
Abstract
Gut homeostasis plays an important role in maintaining animal and human health. The disruption of gut homeostasis has been shown to be associated with multiple diseases. The mutually beneficial relationship between the gut microbiota and the host has been demonstrated to maintain homeostasis of the mucosal immunity and preserve the integrity of the gut epithelial barrier. Currently, rapid progress in the understanding of the host–microbial interaction has redefined toxicological pathology of opioids and their pharmacokinetics. However, it is unclear how opioids modulate the gut microbiome and metabolome. Our study, showing opioid modulation of gut homeostasis in mice, suggests that medical interventions to ameliorate the consequences of drug use/abuse will provide potential therapeutic and diagnostic strategies for opioid-modulated intestinal infections. The study of morphine’s modulation of the gut microbiome and metabolome will shed light on the toxicological pathology of opioids and its role in the susceptibility to infectious diseases.
Gut Homeostasis and Health
The gut is a complex and dynamic network in which the interaction between the host and gut microbiota establishes a balanced, symbiotic, and mutually beneficial relationship (Kau et al. 2011). Gut homeostasis refers to the state of resilience and resistance to external and endogenous disturbances (Lozupone et al. 2012). Gut homeostasis is established and maintained by commensal microbiota, a functional barrier and a tolerant immune response (Brown, Sadarangani, and Finlay 2013). Gut microbiota include all microorganisms within the gastrointestinal (GI) tract, including bacteria, archaea, eukaryotes, fungi, and viruses (Gordon 2012). Microbiome refers to the entire collection of microbial genes in a particular environment (The Human Microbiome Project Consortium 2012). Recent rapid progress in metagenomics has provided powerful tools to determining perturbations of the human microbiome as contributors to diseases (Gordon 2012; Wang and Jia 2016). It is estimated that approximately 1013–1014 bacteria inhabit the GI tract, which exceeds the total number of host cells by two orders of magnitude (Relman 2012). The unborn fetus lives in a basically sterile environment. During birth and thereafter, infants are exposed to the external environment whereby the gut microbial community is initialized, established, and gradually developed (Dominguez-Bello et al. 2010). The human gut microbiota become stable and adultlike at approximately 3–5 years of age (Rodríguez et al. 2015). It has been demonstrated that the gut microbiota play important roles in modulating host neural and immune development, morphogenesis, and resistance to diseases in both human beings and animals (Sommer and Bäckhed 2013). The mechanisms by which the gut microbiota maintain a healthy state and how microbial dysbiosis increases the susceptibility to diseases remain largely unknown (Figure 1).
