
Abstract
Conventional chemotherapy treatments for pancreatic cancer are mainly palliative. RNA interference (RNAi)-based drugs present the potential for a new targeted treatment.
Keywords
Introduction
Pancreatic ductal adenocarcinoma (PDAC) encompasses more than 85% of pancreatic cancers and is currently the fourth leading cause of cancer deaths in the United States (Craven, Gore, and Korc 2015; Ryan, Hong, and Bardeesy 2014). Since PDAC is usually detected at an advanced stage, it is highly resistant to chemotherapy and radiation, lacks a proper screening program, and is estimated to become the second leading cause of cancer deaths by 2030 (Apte et al. 2015; Biankin et al. 2012; Craven, Gore, and Korc 2015; Rahib et al. 2014; Rishi et al. 2015; Waddell et al. 2015). The only option for curative treatment is surgical resection; however, only 15–20% of the patients are suitable for this procedure at presentation (Craven, Gore, and Korc 2015; Ryan, Hong, and Bardeesy 2014). Moreover, as most of the patients undergoing resection experience recurrence of the disease, the 5-year survival rate still is low, standing at 20–25% postresection (Craven, Gore, and Korc 2015). Chemotherapy is advocated as an adjuvant therapy after resection or for patients with locally advanced or metastatic disease (Ryan, Hong, and Bardeesy 2014). The common regimens are gemcitabine plus nab-paclitaxel or fluorouracil plus leucovorin, irinotecan, and oxaliplatin (FOLFIRINOX; Garrido-Laguna and Hidalgo 2015).
In order to increase drug concentrations at tumor sites and to avoid systemic toxicity, regional administration of chemotherapy has been developed and studied in a large number of cancers (Collins 1984). Such an approach has been evaluated in pancreatic cancer, most commonly by arterial infusion and perfusion of chemotherapy (Davis et al. 2011). A more localized delivery technique for chemotherapy was utilized in pancreatic cancer by endoscopic ultrasound (EUS)-guided fine-needle injection of cytotoxic compounds (Bhutani 2003; Klapman and Chang 2005). More recently, new methods of drug delivery are being investigated, and these include the use of polymer-based drug delivery systems (Hoffman 2008; Kim et al. 2009; Rothenfluh and Hubbell 2009; Weinberg, Blanco, and Gao 2008; Wolinsky, Colson, and Grinstaff 2012). A combination of these two methods was introduced with the use of paclitaxel formulations of a poly(lactic-co-glycolic) acid and polyethylene glycol triblock copolymer (PLGA-PEG-PLGA) which is manufactured under the trade name OncoGel (Zentner et al. 2001). OncoGel was tested after EUS injection in a porcine model (Matthes et al. 2007) and in a phase 1 clinical trial in humans (Vukelja et al. 2007), and indeed this delivery method produced minimal systemic paclitaxel concentrations (Vukelja et al. 2007).
Currently, there is an increased understanding of genetic alterations that initiate and propagate multiple cancers. The identification of these cancer-related gene targets paved the way for the development of a promising new anticancer approach which utilizes small interfering RNA (siRNA). The siRNA-based therapeutic modality enables the selective silencing of gene expression. Compared to chemotherapeutic anticancer drugs, siRNA molecules present several prominent advantages as a drug which include (i) high degree of safety, as siRNA acts on the posttranslational stage of gene expression and does not interact with DNA, thereby avoiding the mutation and teratogenicity risks of gene therapy, (ii) high potency, as siRNA can cause strong suppression of gene expression with just a few copies per cell, (iii) unrestricted choice of drug targets and high targeting specificity, and (iv) rapid development, provided an applicable delivery method is demonstrated (Zuckerman and Davis 2015; Xu and Wang 2015).
Most of the siRNA-based investigational therapeutics for cancer are based on systemic administration of siRNA and involve delivery vehicles made of some type of a synthetic component (e.g., lipid or polymers). The siRNA in many cases is modified from its naked natural form (Burnett and Rossi 2012; Egusquiaguirre et al. 2012; Gomes-da-Silva, Simoes, and Moreira 2014; Kanasty et al. 2013; Wu et al. 2014). In PDAC, it has been established that genetic changes in the KRAS signaling pathways are responsible for over 90% of pancreatic cancers (Belda-Iniesta et al. 2008; Jones et al. 2008; Maitra and Hruban 2008). However, all attempts thus far to target KRAS directly have failed, and KRAS is widely assumed to be undruggable (Eser et al. 2014). Silencing of KRAS has been shown to be effective in controlling pancreatic cell line proliferation (Rejiba et al. 2007).
To target mutated KRAS and to realize the potential of siRNA as a very specific and potent drug, Silenseed Ltd. has developed
It is of great importance that biodegradable materials are properly assessed for their safety because they are implanted in the tissue for a long period. Biodegradable polymers should also be evaluated for their biodegradability and biocompatibility (Nyska et al. 2014; Ramot, Nyska, et al. 2015; Ramot, Touitou, et al. 2015). While PLGA has been shown to be biocompatible, some side effects have been reported following its implantation (Eppley et al. 2004). Furthermore, drug incorporation can change its original properties and lead to an inflammatory reaction (Yamaguchi and Anderson 1992). This is especially important when discussing siRNA, which might lead to activation of the alternative pathway of the complement system (Schultheis et al. 2014; Tabernero et al. 2013) and can also be immunostimulatory under certain conditions (Zuckerman et al. 2014).
The aim of this study was to comprehensively evaluate the safety and the toxicity of siG12D-LODER, in repeated dosing regimen in 192 Hsd:Sprague Dawley rats, after subcutaneous administration. The toxicokinetics profile was also evaluated. To the best of our knowledge, this is the first thorough report on the toxicity and safety profile of a biodegradable polymeric matrix releasing siRNA and can serve as a case study for the pathological evaluation and expected results for such materials.
Animals, Materials, and Methods
Test Device
The siG12D-LODER is a millimeter-scale biopolymeric drug delivery system. It contains 0.3 mg of siG12D, siRNA against G12D-mutated KRAS, which is released over the course of 4 months (Shemi et al. 2015; Zorde Khvalevsky et al. 2013). LODER, supplied by Silenseed Ltd, is constructed with a medical grade copolymer of PLGA of high molecular weight > 50 kD, in the form of a cylindrical homogenous rod of diameter 0.9 mm and length 4 mm ± 1 mm. In vivo, the surface area of LODER remains unchanged and constant over the entire release period. For human use (Golan et al. 2015), the dimensions of the LODER were selected to provide a device that can be properly inserted to human pancreatic tumor using EUS procedure and off-the-shelf EUS biopsy needles.
