
Abstract
Dibromoacetic acid (DBAA), a haloacetic acid found in drinking water as a disinfection by-product, can cause many adverse effects, including immunotoxicity. In a previous study, we confirmed that DBAA can induce obvious immunotoxicity in mice but that the underlying mechanisms are not clearly understood. In our current study, we confirmed that DBAA induced cytotoxicity and apoptosis in thymocytes isolated from mice by a range of DBAA concentrations (0, 5, 10, 20, or 40 μM). The data showed that DBAA exposure led to a significant decrease in proliferative responses to T-cell mitogens and obvious inhibition in the production of cytokines interleukin-2 and interleukin-4. We found obvious morphological changes of apoptosis in thymocytes and observed the percentage of apoptotic thymocytes to increase significantly as the DBAA concentration increased. Further investigation showed that DBAA can cause G0/G1 arrest in cell cycle analysis, increase intracellular calcium ([Ca2+]i) levels, increase the expression of Fas/FasL proteins, and decrease the expression of Bcl-2 protein. It is concluded that in vitro exposure to DBAA can lead to marked cytotoxicity and apoptosis among thymocytes, and the mechanism involved is strongly related to blocking cell cycle progression, increasing intracellular calcium, and increasing Fas/FasL expressions.
Introduction
Dibromoacetic acid (DBAA) is one of the haloacetic acids formed when water supplies containing natural organic matter are disinfected with disinfectants. Certain haloacetic acids have been shown to cause many adverse effects in laboratory animals and in several bacterial systems (Kaydos et al. 2004; Pressman et al. 2010; Zhang et al. 2010; Pals et al. 2011). Of the bromoacetic acids, DBAA is usually found in greater concentrations than the monosubstituted form (Richardson et al. 2003). Accurate quantification of human exposure to DBAA via drinking water is currently difficult because the presence of DBAA in water supplies is a complex issue dependent on many factors, such as bromine ion concentration, temperature, pH, the disinfectant dose, and contact time (Liang and Singer 2003). Because little is known about the toxicity of DBAA, several rodent studies have focused on DBAA exposure in juvenile and adult rats, demonstrating many effects including reproductive and developmental toxicity, neurotoxicity, hepatotoxicity, and carcinogenicity (Bodensteiner et al. 2004; Moser et al. 2004; Tao et al. 2004; Melnick et al. 2007). The mammalian cell cytotoxicity assay revealed that the IC50 of DBAA is 0.5 mM and single-cell gel electrophoresis genotoxic potency is 1.756 mM in Chinese hamster ovary cells (Plewa et al. 2002).
It has been suggested that immunosuppression may play a crucial role in DBAA-induced cancers by interfering with normal immunosurveillance against tumors. Some animal studies have documented that DBAA can alter a variety of immune cell functions and affect both the cellular and humoral immune response (Gao et al. 2008; Smith et al. 2010). Yet, there has been little research on the mechanisms of DBAA-induced immunotoxicity; the exact cellular and molecular mechanisms leading to altered function or cell death are not clearly understood. In our previous study, we found that DBAA could induce marked immunotoxicity in mice and increased apoptosis of both splenocytes and thymocytes following DBAA exposure (Gao et al. 2008). In the immune system, many lymphocytes die at the termination of the acute phase of the immune response, and dysregulation of programmed cell death has been found to contribute to the development of autoimmunity and neoplasia. The objective of the present study was therefore to investigate the immunotoxicity mechanisms of DBAA. Specifically, thymocytes isolated from BALB/c mice were assessed for the morphological, biochemical, and molecular biological features of apoptosis following incubation with different concentrations of DBAA. Furthermore, immune responses are regulated by the secretion of cytokines in activated T cells, including interleukin-2 (IL-2), interferon-γ (IFN-γ), interleukin-4 (IL-4), and tumor necrosis factor-α (TNF-α). These pleiotropic factors can modulate cell survival, growth, differentiation, and apoptosis (Lantz et al. 2000). In the current project, the cytokines IL-2 and IL-4 were analyzed in thymocytes activated by mitogenic lectins (concanavalin A [Con A]) after exposure to DBAA.
Material and Methods
Chemicals and Reagents
DBAA (purity 99%) and Con A (Type IV, Cat. No. C-2010) were purchased from Sigma-Aldrich (St. Louis, MO). The Annexin V-FITC Apoptosis Detection kit and Cycletest™ Plus DNA Reagent kit were obtained from Becton Dickinson (San Jose, CA). The CellTiter 96® AQueous One Solution Cell Proliferation Assay was purchased from Promega (Madison, WI). RPMI 1640 and fluo-3 were purchased from Invitrogen (Grand Island, NY). Antibodies specific against Fas, FasL, Bcl-2, β-actin as well as host-/isotype-specific secondary antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA).
Cell Culture
As a source of thymocytes, male BALB/c mice (6–8 weeks) were purchased from Harbin Medical University Laboratory Animal Center (Harbin, China). All mice were housed in specific pathogen-free facilities maintained at 18°C to 26°C, with a 40 to 70% relative humidity and a 12-hr light–dark cycle. All mice had ad libitum access to standard rodent chow and filtered water. All animal experiments in this study were approved by Harbin Medical University Ethics Committee for animal research and were conducted in accordance with the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. Single-cell suspensions of mouse thymocytes from the naive hosts were prepared as follows: the thymus of each mouse was aseptically removed after sacrifice by cervical dislocation, and single-cell suspensions were prepared by forcing each organ through 400-μm stainless steel mesh. Thymocytes were washed with Hank’s balanced salt solution, and viable thymocyte counts were obtained by trypan blue exclusion. The cell suspension was adjusted to different levels as needed to complete RPMI 1640 (RPMI supplemented with 10% fetal bovine serum [FBS; Hyclone, Logan, UT], 2 mM
DBAA Preparation and Exposure
Shortly before addition to the primary thymocyte cultures, DBAA solutions were prepared at a range of concentrations. An initial stock solution was generated by dissolving DBAA in phosphate-buffered saline (PBS, pH 7.4) and then adjusting the pH to 7 with 1 N NaOH. Thereafter, the stock was diluted in standard medium without FBS to final concentrations of 0, 5, 10, 20, or 40 μM in culture medium.