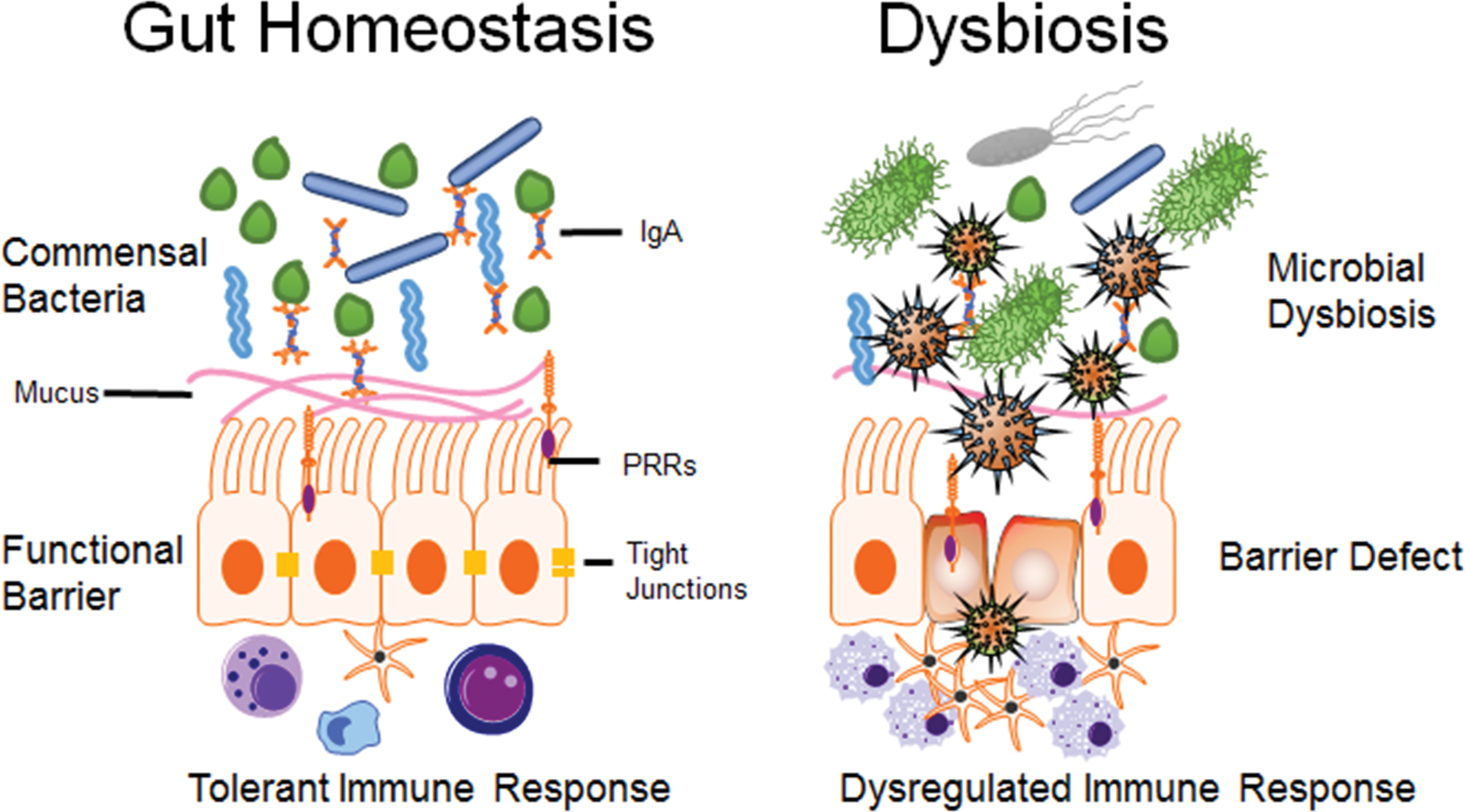
Illustration of gut homeostasis and dysbiosis. Gut homeostasis is maintained by commensal bacteria, functional barrier, and tolerant immune response. The symbiotic relationship between commensal microbiota and the host achieves a balanced, mutually beneficial state. Alteration in gut microbiome has been shown to contribute to dysregulated immune response, bowel dysfunction, and gut barrier disruption.
Microbial Dysbiosis and Diseases
Microbial dysbiosis refers to a change in the structural and/or functional configuration of gut microbiota, which causes disruption of gut homeostasis and is associated with a variety of diseases, such as obesity, diabetes, autoimmune diseases, neurological disorders, allergies, and inflammatory and infectious diseases (Gordon 2012; Sommer and Bäckhed 2013). Changes in the composition or density of the microbiota have been shown to increase the susceptibility to a variety of pathogens and abnormal mucosal immune responses in humans and murine models (Stecher and Hardt 2008; Wells et al. 2011). For example, antibiotic-induced shifts in the mouse gut microbiome and metabolome increase the susceptibility to Clostridium difficile infections (Theriot et al. 2014). In addition, a dysregulation of the host immune defense can change the gut microbiota during host–microbe interactions in mice (Brown, Sadarangani, and Finlay 2013). For instance, Salmonella enterica serotype typhimurium (S. typhimurium), which causes acute gut inflammation, has been associated with a shift in the gut microbiota in favor of pathogen growth in a mouse colitis model (Stecher et al. 2007). In addition, S. typhinurium–induced intestinal inflammation has been demonstrated to promote the production of the respiratory electron acceptor tetrathionate, which provides a growth advantage for S. typhinurium (Winter et al. 2010). Although the specific causal relationships are unclear, a variety of diseases are associated with a vicious circle of microbial dysbiosis, gut barrier dysfunction, and dysregulated immune response. HIV-1 infection–induced alterations of intestinal mucosal microbiome are associated with mucosal and systemic immune activation and endotoxemia in HIV-1-infected humans (Dillon et al. 2014). A dysregulated immune response in inflammatory bowel diseases, such as Crohn’s disease (CD) and ulcerative colitis (UC), has been shown to be associated with a dysfunction of the intestinal microbiome in a clinical study including 121 CD patients, 75 UC patients, and 27 healthy controls (Morgan et al. 2012). Mechanistic links between gut microbial functions and dynamics and host metabolic health are currently important targets of research for clinical therapeutics and in basic research (Ha, Lam, and Holmes 2014).