The LODER test devices were produced using the identical aseptic manufacturing process used to produce the device employed in humans, at the same dimensions and drug load. The relative amount of drug material in the rats was approximately 20 times greater than the amount of material used in human subjects. This conversion factor was calculated to be 18.2 for human weight of 75 kg, rat weight of 250 g, and Km factors of 37 and 6 in human and rat, respectively, and is 19.5 for human weight of 80 kg and similar cited parameters (Guidance for Industry—Estimating the Maximum Safe Starting Dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteers, Center for Drug Evaluation and Research, US Food and Drug Administration [FDA], 2005).
Animals and Housing
Hsd:SD rats, approximately 8–10 weeks old, were obtained from Harlan Italy s.r.l., San Pietro al Natisone (UD), Italy. A commercially available laboratory rodent diet (4 RF 21, Mucedola S.r.l., Settimo Milanese (MI), Italy) was offered ad libitum throughout the study. The animals were allowed free access to drinking water, supplied to each cage via water bottles. During acclimation and throughout the entire study duration, animals were housed within a limited access rodent facility and kept in groups of up to 5 of 1 sex to a cage, in clear polysulfone solid-bottomed cages. Nesting material was provided inside a suitable bedding bag and changed at least twice a week. The rats were allowed an approximately 2-week acclimation period to the facility conditions (20°C–24°C, 40–70% relative humidity, and a 12-hr light/dark cycle) prior to inclusion in the study. The experimental protocol was approved by the ethical committee of the Research Toxicology Centre (RTC), Pomezia, Italy, and the study was conducted in accordance with the principles of Good Laboratory Practice. This experiment was conducted in facilities approved by the Association for Assessment and Accreditation of Laboratory Animal Care International, and the animals were maintained in accordance with the Guide for the Care and Use of Laboratory Animals (1996).
Experimental Design
For the safety and toxicity study, 75 male and 75 female rats were randomized into 5 groups (Table 1). For the toxicokinetics study, 21 male and 21 female rats were randomized into 3 groups (Table 2). All animals were dosed once a day on days 1, 14, and 28 of the study. The first 10 animals/sex/group of the safety and toxicity study were sacrificed on day 29, and the last 5 animals/sex/group were sacrificed on day 42 (day 14 of the recovery phase). The first 3 animals/sex/group of the toxicokinetics study were sacrificed on day 2, the second 3 animals/sex/group on day 15, and the last 3 animals/sex/group on day 29.
Experimental Design for the Safety and Toxicity Study.
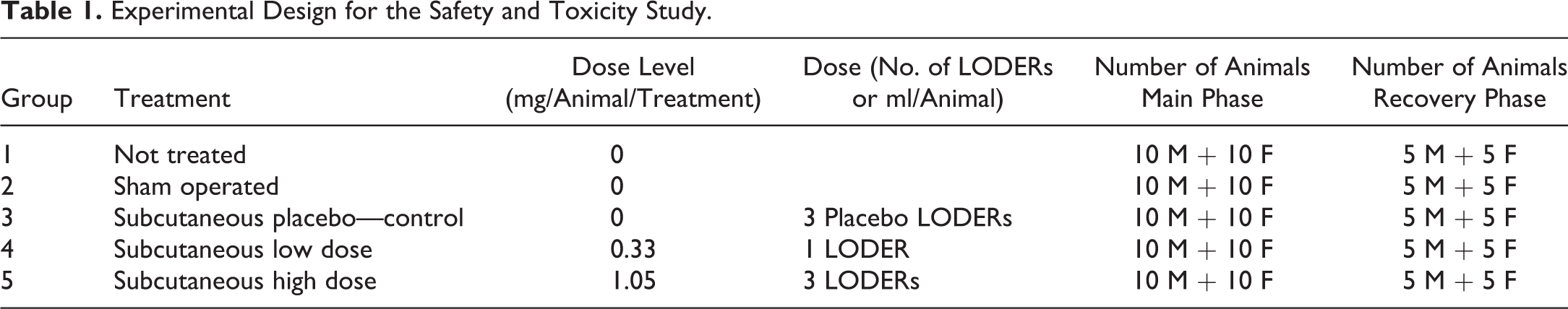
Experimental Design for the Toxicokinetics Study.

Subcutaneous Implantation
Before the start of treatment, the area pertaining to the implant site was shaved and the treatment site was marked with an indelible pen. Skin shaving of the implant site was repeated at approximately 2-week intervals during the study, and the marking of the implant site was repeated as necessary. Animals were anesthetized using isofluorane. Animal viability was rechecked every 2–3 min. The test item was inserted via a minor dissection into the subcutis of the scapular region of the animals by means of a sterile trocar. The LODERs were implanted separately, at a distance of approximately 1 cm from each other. Animals of groups 4 and 7 received 1 siG12D-LODER and animals of groups 5 and 8 received 3 siG12D-LODERs, while control animals (groups 3 and 6) received 3 Placebo LODERs. Animals of group 2 were sham operated, that is, the same dissection procedure for groups 3–5 was performed, but no test item was inserted. Animals of group 1 were not treated.
Viability, Clinical Signs, Weight, and Food Consumption
Each animal was observed and any clinical signs were recorded once before commencement of treatment and twice daily during the study. On each day of dosing (days 1, 14, and 28), animals were observed at approximately 1, 2, 4, and 6 hr after dosing. Body weights were measured on the day of allocation to treatment group, once during the pretreatment period, approximately weekly during the study, including each day of treatment, and prior to scheduled termination. The weight of food consumed by each cage of rats in the safety and toxicology study was recorded at weekly intervals following allocation, and the group mean daily intake per rat was calculated.
Ophthalmoscopy
Both eyes of all animals assigned to the safety and toxicity study were examined just prior to the commencement of treatment using an ophthalmoscope, and by a slit lamp microscope, after the instillation of 0.5% tropicamide (Visumidriatic®; Visufarma, Rome, Italy). The eyes were reexamined during week 4 of treatment.