The 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) Assay
To determine the thymocyte proliferative activity to mitogens after DBAA exposure, a Promega Cell Titer 96 Non-Radioactive Cell Proliferation Assay kit was used to measure the amount of formazan produced by metabolic bioreduction of MTS. For each DBAA concentration (or control with medium alone), isolated thymocytes were seeded at 2 × 106/well in a flat-bottomed, 96-well plate (Corning Inc., Corning, NY) for 24 hr before DBAA treatment. The thymocytes were then treated with various DBAA solutions (0, 5, 10, 20, and 40 μM) and Con A (5 μg/ml and 5 μl) with 8 wells used for each concentration, then time-dedicated plates were incubated for 6, 12, 24, 48, or 72 hr at 37°C in a humidified 5% CO2 atmosphere. At the end of each indicated exposure period, 15 μl of a stock MTS solution was added to each well and the plate was incubated a further 4 hr at 37°C. Thymocytes proliferation was quantified at 490 nm using a microplate reader (Bio-Rad, CA). For each incubation time point, the mean optical density (OD) of the 8 replicate/exposure conditions was compared to the mean OD of the appropriate control to calculate the relative viability of each treated cell culture. Three separate experiments were conducted for each end point measured.
Cytokine Determination
The release of IL-2 and IL-4 in thymocytes was measured using Mouse IL-2 and IL-4 ELISA kits, respectively. Briefly, 2 × 106 cells in 1 ml of complete RPMI 1640 medium were seeded in 24-well plate with 5 μg/ml Con A and DBAA (0, 5, 10, 20, or 40 μM) and incubated for 24 hr at 37°C in a CO2 incubator. The plate was centrifuged, and the supernatant was used to measure the cytokine release by ELISA according to the kit’s instructions. Absorbance was read at 450 nm using a plate reader (Bio-Rad). Three separate experiments were conducted for each cytokine measured.
Transmission Electron Microscopy
Thymocytes were seeded into 6-well plates at a density of 5 × 106/well and then treated with different concentrations of DBAA prior to a 24-hr incubation at 37°C. Thereafter, the treated cells were washed with cold PBS, centrifuged (1,500 rpm/min, 10 min, and 4°C), and fixed in 2.5% glutaraldehyde for 1 hr. The cells were then collected by centrifugation (1,500 rpm/min, 10 min, and 4°C) and rinsed 3 times in 0.1 M cacodylate buffer (pH 7.4). The cells were dehydrated with ethanol and embedded in epoxy resin. Ultrathin sections were cut and collected on gold grids, and 3 slices/dose were examined using a JEM-101 transmission electron microscope (Jeol Electron Inc., Tokyo, Japan).
Flow Cytometric Analysis of Phosphatidylserine Externalization in Apoptotic Cells
To determine the extent of early or late apoptosis and necrosis among treated thymocytes, cells were analyzed by staining with annexin V fluorescein isothiocyanate and propidium iodide (PI). Staining was performed using an Annexin V-FITC Apoptosis Detection kit (Becton Dickinson), according to the manufacturer’s instructions. In brief, thymocytes were treated with different concentrations of DBAA during a 24-hr incubation at 37°C. The treated cells were washed with cold PBS, centrifuged (1,500 rpm/min, 10 min, and 4°C), and then resuspended in a final volume of 100 μl kit-provided binding buffer prior to the addition of 5 μl annexin V and 5 μl PI. The cells were then incubated at room temperature in the dark for 15 min before the addition of 400 μl binding solution and were then analyzed using a FACSCalibur (Becton Dickinson) flow cytometer for 10,000 collected cells. Cells alive or in early apoptosis, late apoptosis, or necrosis were determined as percentages of AV−/PI−, AV+/PI−, AV+/PI+, or AV−/PI+ cells, respectively. All data were analyzed and quantified using Cell Quest software (BD Biosciences, San Jose, CA). All results were obtained from 3 independent experiments.
Cell cycle analysis
A Cycletest Plus DNA Reagent kit was used for cell cycle analysis, according to the manufacturer’s instructions. In brief, thymocytes were seeded into 6-well plates at a density of 5 × 106/well and were incubated for 24 hr and treated with different concentrations of DBAA. Thereafter, the treated cells were washed with cold PBS, centrifuged (1,500 rpm/min, 10 min, and 4°C), and resuspended in nuclear isolation medium. Because nuclear fluorescence is proportional to DNA content, analysis of DNA ploidy and discrimination of cells in the G0/G1, S, and G2/M phases of the cell cycle were performed by measuring the cellular DNA in a FACSCalibur system. Each DNA histogram from each treatment was analyzed with ModFit LT 2.0 software (Verity Software House, ME). All results were obtained from 3 independent experiments.
Confocal Fluorescence Imaging of Intracellular Calcium
Intracellular changes in calcium ion (Ca2+) levels were determined based on changes in the fluorescence intensity of fluo-3-acetoxymethyl (AM). In brief, thymocytes were treated/loaded with 5 μM fluo-3-AM and 5 μM Pluronic F-127 (each diluted in 99.9% DMSO) for 30 min at 37°C in the dark (Locknar et al. 2004). The cells were then washed with D-Hank’s and transferred to 100 μl of RPMI 1640 (containing CaCl2) to a petri dish, and the plate was mounted in a FV300 confocal microscope (Olympus, Tokyo). To measure the changes in intracellular Ca2 +, the cells were scanned by confocal laser microscopy before and just after treatment with 5 μl of DBAA (at final concentrations/dish of 0.1, 0.2, 0.4, 0.8, or 1 mM). Confocal laser scanning microscopy was performed as previously described (Dlugosz et al. 2002), using an excitation wavelength of 480 nm and a scanning time of 5 sec/image. Images were taken through the center of the cell nuclei, and digitized images were captured every 10 sec for 300 sec. Digitized images for each time point following stimulation were analyzed to determine the total cell pixel intensity using FV10-ASW software, version 4.0 (Olympus, Tokyo). For each condition, 10 to 30 cells from different plates (of 3 separate experiments) were analyzed. The measurement of Ca2+ was expressed as F/F0, that is, peak fluo-3 fluorescence (F) normalized to basal fluorescence (F0).
Determination of Apoptotic Proteins by Sodium Dodecyl Sulfate–Polyacrylamide Gel Electrophoresis
After treatment with DBAA for 24 hr, thymocytes were washed with cold PBS and resuspended in lysis buffer containing protease inhibitors. The cells were vortexed briefly and placed on ice for 2 hr. After centrifugation for 10 min (12,000 × g) at 4°C, the supernatant was collected and quantified using a Bicinchoninic Acid Protein Assay kit (Thermo, Rockford, IL). Forty micrograms of total protein of each sample were loaded on a 10 to 12% polyacrylamide gel. After electrophoresis, protein was electrotransferred to a nitrocellulose membrane (0.22 μm pore size). The membranes were blocked with 5% nonfat milk in Tris-buffered saline (containing 0.1% Triton X-100) for 1 hr at room temperature and were then incubated with the same solution containing the primary antibody specific for β-actin, Fas, FasL, or Bcl-2 (1:500 dilution) for 2 hr at 37°C (or alternatively, 4°C overnight). The membranes were washed with Tris-buffered saline with tween again and were then incubated with a solution containing the corresponding secondary antibody (1:5,000 dilution) for 1 hr at room temperature. To detect the extent of antibody binding, bands were visualized using an enhanced chemiluminescence (ECL system; Amersham Biosciences, Piscataway, NJ), according to the manufacturer’s protocols.