The association between microbial signatures and disease demonstrates potential diagnostic application in monitoring disease onset and progression. The microbial signature refers to the interacting biomarkers of diseases and their relationship with the host phenotypes. Such a signature includes not only the taxonomic characterizations of communities but genes or functional configurations. Intestinal Prevotella copri plays a potentially diagnostic role based on its strong correlation with enhanced susceptibility to arthritis in a gut microbiome study of new onset untreated rheumatoid arthritis patients (Scher et al. 2013). After characterization of gut microbiome in patients with liver cirrhosis compared with healthy human subjects, 15 gene markers have been identified as a signature of alterations of the human gut microbiome in liver cirrhosis (Qin et al. 2014). Biomarkers specific for liver cirrhosis are powerful tools in differential diagnosis. The specificity of biomarkers for liver cirrhosis was confirmed by comparisons with those for type 2 diabetes and inflammatory bowel diseases (Qin et al. 2014). Metagenome-wide association studies have produced new advances in the disease prevention, diagnosis, and treatments (Wang and Jia 2016).
An altered microbiome, directly reflecting changes in microbiota, is not only a marker of disease but also actively contributes to disease pathogenesis (Chassaing et al. 2012; Goodrich et al. 2014). The causal relationship is established when the healthy host displays the disease phenotype when the gut microbiota from diseased donors are transplanted into healthy germfree hosts (Turnbaugh et al. 2006; Faith et al. 2014). To determine direct causal relationship between microbiome and disease, the approach of microbiota transplantation has been conducted in studies of multiple diseases, such as colitis, type 1 diabetes, and metabolic diseases (Wen et al. 2008; Elinav et al. 2011; Koren et al. 2012; Garrett 2013).
Another important area of research is the relationship between the microbiome and therapeutic treatment. Probiotics and prebiotics have a long history as therapeutic tools in treating diarrhea, vaginal yeast infections, urinary tract infections, irritable bowel syndrome, and preventing or reducing the severity of colds and flu (Leyer et al. 2009; Vouloumanou et al. 2009; Ciorba 2012; Borges, Silva, and Teixeira 2014). Recently, fecal microbial transplantation has been conducted effectively in treating recurrent C. difficile infection in human patients (Hamilton et al. 2012; Weingarden et al. 2014). The administration of prebiotics (oligofructose) to genetically obese mice decreases the fat to muscle mass ratio, improves glucose and lipid metabolism, reduces plasma Lipopolysaccharide (LPS), improves gut barrier function, and increases enteroendocrine L-cell number in obese mice (Everard et al. 2011). Administration of oligofructose induces changes in gut microbiota and increases the abundance of Akkermansia muciniphila by 100-fold in obese mice (Everard et al. 2011). Another study from the same research group shows that the abundance of A. muciniphila decreased in obese and type 2 diabetic mice, whereas administration of A. muciniphila reduces body weight, improved metabolic disorders, and counteracts mucosal barrier dysfunction in obese mice (Everard et al. 2013). Remedies for gut dysbiosis and restoration of healthy gut homeostasis have become important therapeutic strategies to cure diseases.
Opioids and Opioid Receptors
Opioids refer to a large group of compounds and chemicals that share characteristics with opium (Ninković and Roy 2013). Synthetic opioids resemble morphine in pharmacological effects (Degenhardt et al. 2013). Opioids are powerful analgesics that are relatively inexpensive and are widely prescribed and used worldwide to control pain (Docherty, Jones, and Wallace 2011). However, their clinical use has been impacted by concerns for drug abuse and by adverse effects such as addiction, immunosuppression, and GI symptoms (Hilburger et al. 1997; Gomes et al. 2011; Docherty, Jones, and Wallace 2011; Juurlink and Dhalla 2012). Opioids can induce other central nervous system (CNS) effects, including sedation, euphoria, and dysphoria (Coetzee 2013). Epidemiological evidence indicates increased mortality among older adults who use opioids (Larney et al. 2015). Mortality rates of dependent opioids users are approximately 15-fold higher than age- and sex-matched control subjects (Degenhardt et al. 2011).