Hematology, Biochemistry, and Coagulation
For the safety and toxicity study, blood for hematology, biochemistry, and coagulation parameters was collected just prior to euthanasia on days 29 and 42. Blood samples were obtained from the abdominal vena cava. The samples were assayed for hematology and biochemistry using the Siemens Advia 120 and Siemens Advia 1200, respectively, and for coagulation by Instrumentation Laboratory ACL 3000 PLUS.
Urinalysis and Nephrotoxicity Biomarkers
Overnight urine samples were collected from the animals in the safety and toxicity study prior to euthanasia on days 29 and 42. Urinalysis parameters were analyzed by Menarini AUTION MAX 4280/AUTION ELEVEN AE 4020. The sediment, obtained from centrifugation at approximately 1,900 g for 10 min, was examined microscopically. Nephrotoxicity analyses, including albumin, β-microglobulin, cystatin C, and NGAL, were carried out on samples from 3 males and 3 females from each group in the safety and toxicity study. The samples were analyzed using Multiplex kits supplied by Merck Millipore (Milan, Italy).
Toxicokinetics
Blood samples were collected at the following 7 time points: 0.25, 0.5, 1, 2, 4, 8, and 24 hr after dosing on days 1, 14, and 28 of the toxicokinetics study from the animals in groups 7 and 8 (Table 2). Blood samples from group 6 (control) were taken at the same time points only on day 1. At each sampling time, up to 8-hr postdose, approximately 0.4 ml blood samples were collected from the tail vein of 3 males and 3 females of each group. At the last time point (24-hr postdose), blood samples were taken at necropsy, from the abdominal vena cava under isofluorane anesthesia. Samples were transferred into tubes containing K3EDTA treated with freshly prepared 10% diethylpyrocarbonate solution in phosphate-buffered saline (0.4 ml/tube) and centrifuged at +4°C. The plasma samples were stored at −80°C pending analyses.
Necropsy and Tissue Handling
Tissues for microscopic evaluation were fixed and preserved in 10% neutral-buffered formalin (except eyes, optic nerves, Harderian glands, testes, and epididymides which were fixed in modified Davidson’s fluid and preserved in 70% ethyl alcohol), processed and trimmed, embedded in paraffin, sectioned to a thickness of approximately 5 µm, and stained with hematoxylin and eosin. In groups 2, 3, 4, and 5, the implantation site was taken and preserved for further histopathology observation. The implantation site was taken deep enough with sufficient surrounding tissue which enabled evaluation of the local tolerability.
The following tissues were examined microscopically: adrenal gland, brain, esophagus, eyes, femur (including marrow), Harderian gland, heart and aorta, implant site, implant insertion area, large intestine (cecum, colon, and rectum), small intestine (duodenum, jejunum, and ileum), kidney, larynx, liver, lung (and main stem bronchi), lymph nodes (cervical and mesenteric), mammary area, nasal cavity, optic nerve, ovary, pancreas, parathyroid gland, pharynx, pituitary gland, prostate gland, salivary gland, skeletal muscle, skin, spinal cord and sciatic nerve, spleen, stomach (including fore stomach and glandular stomach), testis (with epididymis and seminal vesicle), thymus, thyroid gland, tongue, trachea, ureters, urinary bladder, uterus, and vagina.
A semiquantitative grading scheme was used to evaluate the extent of the lesions in the tissue, generally using the criteria presented by Shackelford et al. (2002), using 5 grades, as follows: no lesion (grade 0), minimal (grade 1), mild (grade 2), moderate (grade 3), and marked (grade 4).
Trimming of the Implantation Sites
A thorough histopathological evaluation was done on the implantation sites in groups 3, 4, and 5, as follows: 3 longitudinal slides were trimmed—1 from the middle (i.e., center) of the implantation site, and 2 others, 1 right and 1 left to the middle of the implantation site, respectively. In addition, a slide was trimmed from the implant insertion site (Supplementary Material, Figure S1).
Statistical Analysis
Standard deviations were calculated as considered appropriate. For continuous variables, the significance of the differences among groups was assessed by analysis of variance. Differences between each treated group and the control group (group 3) were assessed by Dunnett’s test using pooled error variance. The homogeneity of the data was verified by Bartlett’s test before Dunnett’s test. If the data were found to be inhomogeneous, a modified t-test (Cochran and Cox) was applied. Statistical analysis of histopathological findings was carried out by means of the nonparametric Kolmogorov–Smirnov test.
Results
Survival, Clinical Observations, and Body and Organ Weight
No unscheduled sacrifice or death occurred during the study. No clinical signs were observed in any animal, during the treatment and recovery periods, except for 1 male from group 3, which presented a damaged left ear from day 7 up to the end of the recovery phase. No remarkable differences in body weight or food consumption were observed in treated male and female animals, when compared to the control group. No relevant changes were observed in terminal body weights and organ weights at the end of dosing and recovery phases. Kidney relative weights from males of group 1 (not treated) and group 2 (sham operated) showed a slight significant difference at the end of the recovery period, when compared to animals of group 3, treated with the Placebo LODERs. Due to the absence of dose relation and other related changes, this finding was considered of no toxicological importance.
Ophthalmoscopy
No findings were detected in both eyes of all animals in the ophthalmic examinations performed during the study.
Hematology and Clinical Chemistry
During the dosing phase, slight leukocytosis was observed in some males of group 4. Mean group values were 26% above controls. Due to the low incidence/severity and the absence of dose relation, this change was considered unrelated to treatment. Additionally, statistically significant differences were noted in erythrocytes and mean corpuscular hemoglobin between controls and females of group 5. These changes were of minimal severity (8% and −3%, respectively), therefore considered of no toxicological importance. No relevant changes were recorded during the recovery phase.
No changes were recorded in the coagulation parameters during the dosing phase. Decreased prothrombin time in the males of group 4 and increased activated thromboplastin time in the females of group 5 were noted during the recovery phase. However, these changes were considered incidental, since they were not observed during the dosing phase.