Statistical Analysis
All of the data presented in this article were analyzed using SPSS 17.0. All values are shown as the mean ± SE. The significant differences were determined by one-way ANOVA followed by post hoc Dunnett’s test. p Values of <.05 were considered significant, and p values of <.01 were considered highly significant.
Results
Exposure to DBAA Led to Significant Reduction in Thymocyte Proliferation In Vitro
Con A is a T-cell mitogen used to activate T cells. The likely mechanism is that Con A indirectly cross-links the T-cell receptor (TCR) and sends the activation signals. Thymocytes from BALB/c mice were exposed in vitro to various doses of DBAA (0, 5, 10, 20, or 40 μM) for different lengths of time in the presence of 5 μg/ml Con A. Cell proliferation was evaluated by the MTS assay, which determines the number of live, metabolically active cells based on mitochondrial activity. Exposure of thymocytes to DBAA led to a significantly decreased cell proliferative response to T-cell mitogen for 6 hr or longer (Figure 1). At 6 hr, significant inhibition was observed only at 40 μM, and significant inhibition was observed for all concentrations at 24, 48, and 72 hr.
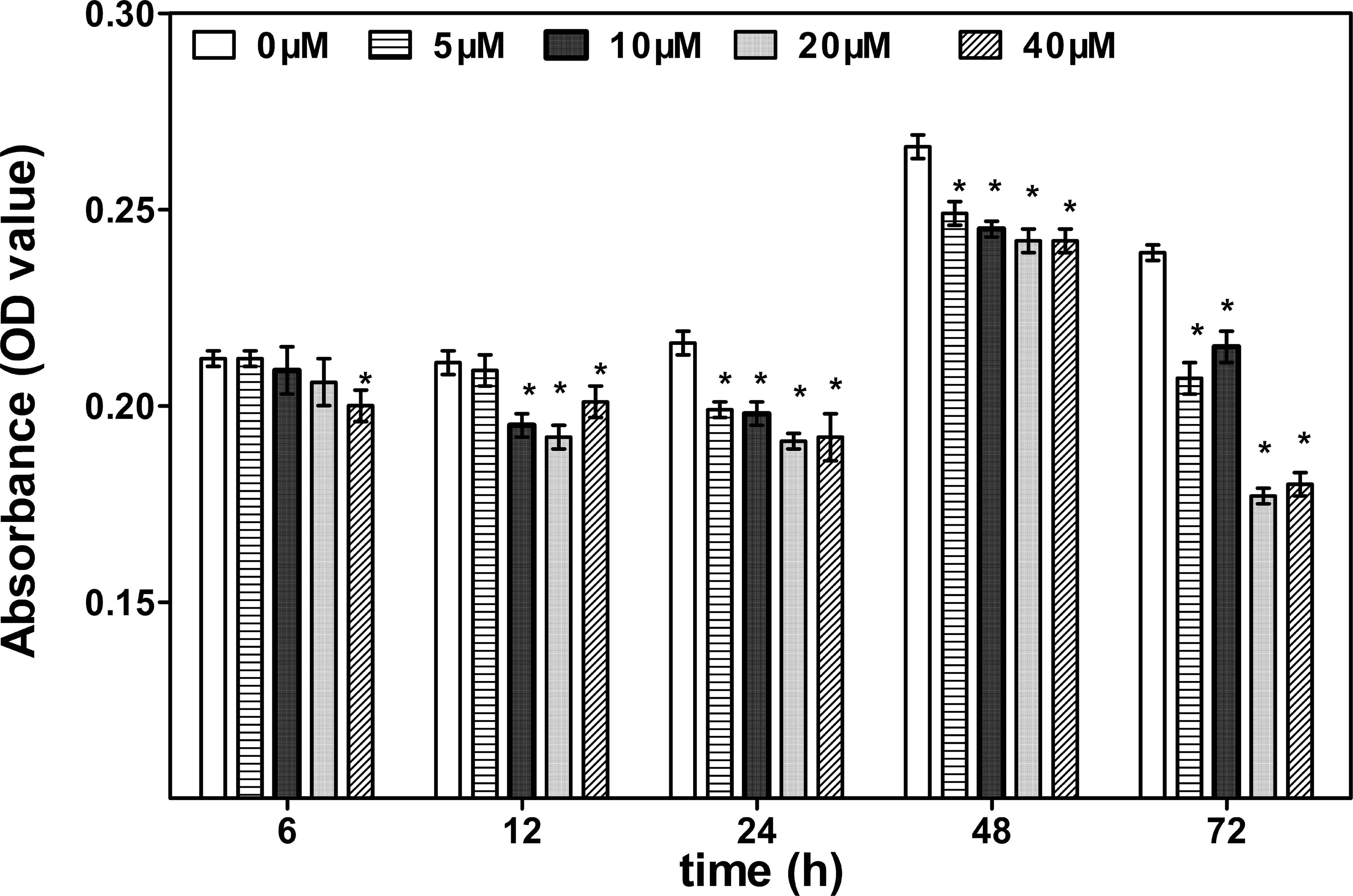
Reduction in thymocyte proliferation after dibromoacetic acid (DBAA) treatment. The proliferation of thymocytes treated with different concentrations of DBAA (0, 5, 10, 20, or 40 μM) for various periods (6, 12, 24, 48, and 72 hr) was measured by MTS assay. Values shown are mean ± SE of 8 wells per exposure condition. *p < .05 as compared to control.
Effect of DBAA Treatment on the Cytokines Release
To investigate the role of DBAA on cytokine production, IL-2 and IL-4 were analyzed in thymocytes treated with DBAA for 24 hr. IL-2 cytokine production was slightly increased at DBAA concentration 5 μM but significantly reduced at other concentrations. Significant reduction in IL-4 was observed at all concentration of DBAA in Con A stimulated thymocytes (Figure 2).