Opioid analgesics alter the transmission and perception of pain by binding to cellular μ-, κ-, and δ-opioid receptors (Chan 2008). Most of the clinical effects of opioids are µ receptor dependent (Ninković and Roy 2013). Opioids may exert their effects as “pure” μ-opioid receptor agonists (e.g., morphine, hydromorphone), partial μ agonists (e.g., buprenorphine), and agonist–antagonists (e.g., butorphanol; Epstein 2013). Although the primary pharmacological effects of opioids are dependent on the distribution of opioid receptors in the CNS, opioid receptors are also expressed in the peripheral nervous system, the GI tract, and the myenteric plexus of the enteric nervous system (Farzi et al. 2015). In addition, opioid receptors are expressed by the cells of the immune system, such as T cells, B cells, and macrophages (Roy et al. 2011; Ninković and Roy 2013). Thus, activation of opioid receptors not only affects the perception of pain but also induces a variety of side effects attributed to regulation of neurological and immune systems.
Opioids and Gut Homeostasis
Opioids Modulate Intestinal Function
Primary functions of GI tract include digestion, absorption, secretion, motility, immune surveillance, and tolerance (Leppert 2015). Opioid administration is associated with multiple GI symptoms, such as constipation, leaky intestinal barrier function, bloating, nausea, and vomiting (Harari, Weisbrodt, and Moody 2006). Morphine inhibits protective mucus and bicarbonate secretion from the intestinal epithelium and human bronchi (Rogers and Barnes 1989). Opioid treatment attenuates intestinal motility by inhibiting coordinated myenteric activity, thereby delaying transit time and potentially increasing the risk for bacterial translocation in humans (Balzan et al. 2007). Morphine has also been shown to attenuate epithelial immune function by decreasing cytokine secretion from porcine jejunal epithelial IPEC-J2 cells in response to enteroadherent Escherichia coli O157: H7 and enteroinvasive S. enterica serovar typhimurium (Brosnahan et al. 2014). Human clinical data and mouse experimental data indicate that opioid administration leads to a dysregulated immune response, increased intestinal barrier permeability, bacterial translocation, increased risk of enteric infection, and gut-derived sepsis (Mora et al. 2012; Babrowski et al. 2012).
Opioids Impair Gut Epithelial Integrity
Disruption of gut epithelial integrity has severe consequences, including bacterial translocation from the gut, leading to proinflammatory immune responses (Schulzke et al. 2009). Well-organized transmembrane and paracellular tight junction proteins in polarized intestinal epithelium facilitate their selective barrier function. Meng et al. demonstrated that morphine disrupts intestinal barrier function and damages tight junction protein organization via modulation of myosin light-chain kinase in a TLR-dependent manner (Meng et al. 2013). Naltrexone, an opioid antagonist, has been shown to have a therapeutic effect on mucosal healing in active CD, thus implicating opioid receptors in the maintenance of gut epithelial integrity (Smith et al. 2011).
Opioids Modulate Immune Systems
Studies on modulation of immune systems by chronic opioid use and abuse are well documented and reviewed (Roy and Loh 1996; Hutchinson, Shavit, and Grace 2011; Roy et al. 2011; Ninković and Roy 2013). Opioid receptors are expressed on cells of the immune system, such as B cells, T cells, and macrophages (Eisenstein 2011). Thus, opioids exert their pharmacological effects not only as analgesics but also as regulators of immune function. Roy et al. (1991, 1998) demonstrated that morphine suppresses macrophage colony formation in bone marrow and modulates NFκB activation in macrophages. Morphine produces immunosuppressive effects, attenuates T-cell maturation, alters cytokine secretion, and decreases the production of protein mediators of energy metabolism, signaling, and cell structure maintenance in nonhuman primates (Brown et al. 2012). Morphine-induced neuro-immune interactions cause direct intestinal functional consequences. Meng et al. (2013) demonstrated that morphine disrupts gut barrier function via a toll-like receptors (TLR)-dependent manner. Chronic morphine treatment inhibits the innate immune response, decreases Th1 cytokine production and T-cell activation, shifts to Th2 differentiation, and reduces antibody production and the major histocompatibility complex, class II (MHC-II) expression, leading to an increased risk of opportunistic infection and impaired pathogen elimination (Roy et al. 2011).