Total bilirubin was increased in some animals from groups 2 and 4 (2.4- to 2.9-fold; Table 3). Due to the absence of dose relation and other related changes, this finding was considered of no toxicological importance. Additionally, sodium was significantly decreased in females of group 5 (Table 3). Due to the low severity (1%), this finding was considered of no toxicological relevance. Other significant changes that were recorded in the clinical chemistry analysis are listed in Table 3, but these findings were considered incidental.
Significant Changes from Control in the Clinical Chemistry Analyses.
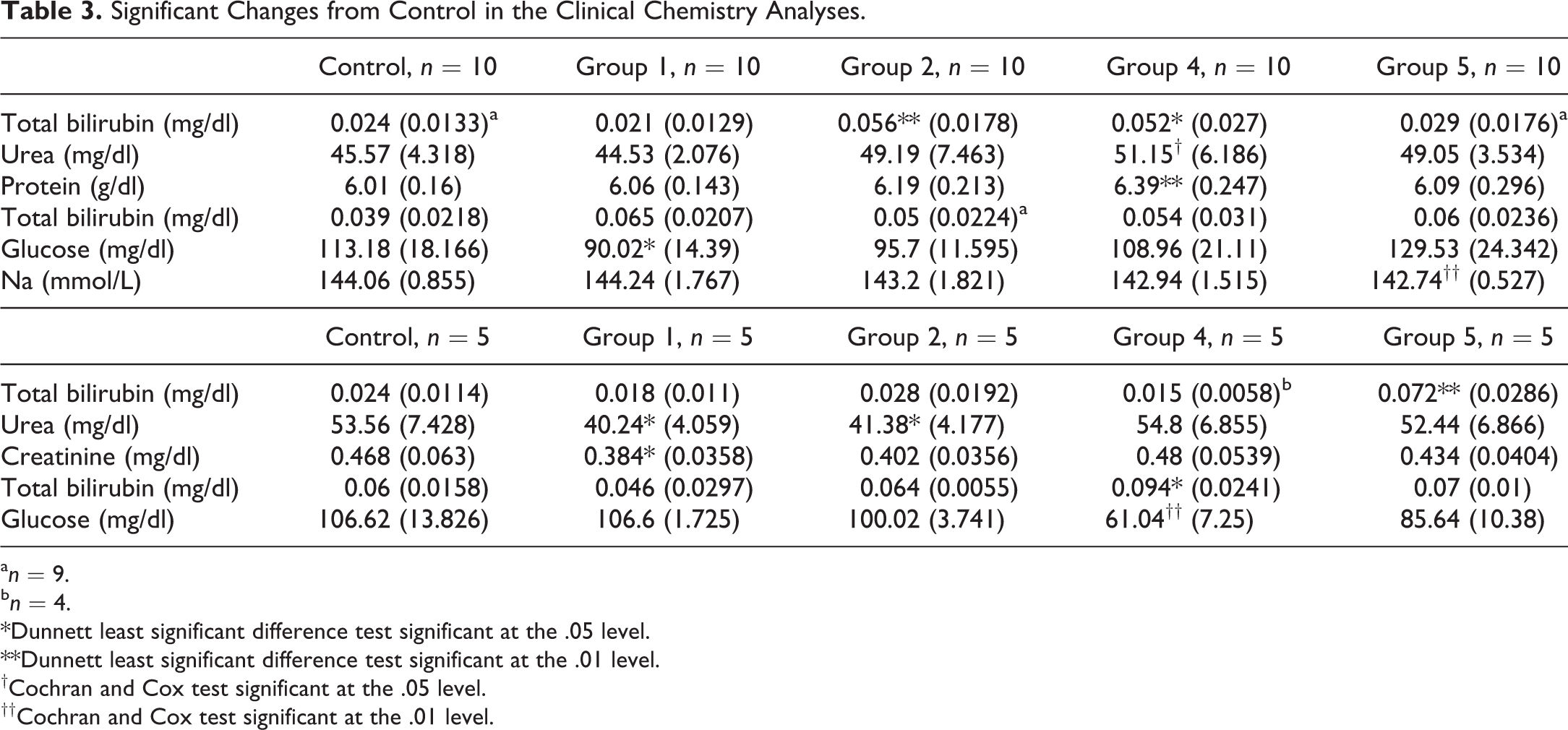
an = 9.
bn = 4.
*Dunnett least significant difference test significant at the .05 level.
**Dunnett least significant difference test significant at the .01 level.
†Cochran and Cox test significant at the .05 level.
††Cochran and Cox test significant at the .01 level.
Urinalysis and Nephrotoxicity Biomarkers
No relevant differences between the different groups were observed in both urinalysis parameters and nephrotoxicity biomarkers evaluated.
Toxicokinetics
Plasma levels of siG12D were below the limit of quantitation in all plasma samples from the group receiving the 3 placebo LODERs (group 6). Small amounts of the test item were found in group 8 plasma samples until 30 min after the subcutaneous administration of 3 siG12D-LODERs.
Macroscopic and Microscopic Observations
No treatment-related changes were noted locally or systemically in any organ, and all observed abnormalities were considered as unrelated to treatment. In particular, in the skin of animals of group 2 (sham operated), as well as in groups 3, 4, and 5 (i.e., subcutaneous placebo, low- and high-dose-treated groups, respectively), red areas were noted in the implant insertion sites. These correlated histologically with focal areas of epidermal ulcer and/or hyperplasia, associated with subcutaneous, subchronic inflammation and were considered as related to the technical procedure of the subcutaneous insertion of the LODER (Table 4; Figures 1 and 2).
Incidence of Microscopic Observations in the Skin.