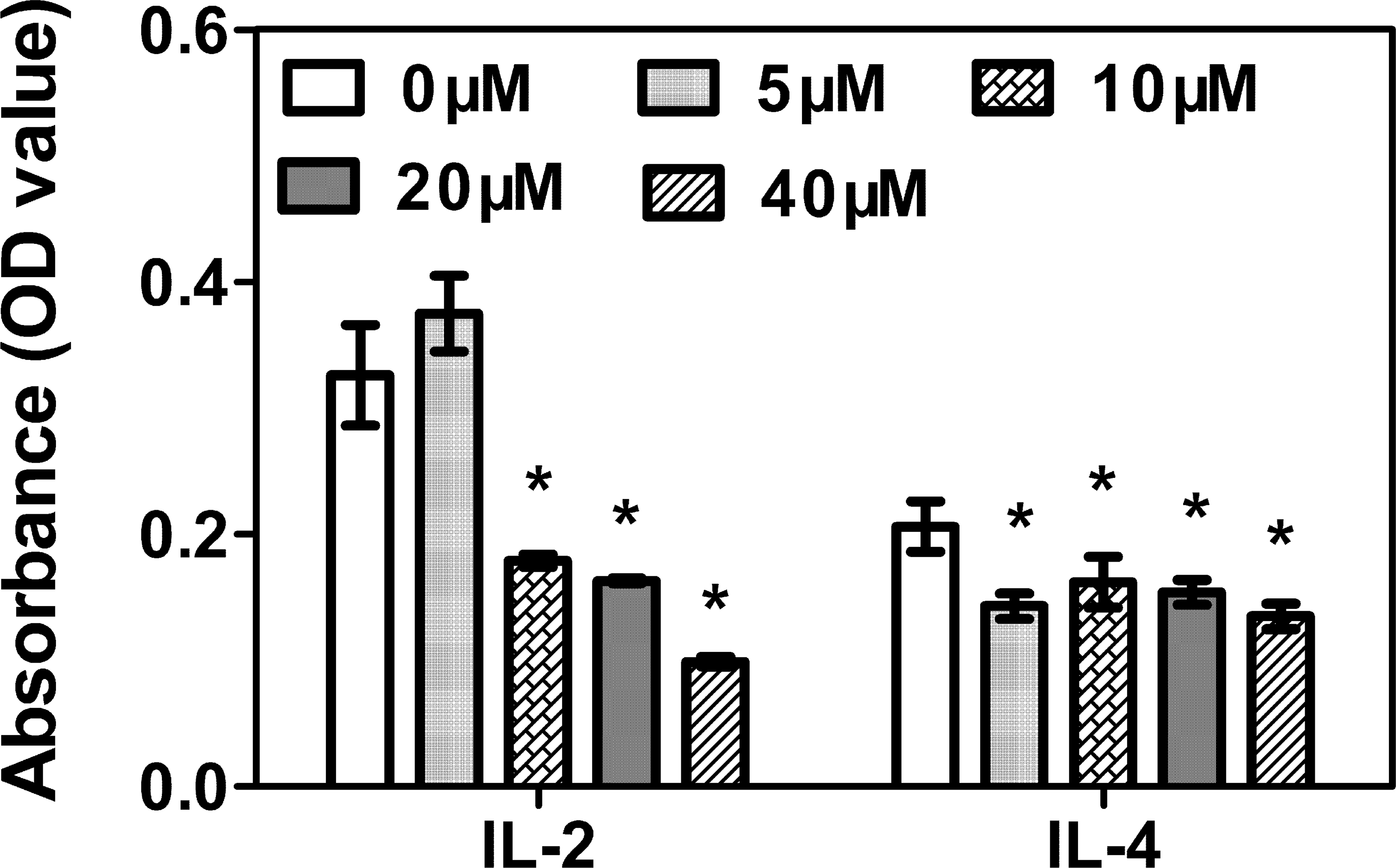
Effect of dibromoacetic acid (DBAA) on cytokine release. Freshly isolated thymocytes at a density of 2 × 106 were treated with different concentrations of DBAA (5, 10, 20, or 40 μM) and concanavalin A (5 μg/ml) for 24 hr at 37°C. The cytokines (IL-2 and IL-4) were measured by ELISA. Each bar represents mean ± SE. *p < .05; **p < .01 as compared to control, using one-way ANOVA.
Electron Microscopy
Electron micrographs of control cells showed integrated nuclear membranes, relatively homogeneous chromatin and extensive membrane interdigitations, and microvilli (Figure 3). The thymocytes treated with different concentrations of DBAA (0, 5, 10, 20, or 40 μM) for 24 hr were characterized by karyopyknosis, mitochondrial degeneration, perinuclear cisternal augmentation, many vacuoles in the cytoplasm, and nuclear fragmentation. All of the characteristics indicated the thymocytes were undergoing apoptosis, and the extent of the changes was dose dependent.
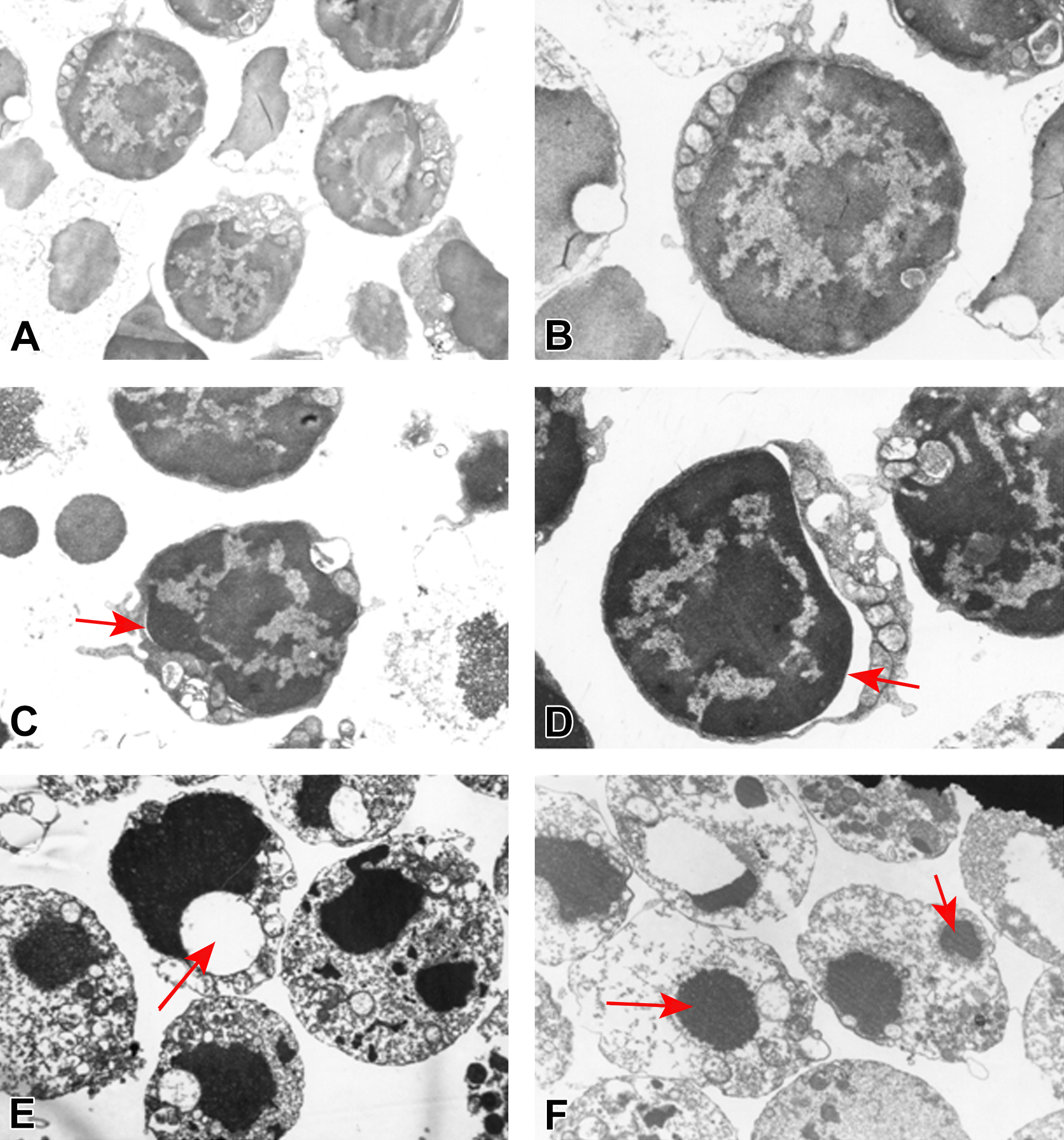
Electron microscopic characterization of thymocytes after treatment with dibromoacetic acid (DBAA). (A) and (B) Control (A magnification 8,000× and B magnification 12,000×) showed integrated nuclear membranes, relatively homogeneous chromatin and extensive membranes interdigitations, and microvilli. (C) Thymocytes treated with DBAA 5 μM (magnification 12,000×) show chromatin condensation into dense granules and karyopyknosis (arrow). (D) Thymocytes treated with DBAA 10 μM (magnification 12,000×) show cytoplasmic vacuolization (arrow). (E) Thymocytes treated with DBAA 20 μM (magnification 8,000×) show many cytoplasmic vacuoles and apoptotic bodies (arrows). (F) Thymocytes treated with DBAA 40 μM (magnification 8,000×) show many cytoplasmic vacuoles and apoptotic bodies (arrows).
DBAA-induced Apoptotic Cell Death