Opioids Induce Gut Microbial Dysbiosis and Bile Dysregulation
Alterations of the gut microbiome have been strongly correlated with a disruption in host metabolic homeostasis (Weingarden et al. 2014). Before secretion into the small intestine, primary bile acids are conjugated with taurine or glycine to form bile salts in the liver (Hofmann and Hagey 2008). Approximately 95% of secreted bile salts are reabsorbed via the enterohepatic recirculation pathway, which occurs in the small intestine. The remaining 5% of secreted bile salts reach the colon, where they are deconjugated and dehydroxylated by the actions of intestinal bacteria and become secondary bile acids such as deoxycholate and lithocholate (Hofmann and Hagey 2008; Nicholson et al. 2012). Secondary bile acids impair C. difficile growth in vitro (Sorg and Sonenshein 2010). Notably, bile acid–mediated resistance to C. difficile can be restored by precision microbiome reconstitution in a murine C. difficile infection model (Buffie et al. 2014). We recently demonstrated in mice that opioids induce gut microbial dysbiosis, disrupt bile acid metabolism, and disrupt barrier function, leading to sustained systemic inflammation. However, the sequence of events remains unknown (Banerjee et al. 2016).
Opioid-induced Microbial Dysbiosis Alters Xenobiotic Metabolism
Morphine metabolism exhibits a variety of host–microbial interactions (Stain-Texier, Sandouk, and Scherrmann 1998). The morphine metabolic pathway is primarily focused on glucuronidation to morphine-3-glucuronide (M3G) and morphine 6-glucuronide (M6G) in the liver (Pacifici et al. 1982). Although M3G exhibits no analgesic effect, M6G is more potent than morphine (Frances et al. 1992). M6G and M3G are hydrolyzed by β-glucuronidase in both intestinal mucosal cells and gut bacteria and subsequently reabsorbed as morphine (Hawksworth, Drasar, and Hill 1971; Walsh and Levine 1975; Koster, Frankhuijzen-Sierevogel, and Noordhoek 1985). Anaerobes, such as Bacteroides and bifidobacteria, are major sources of β-glucuronidase (Walsh and Levine 1975). Given that β-glucuronidase-mediated hydrolysis is dependent on the bacterial composition of the gut, gut microbiota alterations may affect the rate and extent of the hydrolysis of morphine glucuronide. Diet has been shown to significantly influence intestinal bacterial β-glucuronidase activity (Reddy, Weisburger, and Wynder 1974). Fecal microbial β-glucuronidase activity in human subjects consuming Western high-meat diet is increased compared with nonmeat diet (Reddy, Weisburger, and Wynder 1974). However, how gut microbiota alterations affect β-glucuronidase activity and subsequent hydrolysis of morphine glucuronide remains unknown.
Morphine metabolism and elimination play an important role clinically in determining drug pharmacokinetics and assessing their efficacy and its adverse effects. Thus, studies investigating the role of the gut microbiome and its impact on morphine metabolism have significant clinical relevance. Our unpublished studies indicate that the ratio of M3G/morphine sulfate (MS) serum concentration increases in a time-dependent manner. Similar increases are also observed in the feces postmorphine treatment, indicating decreased M3G deconjugation in the gut. The major glucuronide deconjugating bacteria are the strict anaerobes, Bacteroides, and bifidobacteria, which exhibit β-glucuronidase activity (Stain-Texier, Sandouk, and Scherrmann 1998). We demonstrate that morphine treatment results in a decrease in Bacteroidales, suggesting that decreased M3G-deconjugation is a consequence of a reduction in deconjugating bacteria. Cross-correlation associations are noted between intestinal bacterial communities and functional metabolites. Moreover, we demonstrated an increased M3G/MS ratio in the gut, suggesting that alteration of M3G deconjugating microbes may influence opioid metabolism and elimination. Collectively, these results reveal opioid-induced distinct alterations of the gut microbiome and metabolome that may contribute to opioid-induced pathogenesis and morphine pharmacokinetics (Figure 2).