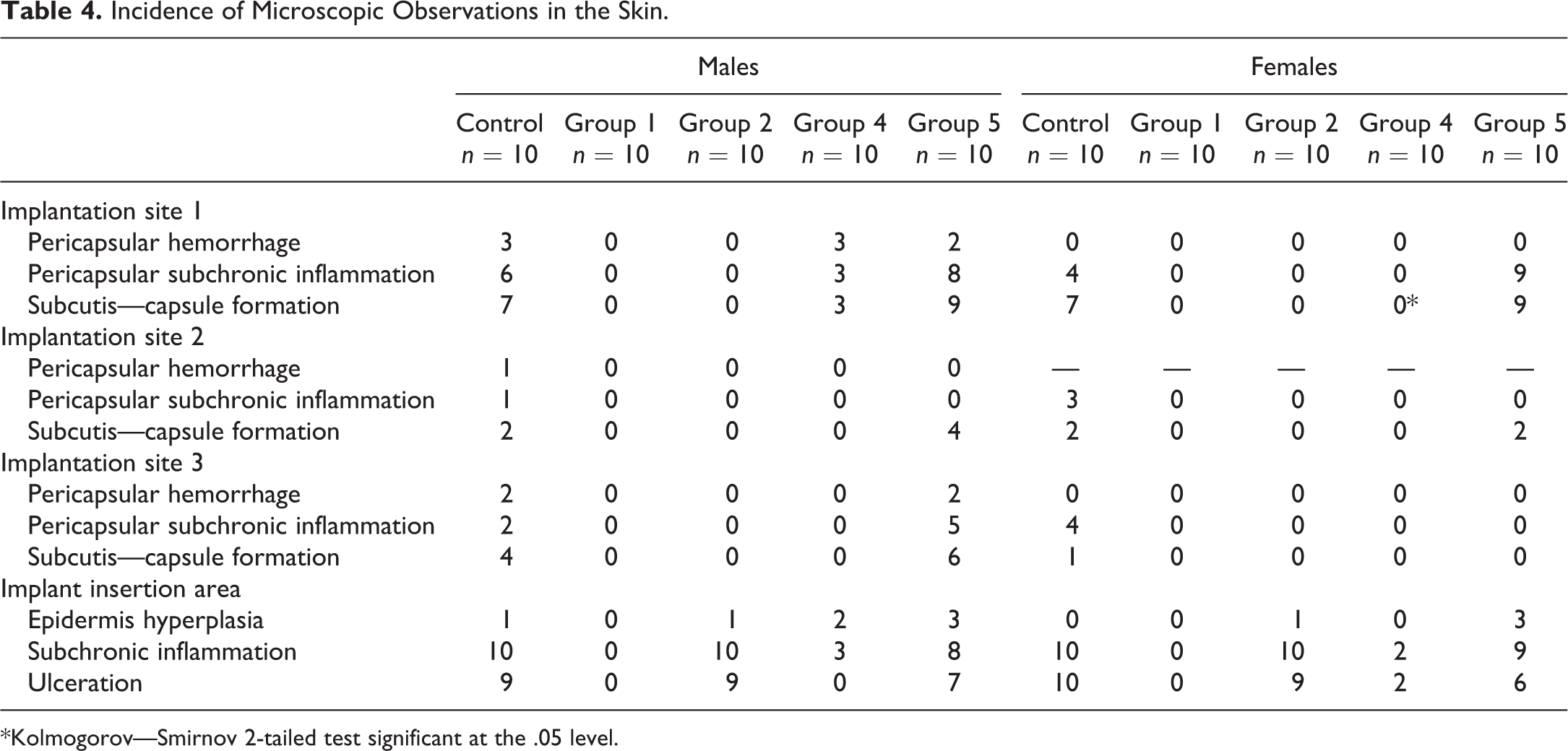
*Kolmogorov—Smirnov 2-tailed test significant at the .05 level.
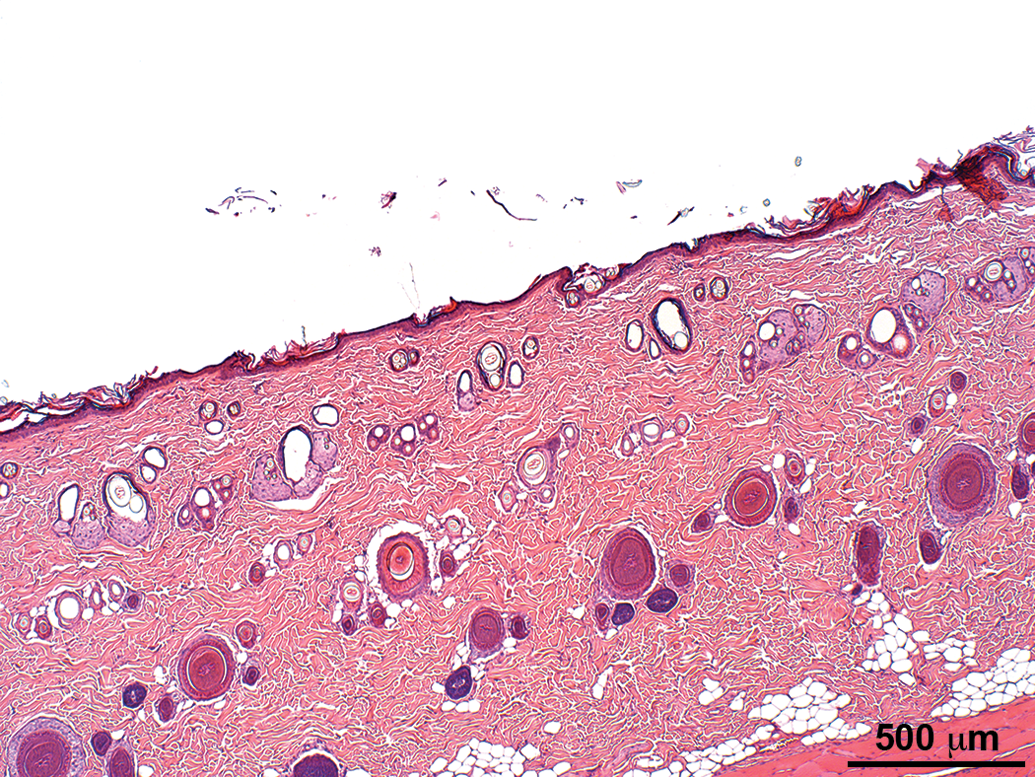
Normal skin section of an animal from group 1 (control). Hematoxylin and eosin stain.