The percentages of apoptotic and necrotic thymocytes due to DBAA treatment were determined using flow cytometry. The annexin V binding assay, which measures the fluorescence generated by the binding of annexin V to externalized phosphatidylserine on apoptotic cells, was used to quantify apoptosis, together with PI to determine the percentage of necrotic and late apoptotic cells. The number of early apoptotic thymocytes (annexin V+PI−) increased progressively with increasing DBAA concentration. The percentage of late apoptotic thymocytes (annexin V+PI+) after DBAA treatment increased significantly at all concentrations. However, there were no differences in the percentage of necrotic cells (annexin V−PI+) between groups (Table 1 and Figure 4).
Effect of DBAA on the Apoptotic Percentage of Thymocytes.

Note: DBAA = dibromoacetic acid; UL = necrosis; UR = late apoptosis; LL = live cells; LR = early apoptosis.
**Indicates significant differences p < .01 compared with control thymocytes (DBAA 0 μM).
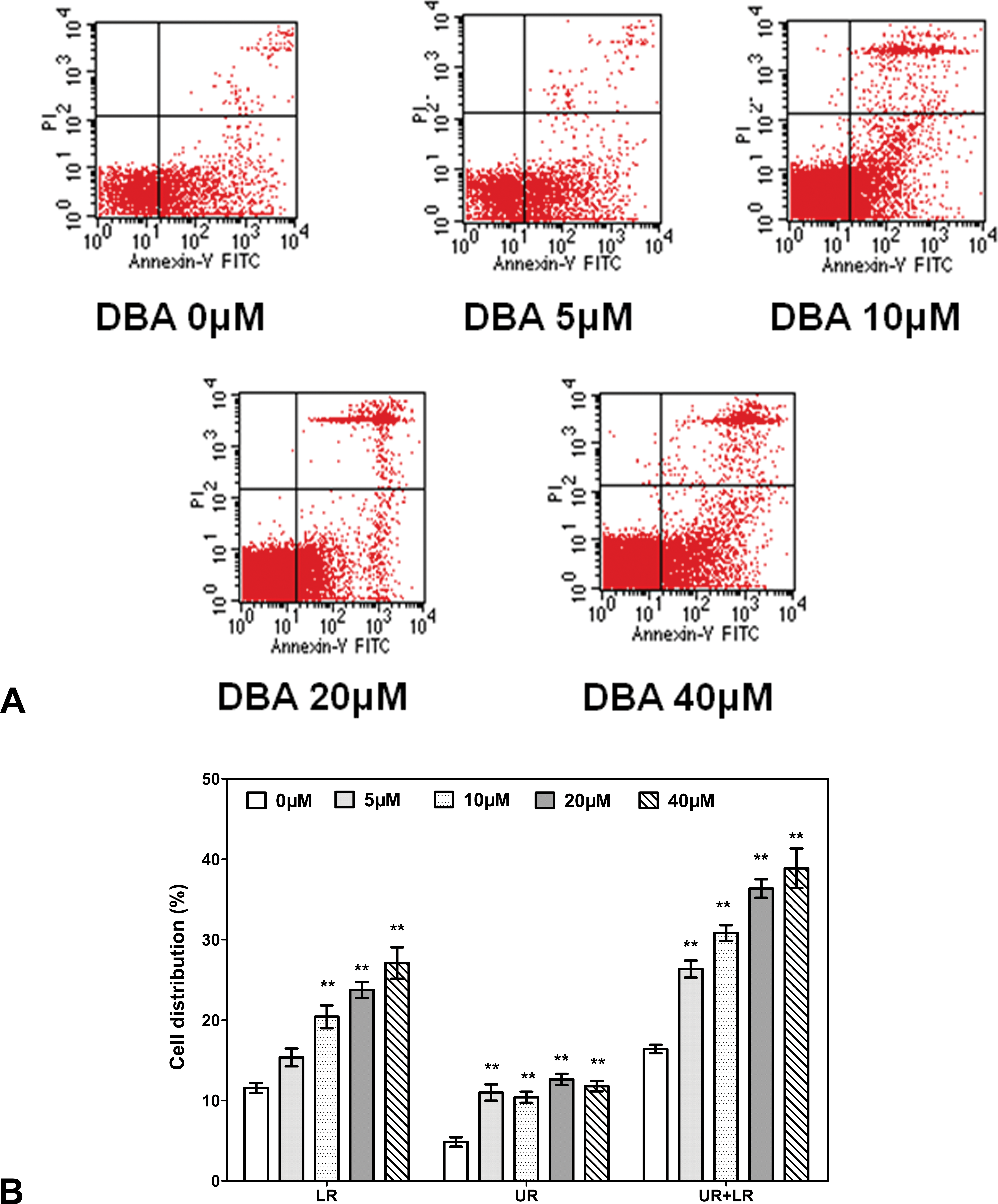
Effect of dibromoacetic acid (DBAA) on thymocyte apoptosis and necrosis. Thymocyte isolated from BALB/c mice were treated with different concentrations of DBAA (0, 5, 10, 20, or 40 μM) for 24 hr; cell viability and apoptosis were analyzed using propidium iodide (PI) staining, annexin V binding, and fluorescence-activated cell sorting analysis. (A) Representative results for each DBAA concentrations expressed as dot plots showing live cells (lower left; annexin V−/PI−), early/primary apoptotic cells (lower right [LR]; annexin V+/PI−), late/secondary apoptotic cells (upper right [UR]; annexin V+/PI+), and necrotic cells (upper left [UL]; annexin V−/PI+). (B) Mean percentages of late (UR), early (LR), and total (UR + LR) apoptotic cells after treatment of thymocytes with different DBAA concentrations. Results are expressed as mean ± SE of 3 experiments. *p < .05; **p < .01 compared to control.
