after PHX, 28 –30 but estrogen receptor signaling also provides protection against the development of HCC, 20 –23,31 where cell proliferation is an important component. Furthermore, apart from these seemingly contradictory results, the role of ESR1 in liver regeneration after chemical-induced liver injury has not been evaluated.
We investigated the role of estrogen-mediated signaling using the novel ESR1-KO rats and the CCl4 model of liver injury and regeneration. The CCl4 model provides several advantages over the PHX model including the development of intrahepatic injury, inflammation, and compensatory proliferation, all of which are part of HCC pathogenesis. Our studies indicate that the disruption of ESR1-KO rats developed a significantly higher liver injury after CCl4 treatment. The higher injury was accompanied by higher inflammatory response in ESR1-KO rats than in the WT rats. Consistent with these data, we also observed increased neutrophil numbers in ESR1-KO rats after CCl4 administration. This is further supported by higher expression of phosphorylated p65, which indicates higher pro-inflammatory NF-κB signaling. These data are consistent with previous observations that estrogen-mediated signaling via ESR1 is anti-inflammatory. 19,21,23,31 Other studies indicate that estrogen may block neutrophil infiltrations and activation. 32 Taken together, these data indicate that the disruption of ESR1-mediated estrogen signaling results in higher liver injury and increased postinjury inflammatory response after acute exposure to hepatotoxicants.
It is well established that liver injury induced by chemicals results in compensatory liver regeneration. 4,5,7 It is also known that the compensatory liver regeneration is inhibited by higher liver injury. Very high tissue injury can inhibit tissue repair by various mechanisms including significant cell stress and lack of critical promitogenic signaling. 5,27 Interestingly, we observed that in spite of 2-fold higher liver injury, compensatory liver regeneration was completely unaffected in the ESR1-KO rats. Our data indicate that the disruption of ESR1 may result in removal of estrogen-mediated inhibitory effect of hepatocyte proliferation, which allows the ESR1-KO hepatocytes to enter cell cycle despite high cellular injury. Estrogen receptor alpha knockout livers had higher activation of cyclin D1 and pRb, the 2 critical regulators of cell cycle entry. Interestingly, while expression of the S phase cyclin, cyclin E was upregulated in both WT and ESR1-KO rats, we did not observe any difference between the genotype. These data indicate that the deletion of ESR1 affected mainly the cell cycle entry rather than cell cycle progression following CCl4 administration. This suggests that estrogen-mediated signaling may inhibit cell cycle entry of the hepatocytes. Overall, the faster increase in cyclin D1 and pRb in ESR1-KO rats resulted in equal compensatory cell proliferation in ESR1-KO rats despite much higher liver injury.
Further studies demonstrated that the higher cyclin D1 protein induction in ESR1-KO rats might be due to increased Wnt/β-catenin and NF-κB signaling. Others and we have previously shown that cyclin D1 is a target gene of β-catenin 33 –36 and is activated by canonical Wnt signaling during regeneration after acetaminophen overdose. 27 We observed an increase in activated (dephosphorylated) β-catenin in ESR1-KO rat liver much earlier than the WT liver after CCl4 treatment. We also observed a concomitant increase in phosphorylation of GSK3β, which is the inactive form of GSK3β involved in β-catenin activation. Similarly, we observed an increase phosphorylation of p65 indicating increased NF-κB signaling. 37 –39 NF-κB is also known to stimulate cyclin D1 expression. The exact mechanism(s) by which ESR1 disruption upregulates Wnt signaling remains to be investigated. Previous studies have shown that ESR1 and β-catenin form a regulatory complex. 40 It is possible that lack of ESR1 results in increased free β-catenin in hepatocytes. Alternatively, the lack of ESR1-mediated signaling may result in increased Wnt signaling that further induces inactivation of GSK3β and activation of β-catenin.
Our study has shown that the disruption of ESR1 in rat liver results in significantly higher cell injury but equal cell cycle activation, which is contrary to the role of estrogen and ESR1 in liver regeneration after PHX. One of the reasons for this discrepancy is the inherent difference in the models. In PHX model, where two-thirds of the liver is surgically removed and the remaining liver is allowed to regenerate, there is minimal cell death in the regenerating lobes and no inflammation. In contrast, there is significant cell death after CCl4 treatment, and cells that are next to the necrotic zone undergo proliferation. Additionally, there is significant inflammation as shown in our studies. It is known that estrogen can inhibit inflammatory signaling, and the lack of ESR1 seems to exacerbate the inflammatory response. The inflammatory cells have a dual function in regeneration after toxicant injury. They are involved in phagocytosis of necrotic cell debris. They are also involved in secretion of promitogenic cytokines and growth factors that further stimulate surrounding hepatocytes to proliferate. In our model, the significant inflammatory cell infiltration may be involved in secretion of promitogenic signals, which further induce cell proliferation. Thus, the lack of injury and inflammation in PHX model may result in a differential role of estrogen signaling in liver regeneration after PHX.
In summary, our studies indicate that the disruption of ESR1 in a model of toxicant-induced injury results in higher injury and inflammation. However, the disruption of ESR1 removes inhibitory effects of estrogen signaling on compensatory cell proliferation, and the dynamics of liver regeneration remains unaffected. These data also partially explain the protective effect of estrogen-mediated signaling on HCC as inflammation is known as a major component of HCC pathogenesis. The ESR1-KO rat is an innovative experimental model and could be further used to determine the mechanisms of hepatic injury and regeneration in the context of chemical cancer pathogenesis.
Footnotes
Author Contributions
S. McGreal and U. Apte contributed to conception and design, contributed to acquisition, analysis, and interpretation, drafted the manuscript, and critically revised the manuscript; M. A. K. Rumi, M. Soares, and H. Jaeschke contributed to analysis and interpretation and critically revised the manuscript; J. Woolbright contributed to acquisition, analysis, and interpretation and critically revised the manuscript. All authors gave final approval and agree to be accountable for all aspects of work ensuring integrity and accuracy.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by NIH grants 8P20 GM103549, 5T32ES007079-34, R01OD01478, R01DK102142, and 1R01DK098414.