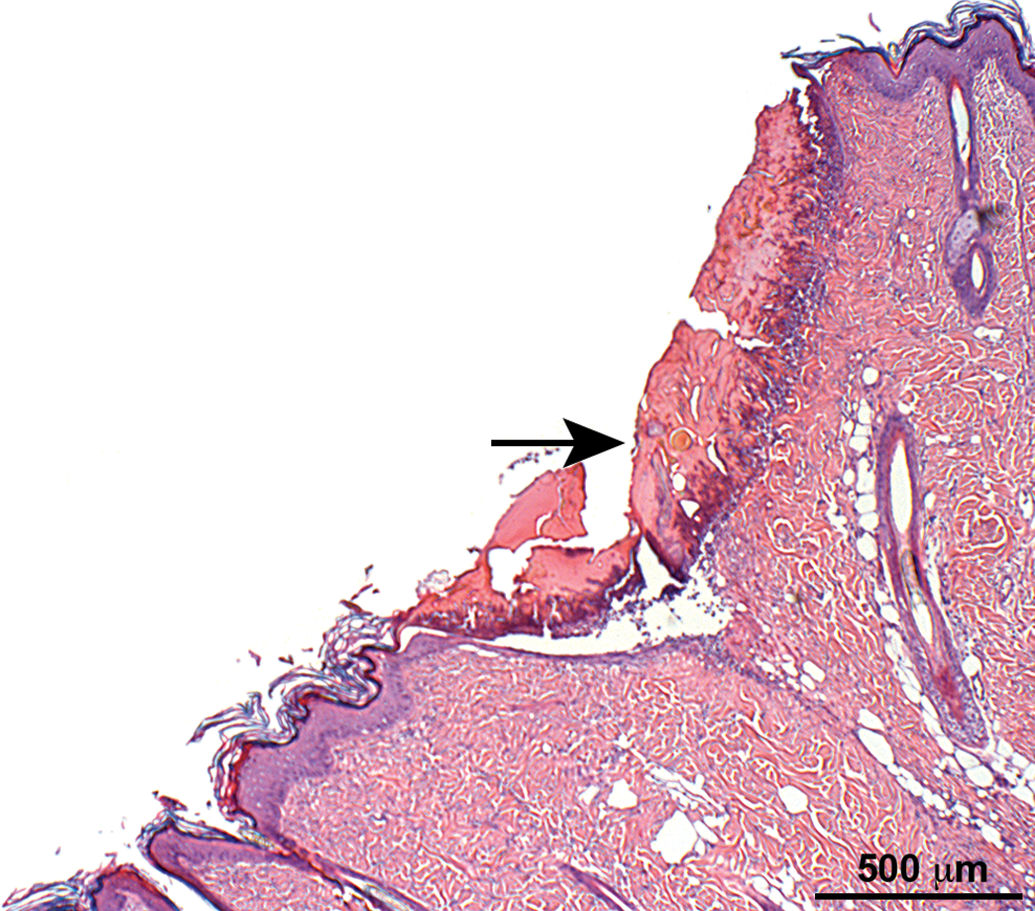
Skin section of an animal from group 2 (sham operated) showing focal area of epidermal ulcer (arrow) and hyperplasia associated with subcutaneous subchronic inflammation. Hematoxylin and eosin stain.
In addition, in groups 3, 4, and 5, red areas were noted in implantation sites 1 and/or 2 and 3. These correlated histologically with local tissue changes (i.e., capsule formation, pericapsular inflammation, and/or hemorrhage), all having a comparable incidence and/or severity in all these groups and therefore were not considered as related to the test compound but rather due to the presence of the LODER in the subcutaneous tissue (Table 4; Figures 3 –6). The capsule was typically composed of a relatively thin mature fibrotic tissue, having within the cavity red amorphous granular material, and on the interface between the cavity and the capsule a single layer composed of macrophages and multinucleated giant cells.
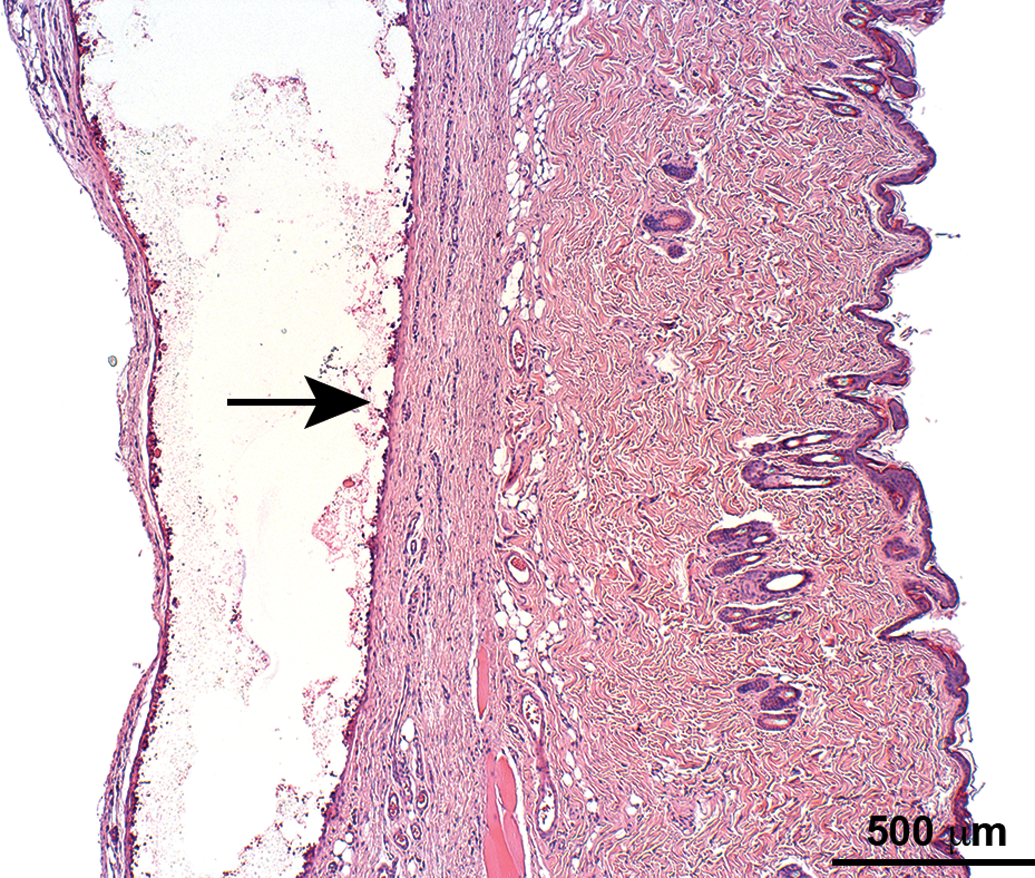
Skin section of an animal from group 3 (subcutaneous placebo LODERTM implantation) showing subcutaneous capsule formation (arrow). Hematoxylin and eosin stain.