DBAA-induced Cell Cycle Arrest
Cell cycle analysis of thymocytes confirmed that DBAA treatment induced cell cycle arrest (Table 2). The data showed at least 40% increase in the G0/G1 phase and 50% decrease in the S phase in thymocytes treated with different concentrations of DBAA.
Effect of Dibromoacetic Acid (DBAA) on Cell Cycle of Thymocytes after Exposure for 24 hr.
*Indicates significant differences p < .05.
**Indicates significant differences p < .01 compared with control thymocytes (DBAA 0 μM).
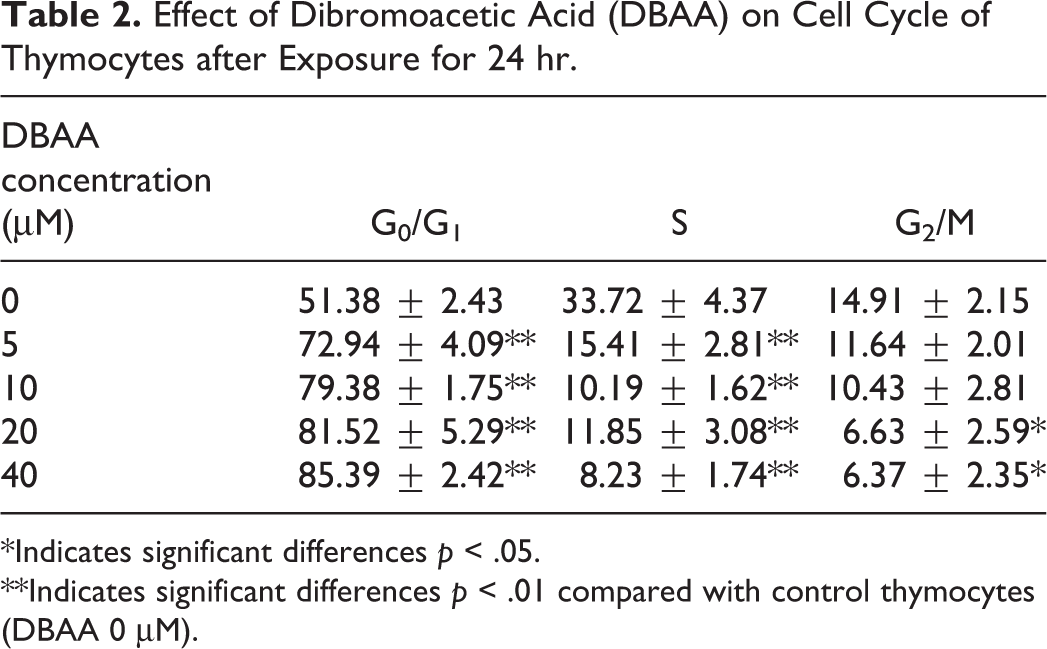
Measurements of Intracellular Free Ca2+ in Single Permeabilized Thymocytes
To identify whether DBAA treatment of thymocytes was associated with the release of intracellular stored Ca2+, we used a method based on the ability of the Ca2+-sensitive fluorescent indicator fluo-3-AM to compartmentalize into thymocytes. An increase in intracellular Ca2+ was induced by DBAA treatment at doses≥5 μM (Figure 5).
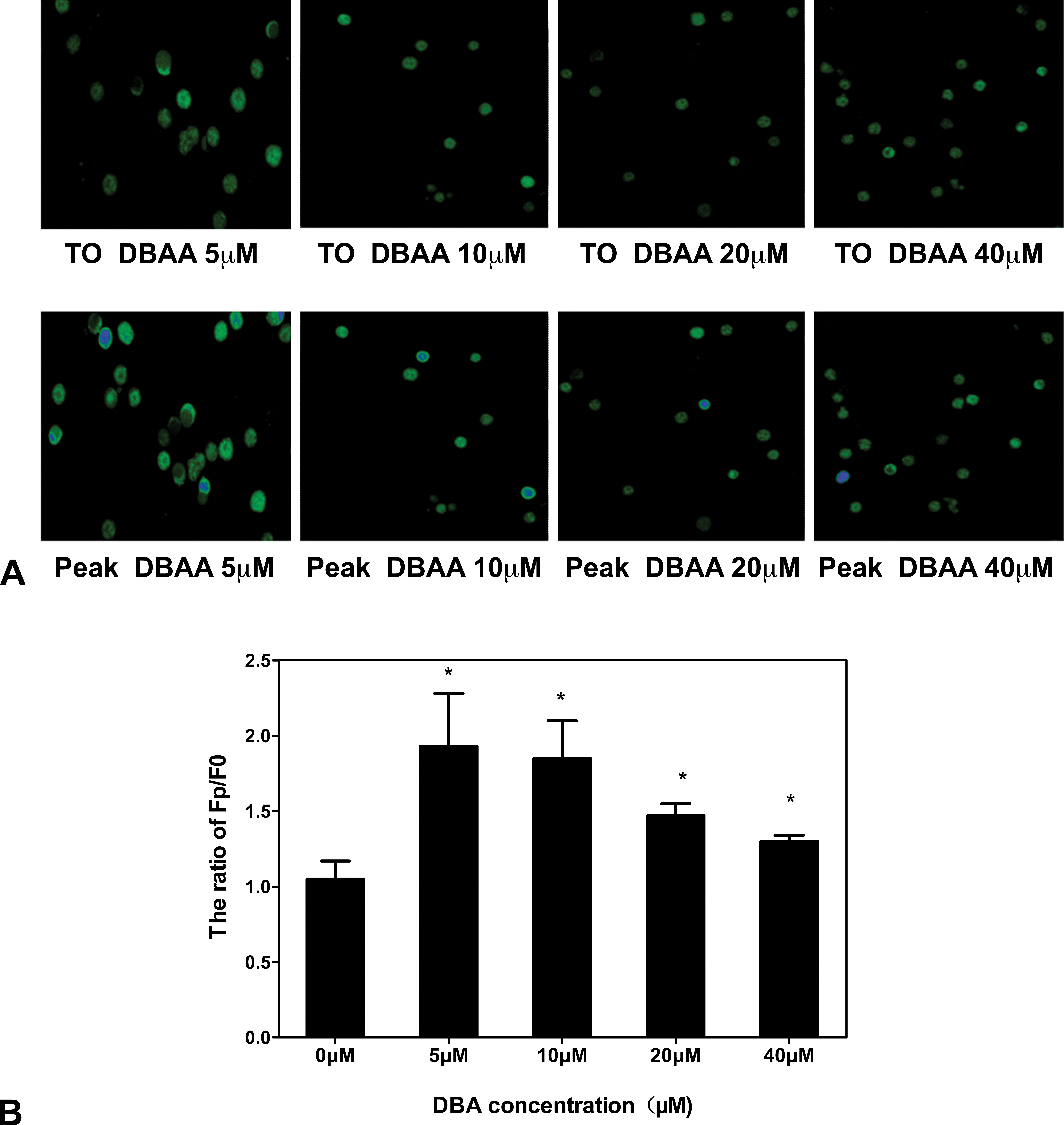
Elevation of intracellular Ca2+ of thymocytes in response to stimulation with different concentrations of dibromoacetic acid (DBAA). (A) Digitized images of cells from confocal fluorescence imaging selected at t = 0 sec and at peak fluorescence. (B) Graphical analysis of Ca2+ signaling from measurements of total pixel intensity per cell; 10–30 cells were analyzed from 3 independent experiments for each DBAA concentration. *p < .05 compared to control.