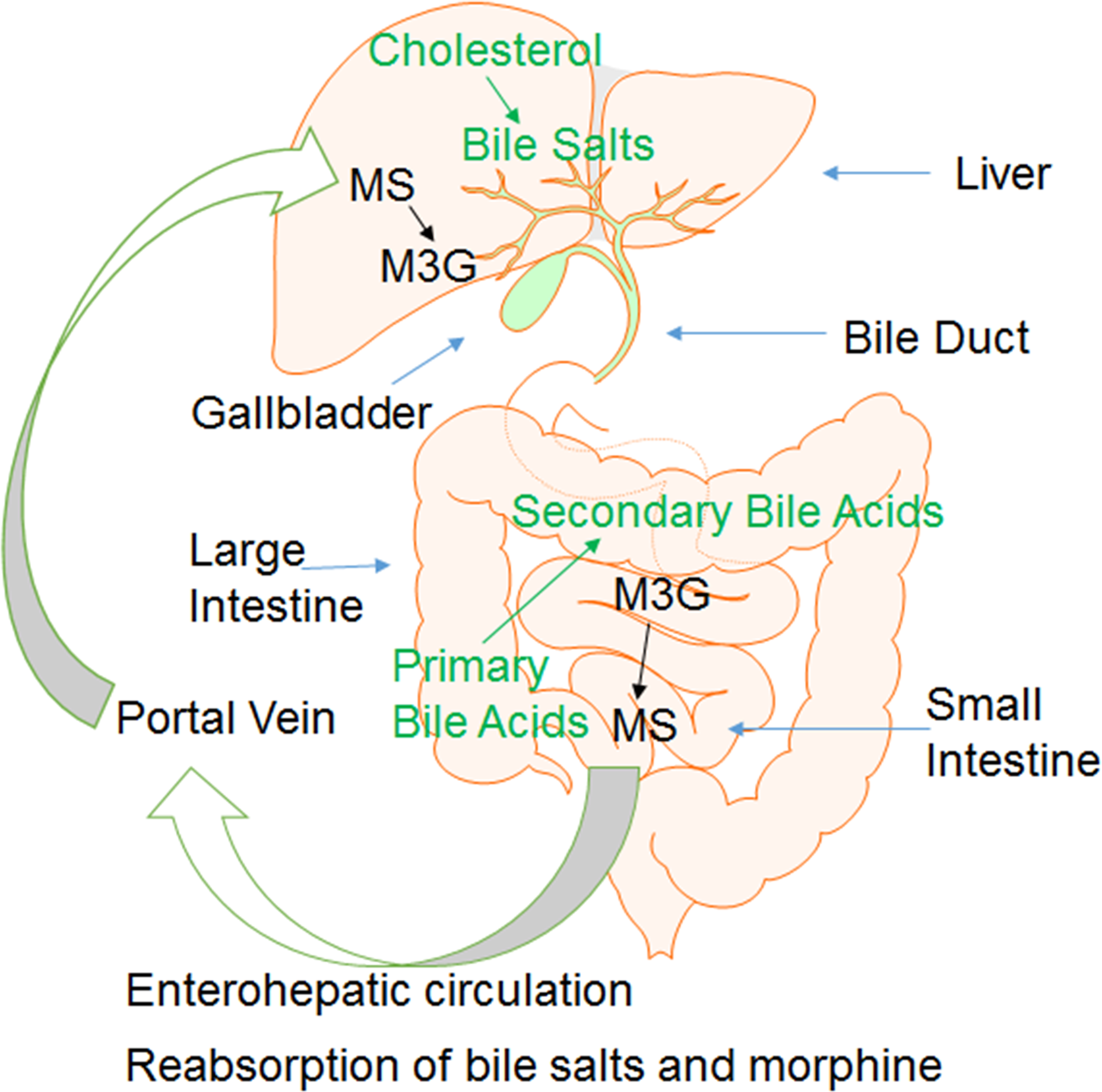
Model of metabolism and biotransformation of morphine and bile acids. In liver, cholesterol was transformed to primary bile acids, and morphine was conjugated to M3G. In gut, intestinal bacteria transform primary bile acids and M3G into secondary bile acids and morphine, respectively. Bile acids and morphine are reabsorbed and recycled via enterohepatic circulation. MS = morphine sulfate; M3G = morphine-3-glucuronide.