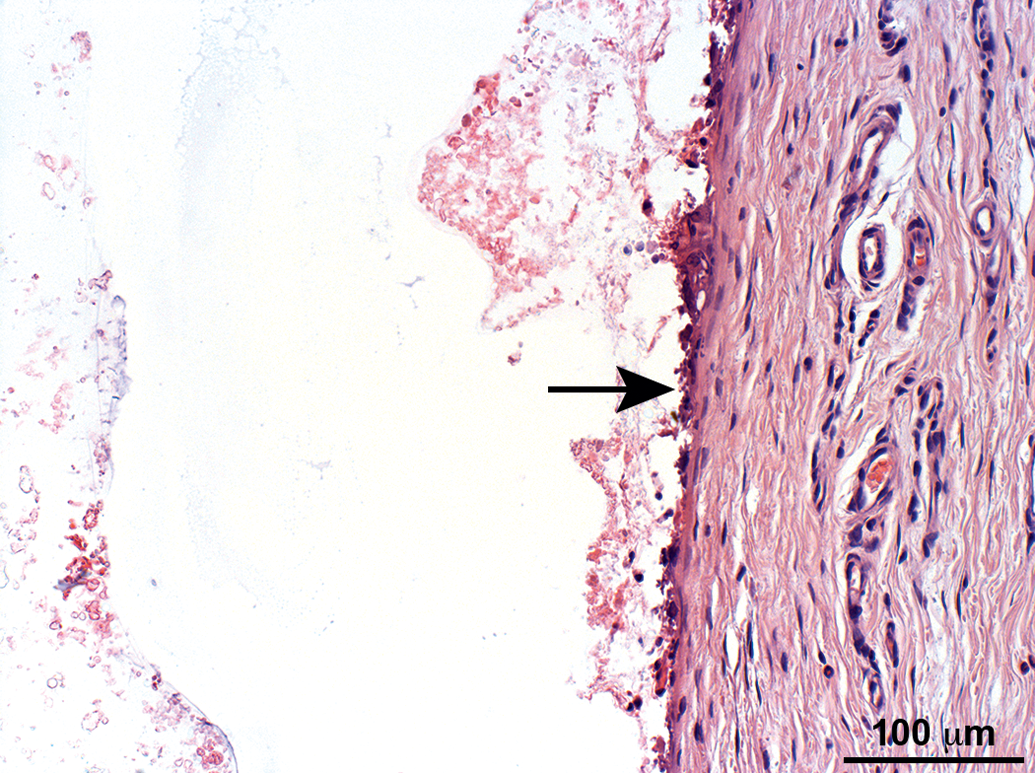
Higher magnification of the subcutaneous capsule from Figure 3 (arrow). The capsule is composed of a relatively thin mature fibrotic tissue, having within the cavity red amorphous granular material, and on the interface between the cavity and the capsule a single layer composed of macrophages and multinucleated giant cells. Hematoxylin and eosin stain.
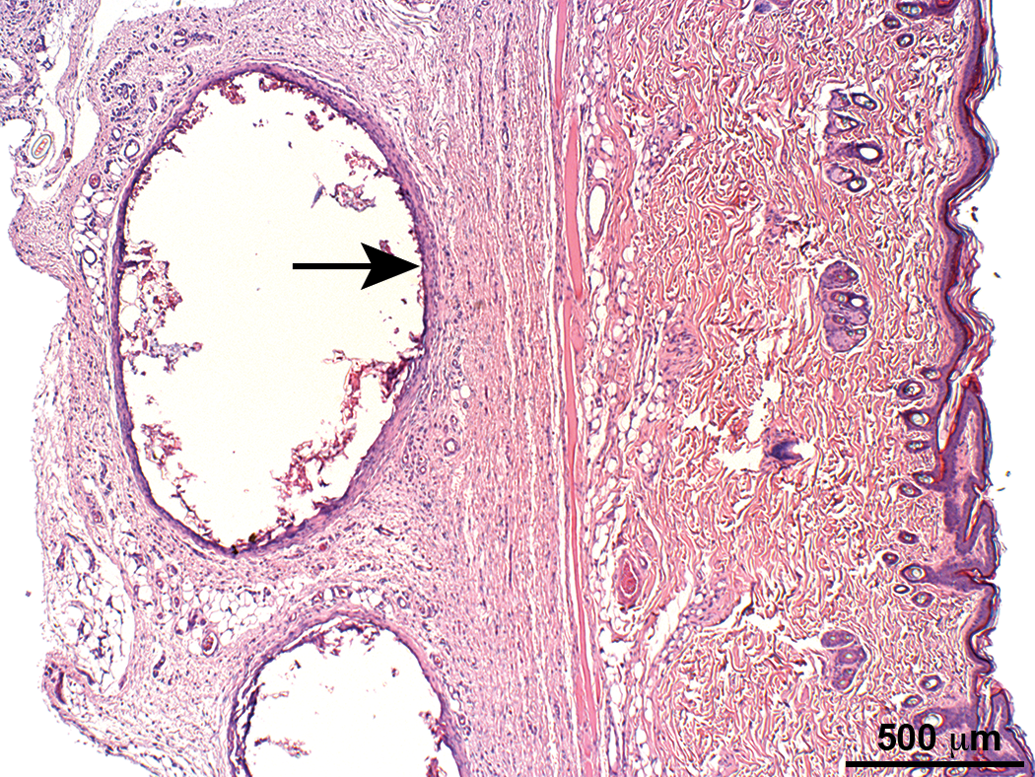
Skin section of an animal from group 5 (high dose of LODERTM implantation) showing subcutaneous capsule formation (arrow). Hematoxylin and eosin stain.