Expression of Fas/FasL and Bcl-2 Proteins
To determine the signaling pathways mediating DBAA-induced apoptosis, the protein expression of Fas-related apoptotic signaling molecules was examined after 24 hr of DBAA exposure. Western blot analysis of Fas/FasL and Bcl-2 expression in the lysates of the thymocytes is shown in Figure 6. The expression of Fas/FasL increased significantly from the DBAA concentration of 10 μM, and the expression of Bcl-2 decreased at all concentration of DBAA.
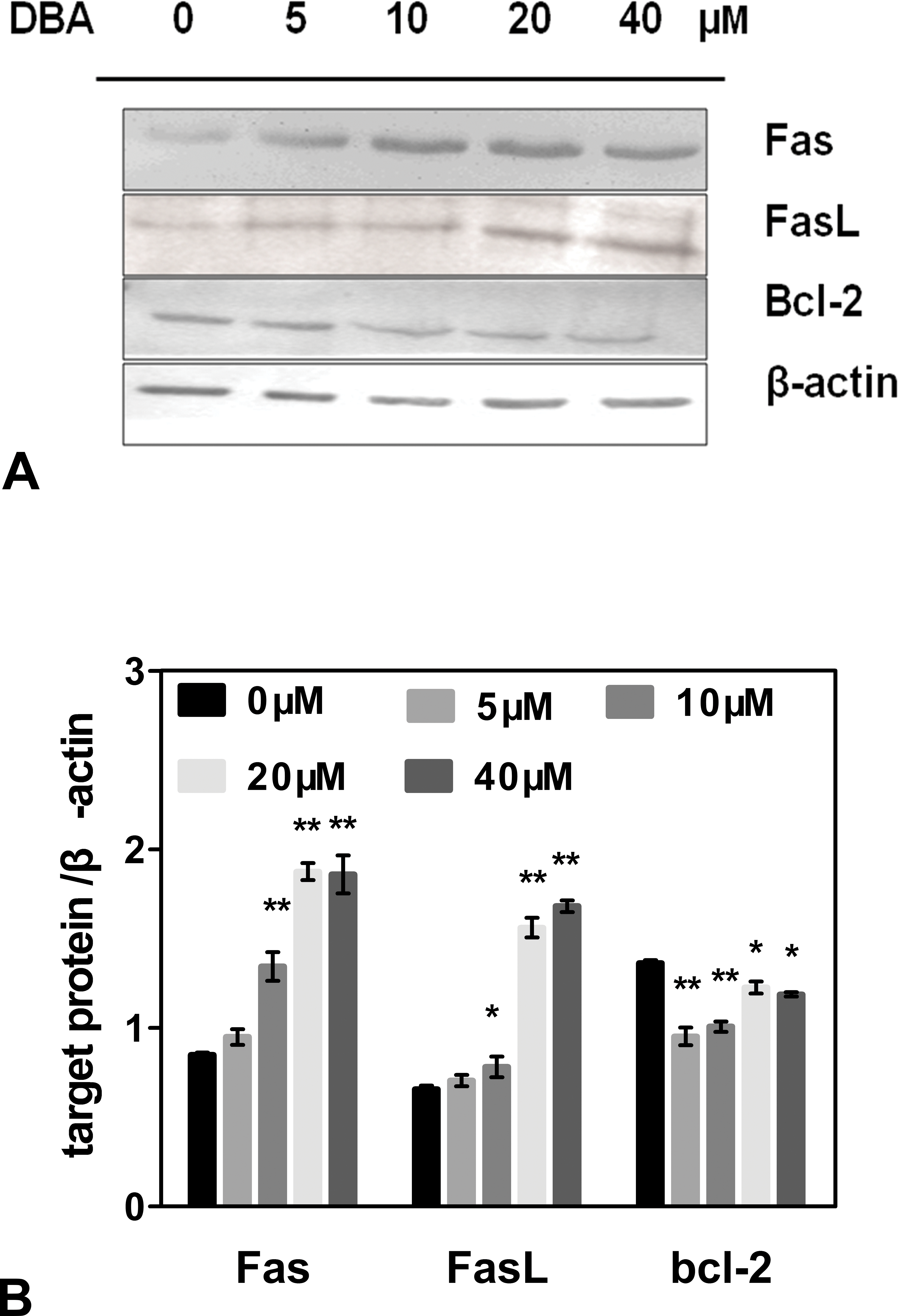
Effects of dibromoacetic acid (DBAA) on expression of Fas/FasL and Bcl-2 proteins in thymocytes after treatment with DBAA for 24 hr. Western blotting was used to analyze the protein expression of Fas/FasL and Bcl-2. (A) Representative blots are shown for Fas /FasL, Bcl-2, and β-actin used as a control. (B) The quantification results are expressed as the ratio of target protein/β-actin in each group. Data represents means ± SE of 3 independent experiments. The stars illustrate a statistically significant difference compared to control (*p < .05 and **p < .01).