Opioids and Diseases
Opioids Induce Sepsis
Morphine treatment has been shown to cause lethal gut-derived sepsis in mice (Hilburger et al. 1997; Babrowski et al. 2012). Morphine-induced sepsis is implicated in the disruption of gut barrier function, increased bacterial translocation and bacterial virulence expression, and dysregulated immune responses in mice (Banerjee et al. 2013). Chronic morphine administration directly activates Pseudomonas aeruginosa virulence expression and leads to lethal gut-derived sepsis in mice (Babrowski et al. 2012). Banerjee et al. (2013) demonstrated that chronic morphine treatment–induced exacerbation of septicemia in mice is mediated by miR-146a regulation.
Opioids Delay Wound Healing
Opioids play an important role in the relief of chronic pain and postsurgical pain (Horn et al. 2002). Medical prescription of opioids in terminal patients is associated with increased risks of diabetic ulcers and bacterial infections (Egydio et al. 2012). Martin et al. (2010a, 2010b) found that chronic morphine treatment is implicated in delayed wound healing by inhibiting LPS-induced angiogenesis and immune cell recruitment to the wound site. A well-powered human clinical trial demonstrates that naltrexone, an opioid antagonist, promotes mucosal healing in active CD (Smith et al. 2011). Topical treatment of naltrexone also improves wound healing in type 1 diabetic rats (Immonen et al. 2013). Opioid-induced delayed wound healing may be implicated in the unrepairable state of intestinal barrier dysfunction following morphine treatment.
Opioids Increase Susceptibility to Infectious Diseases
Opioid treatment has been correlated with increased susceptibility to bacterial and opportunistic infections in mice and humans (MacFarlane et al. 2000; Ross et al. 2008; Mora et al. 2012). For instance, morphine increases susceptibility to Streptococcus pneumoniae lung infection by impairing host innate immune response in mice (Wang et al. 2005). Human clinical evidence indicates that opioid administration is associated with an increased risk of C. difficile infection (Mora et al. 2012; Keller 2013). In our unpublished study in mice, we demonstrate that morphine promotes pathogen dissemination in the context of intestinal Citrobacter rodentium infection, indicating that morphine modulates virulence factor–mediated adhesion of pathogenic bacteria, exacerbates gut dysbiosis, and induces disruption of the mucosal host defense during C. rodentium intestinal infection in mice.
Opioids are also known to exacerbate viral pathogenesis. For example, morphine together with HIV-1 Tat, a viral protein crucial for HIV replication, can regulate the tight junction function and modulate the blood–brain barrier’s permeability, which is correlated with an increased risk of developing HIV dementia in HIV-1 patients using opiate (Mahajan et al. 2008). The neuropathogenesis of simian immunodeficiency virus infections in rhesus macaques has been shown to be exaggerated following morphine treatment (Bokhari et al. 2011). It has been demonstrated in an HIV-1 model of coinfection with pneumococcal pneumonia that morphine potentiates neuropathogenesis through modulating toll-like receptors in microglial cells (Dutta et al. 2012). To better study the effects of morphine on early HIV pathogenesis in the gut, Sindberg et al. (2015) established a mouse model of EcoHIV infection following opioid treatment. In this infectious murine model, EcoHIV infection with opioid treatment induces bacterial translocation, disrupts intestinal barrier function, and activates a proinflammatory immune response, resembling the effects of opioids on early HIV pathogenesis in the gut (Sindberg et al. 2015).
Conclusion
There is ample evidence demonstrating that treatments with opioid analgesics disrupt gut homeostasis, leading to increased susceptibility to infections (Das et al. 2011; Mora et al. 2012; Meng et al. 2013; Larney et al. 2015). The interaction of gut microbiota with the host immune system has been shown to be required to maintain the homeostasis of the mucosal immunity and preserve the integrity of the gut epithelial barrier (Hooper, Littman, and Macpherson 2012). However, it is unclear how opioids modulate the gut microbiome and metabolome. Current rapid progress in the understanding of the host–microbial interaction has redefined pathogenesis and pharmacokinetics of drug metabolism, thus paving the way to improved medical intervention in the drug users/abusers population. The study of morphine modulation of gut microbiome and metabolome will shed light on the toxicological pathology of opioids and its role in infectious disease scenarios. The disruption of gut homeostasis increases the risk of infections and represents another potential adverse effect of opioid use or overuse (Mora et al. 2012; Babrowski et al. 2012).
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported in part by the NIH grants RO1 DA 12104, RO1 DA 022935, RO1 DA031202, K05DA033881, 1R01DA034582 and 1R01DA037843 to SR.