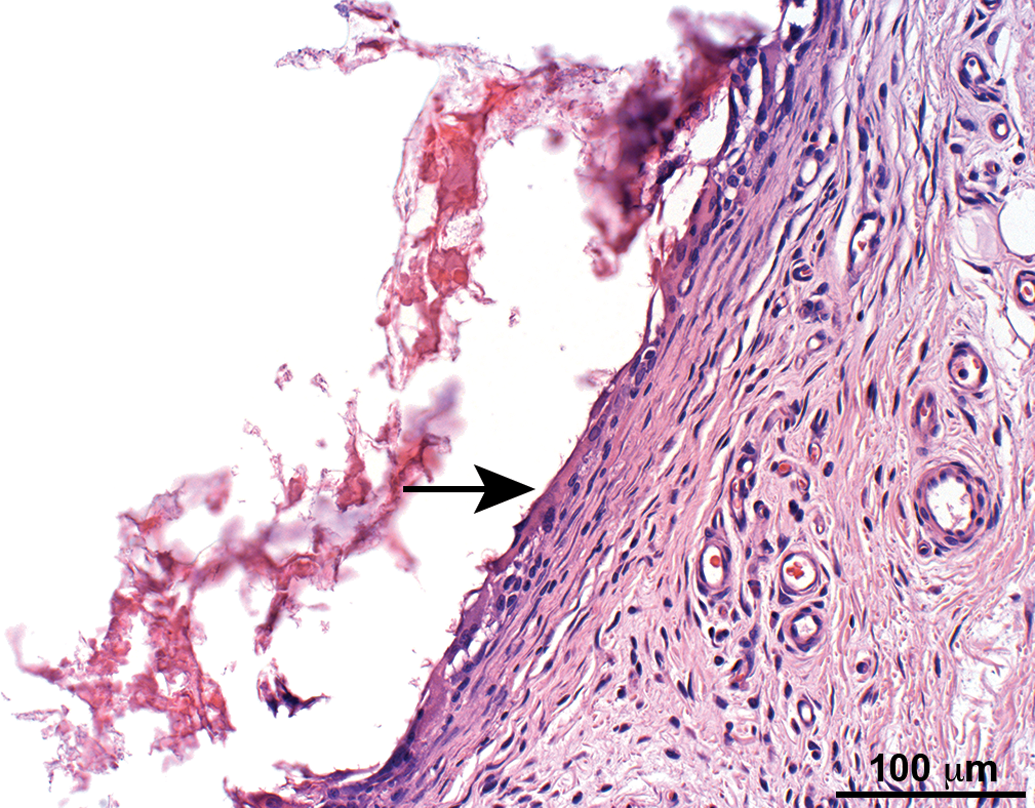
Higher magnification of the subcutaneous capsule from Figure 5 (arrow). The capsule is composed of a relatively thin mature fibrotic tissue, having within the cavity red amorphous granular material and on the interface between the cavity and the capsule a single layer composed of macrophages and multinucleated giant cells. Hematoxylin and eosin stain.
All other observed changes were seen with a comparable incidence in control and treated animals or were characteristically seen in untreated rats of the same age and strain and therefore were not considered as related to treatment.
Discussion
The aim of the present study was to comprehensively evaluate the safety and the toxicity of siG12D-LODER in Hsd:SD rats, after subcutaneous administration.
According to the Evaluation of Medicinal Products (EMA) guidance on the nonclinical local tolerance testing for medicinal products (2015, page 3): Non-clinical local tolerance is intended to support human exposure to a drug product (both active substance and excipients at contact sites of the body following clinical use. Such local tolerance testing should aim to support initial testing in clinical trials, as well as intending to support the final product.
Even though the primary goal of siG12D is to act locally on solid pancreatic tumors, the preclinical testing aimed to address the potential for any systemic toxicity. The toxicokinetics data confirmed that only small amounts of the test item were measured after 30 min or 1 hr postdose after IV administration.
Other investigations with the LODER loaded with the active drug indicated that the dimensions and surface area of this drug delivery system remain unchanged and constant over the entire study period. It was demonstrated that the LODER surface remains clear, without the formation of a strong stromal and/or protein blocking layer (Zorde Khvalevsky et al. 2013).
This notion was clearly supported by the histopathology data obtained from the current experiment (i.e., groups 3, 4, and 5), indicating that no toxicity related to the test compound was noted. The capsule formation typically composed of a relatively thin mature fibrotic tissue, which was of comparable thickness in all these groups, as well as without any evidence of chronic active inflammation, necrosis, and/or mineralization, which may suggest potential adverse effects (Northup 1999; Nyska et al. 2014; Onuki et al. 2008; Ramot, Nyska, et al. 2015; Ramot, Touitou, et al. 2015; Schuh 2008). The presence of a relatively thin layer of macrophages and multinucleated giant cells at the interface between the inner surface layer of the LODER and the capsule, without extension of the granulomatous reaction to the adjacent tissue as well as the pharmacokinetics data, all confirm that the tissue reaction to the LODER with the drug was local without leakage or involvement of adjacent tissues.
In systemic administration, using (nonviral) formulated siRNA that is based in general on different types of nanoparticles, in 1 case the safety concerns led the FDA in 2008 to study termination, while more recent phase 1 studies demonstrated an encouraging level of safety profiles for humans (Xu et al. 2014; Zuckerman and Davis 2015), and most of the treatments were well tolerated (Schultheis et al. 2014; Tabernero et al. 2013; Tolcher et al. 2014; Zuckerman et al. 2014). Most of the toxicities that were reported in these trials were related to infusion reactions and cytokine release symptoms. Hepatotoxicities were often reported as a side effect of siRNA treatment in preclinical trials but significantly less observed in humans (Zuckerman and Davis 2015). Concerns regarding accumulation of nanoparticles also were related to tissue other than the liver, although in lower amounts, including kidney and spleen (Zuckerman and Davis 2015).
In this study, that is focused on local and prolonged delivery, no adverse reactions or laboratory test abnormalities were evident, confirming the safety of the delivered drug. In summary, based on this comprehensive toxicity study, we can conclude that siG12D-LODER is safe for use and does not lead to any local or systemic side effects.
Footnotes
Author Contributions
Authors contributed to conception or design (EZ, GM, AS, AN); data acquisition, analysis, or interpretation (YR, SR, RG, EZ, SM, GM, AS, AN); drafting the manuscript (YR, AN); and critically revising the manuscript (YR, SR, RG, EZ, SM, GM, AS, AN). All authors gave final approval and agreed to be accountable for all aspects of work in ensuring that questions relating to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Dr. Abraham Nyska served as a consultant to Silenseed Ltd.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The study was funded by Silenseed Ltd.