Discussion
DBAA is a disinfection by-product commonly found in drinking water as a result of chlorination/oxidation processes. It is critical for public health to evaluate DBAA at or near levels that occur in drinking water, but epidemiology studies suffer from the difficulty of establishing sound exposure measures for people who are exposed to low concentrations of DBAA that varies widely over the years (Boorman 1999). So the toxicity data on DBAA are obtained from animal tests and in vitro experiments at much higher concentrations to find adverse effect.
In a previous study, we confirmed that DBAA induced marked immunotoxicity in mice characterized by thymus atrophy and splenomegaly, reduction in the response to polyclonal mitogens, and increased apoptosis (Gao et al. 2008). To explore the mechanisms of DBAA-induced immunotoxicity, thymocytes isolated from mice were incubated with different concentrations of DBAA in vitro, and the toxic effects were examined in multiple ways, focusing mainly on apoptosis.
Cell viability and morphology are crucial indicators for in vitro toxicity assays, which can provide a first insight into the cellular response to a toxicant. The results in the current study indicated that low-concentration DBAA was cytotoxic, that is, it was able to induce cell death and inhibit proliferation in thymocytes. In the process of modulation of cellular immune responses, T-cell cytokines play important roles to maintain immune homeostasis (Belkaid and Rouse 2005). In thymus, naive CD4+ T cells are separated into T-help 1 (Th1) and T-help 2 (Th2) cells depending upon the specific cytokines the cells secrete in response to antigenic stimulation. Th1 cells secrete IL-2 and IFN-γ, whereas Th2 cells secrete IL-4 and IL-5 (Lorré, Van Damme, and Ceuppens 1990). In our study, the data of cytokine release indicated that IL-2 was inhibited from 10 μM to 40 μM, and IL-4 was inhibited at all concentrations of DBAA (Figure 2). In the process of immune response, activation of CD4+ T cells is characterized by a strong proliferative response accompanied by secretion of cytokines in response to antigen stimulation; but CD4+ hyporesponsiveness is caused by exhaustion due to the presence of high levels or repeated stimulation of antigen (Wherry 2011). Furthermore, persistent stimulation of T cells through TCR and IL-2 signaling eventually induces apoptotic pathways, resulting in activation-induced cell death (AICD; Boyman and Sprent 2012).
It was confirmed in our study that the immunosuppressive effects of DBAA were associated with an induction of apoptosis. Ultrastructural cell morphology showed a number of morphological changes consistent with apoptosis, including karyopyknosis, karyorrhexis, mitochondrial degeneration, and perinuclear cisternal augmentation (Figure 3). Flow cytometry analysis revealed that the percentage of thymocytes identified in early and late apoptosis showed significant increase after DBAA treatment for 24 hr (Figure 4).
To investigate possible mechanisms for DBAA-induced apoptosis, we investigated its effects on the cell cycle, intracellular Ca2+ release, and expression of proteins related to apoptosis. The cytometric analyses revealed a dose-related increase in thymocytes in the G0/G1 phase of the cell cycle (Table 2). This result may be indicative of ongoing cell cycle arrest and was consistent with the results of Annexin V/PI staining, showing an increase in apoptotic cells. It is therefore plausible that the increase in apoptosis and cell cycle arrest in thymocytes are responsible for the inhibition of cell proliferation.
Calcium is one of the most crucial intracellular messengers and regulates cellular development, survival, and differentiation (Berridge, Bootman, and Roderick 2003). The intracellular calcium concentration must be precisely regulated for the increased cytosolic Ca2+ concentration to activate the intrinsic apoptotic pathway. In our study, a marked increase in intracellular Ca2+ release was detected at DBAA concentrations of 5 μM and above (Figure 5). In T cells, Ca2+ is a versatile second messenger mediating a variety of responses to TCR activation, including cell proliferation and apoptosis (Winslow, Neilson, and Crabtree 2003). Here, DBAA led to excessive Ca2 + influx into the cells, a process known to sensitize cells to apoptosis and enhance mitochondrial Ca2+ uptake (Oakes, Lin, and Bassik 2006; Pinton and Rizzuto 2006). Furthermore, apoptosis can be influenced by subtle changes in intracellular Ca2+ concentration, and cellular Ca2+ overload or perturbations of intracellular Ca2+ stores can lead to cytotoxic stress and trigger cell death (Orrenius, Zhivotovsky, and Nicotera 2003). In contrast, apoptosis can be inhibited by the overexpression of antiapoptotic Bcl-2, which diminishes intracellular Ca2+ stores and protects cells from Ca2+-dependent apoptotic stimuli (Foyouzi-Youssefi et al. 2000). In the present study, the expression of Bcl-2 decreased and the expression of Fas/FasL increased after treatment with DBAA (Figure 6). One possible mechanism for this is that Bcl-2 was degraded, causing inactivation of Bcl-2 during the G2/M phase, thus increasing the susceptibility of the cells to apoptosis during the cell cycle (Yamamoto, Ichijo, and Korsmeyer 1999; Bassik et al. 2004). Fas/FasL not only is essential for the induction of apoptosis but also has a critical role in cellular activation, proliferation, and differentiation within the immune system (Nagata and Goldstein 1995; Opferman 2007; Bell et al. 2008). One of the mechanisms for T-cell hyporesponsiveness to antigen is the induction of AICD or apoptosis in the T-cell population, particularly through the engagement of Fas/ FasL (Lenardo et al. 1999; McKinstry, Strutt, and Swain 2010).
Conclusions
In summary, DBAA inhibited thymocyte proliferation and induced apoptosis in vitro. The underlying mechanisms were found to include the inhibition of cytokines release, the increase in intracellular Ca2+, G0/G1 cell cycle arrest, and extrinsic (Fas/FasL) apoptotic signaling pathways. These results provide new insight into the mechanism of immunotoxicity induced by DBAA.
Footnotes
Author Contributions
Authors contributed to conception or design (SG, XZ, TG, MJ, and BL); data acquisition, analysis, or interpretation (SG, XZ, TG, MJ, and BL); drafting the manuscript (SG, XZ, TG, MJ, and BL); and critically revising the manuscript (SG). All authors gave final approval and agreed to be accountable for all aspects of work in ensuring that questions relating to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the grant from National Natural Science Foundation of China (grant number 30901206) and a project funded by Scientific and Technological Innovation in Harbin (grant number 2009RFQXS025).