
Abstract
Perfluorooctanoic acid (PFOA) is a ubiquitous pollutant that causes liver toxicity in rodents, a process believed to be dependent on peroxisome proliferator–activated receptor-alpha (PPARα) activation. Differences between humans and rodents have made the human relevance of some health effects caused by PFOA controversial. We analyzed liver toxicity at 18 months following gestational PFOA exposure in CD-1 and 129/Sv strains of mice and compared PFOA-induced effects between strains and in wild type (WT) and PPARα-knockout (KO) 129/Sv mice. Pregnant mice were exposed daily to doses (0.01–5 mg/kg/BW) of PFOA from gestation days 1 to 17. The female offspring were necropsied at 18 months, and liver sections underwent a full pathology review. Hepatocellular adenomas formed in PFOA-exposed PPARα-KO 129/Sv and CD-1 mice and were absent in untreated controls from those groups and WT 129/Sv. Hepatocellular hypertrophy was significantly increased by PFOA exposure in CD-1, and an increased severity was found in WT 129/Sv mice. PFOA significantly increased nonneoplastic liver lesions in PPARα-KO mice (hepatocyte hypertrophy, bile duct hyperplasia, and hematopoietic cell proliferation). Low-dose gestational exposures to PFOA induced latent PPARα-independent liver toxicity that was observed in aged mice. Evidence of liver toxicity in PPARα-KO mice warrants further investigation into PPARα-independent pathways.
Introduction
Perfluorooctanoic acid (PFOA) is an 8-carbon perfluoroalkyl acid, a member of a group of chemicals called perfluoroalkyl and polyfluoroalkyl substances (PFAS). It is produced both synthetically and through the degradation of other PFAS. PFOA is commonly used as a water and oil repellent for fabric coatings, food storage containers, lubricants, and fire extinguishing foams. It is persistent in the environment and is bioaccumulative. The half-life in humans is estimated to be between 2.3 and 3.8 years (Lau 2012). Because of its environmental persistence, it is found ubiquitously in human serum (Kato et al. 2011). Children under the age of 11 years have the highest concentrations of PFOA in their serum compared with teenagers, adults, and seniors (Kato et al. 2009; Schecter et al. 2012). PFOA transfers to the fetus and is found in umbilical cord serum and breast milk, thus posing a risk for developmental exposure (von Ehrenstein et al. 2009; Ode et al. 2013; Fromme et al. 2010).
There are many differences in response to PFOA that are dependent on species. A recent review details species differences in elimination half-lives, with humans >> dogs = monkeys > mice > rabbits > rats and reported differences in sex-specific elimination rates for several PFAS (Lau 2012). Sex-specific differences in rat PFOA elimination half-lives are large; that is, 2 to 4 hr in females and 4 to 6 days in males. Other species, including humans and mice, have closer to equal half-lives with regard to sex (Lau 2012). To avoid daily episodic exposure situations due to a short PFOA elimination half-life in female rats, mice are the preferred laboratory model for developmental exposure studies. The half-life in mice is 17 to 19 days (Lau 2012). There are strain and/or developmental window differences in PFOA sensitivity observed in mice. Peripubertal PFOA exposure inhibited mammary gland growth in both Balb/c and C57Bl/6 wild-type (WT) mice; however, Balb/c mice were more sensitive to PFOA inhibition (Zhao et al. 2012). Furthermore, PFOA exposures that were one-fifth those needed in Balb/c and C57Bl/6 mice induced mammary epithelial delays in CD-1 mice following prenatal exposure (Macon et al. 2011).
Using all available published information, a mode of action (MOA) analysis of PFOA-induced tumorigenicity and its related human relevance was performed recently (Klaunig, Hocevar, and Kamendulis 2012). In the rodent analyses, the peroxisome proliferator–activated receptor-alpha (PPARα) pathway was the proposed mode of carcinogenic action for PFOA in the rodent liver, based primarily on data from adult PFOA-exposed male rats or mice. While liver tumor induction is plausible in humans since the gene is expressed in the liver, the weight of evidence suggested that PPARα induction was not a primary MOA in humans. There is approximately 10-fold less mRNA for PPARα found in human liver compared to rodent livers (Palmer et al. 1998). Human PPARα mediates pathways controlling lipid metabolism and those pathways are reported to be independent of cell proliferation pathways in adult dosed male mice (Cheung et al. 2004). In COS-1 cells transfected with human- or mouse-derived PPARα, PFOA activated the mouse PPARα to a greater extent than the human PPARα (Maloney and Waxman 1999; Takacs and Abbott 2007). This suggested that PPARα in human hepatocytes would be less responsive to PFOA than rodent hepatocytes. PFOA increased liver weight at 3 and 10 mg/kg in adult male cynomolgus monkeys, but microscopic effects were not evident (Butenhoff et al. 2002). PPARα may not be the MOA of PFOA-induced liver toxicity in human liver, but PPARα-independent effects may exist in rodents/humans, thus it is important to evaluate the relevance of these findings to human health.
PPARα activation has been suggested to protect against chemically induced hepatobiliary injuries, and this function could mask the potential toxicities of PFOA (Minata et al. 2010). In adult male 129/Sv mice (both WT and PPARα-knockout [KO]) given PFOA daily for 4 weeks, PFOA induced a greater occurrence of cholestatic lesions in PPARα-KO mice than in WT mice. Another weak PPARα ligand, Bezafibrate, induces cholestasis without neoplastic changes in PPARα-KO mice (Hays et al. 2005). In mice, PFOA induced different hepatocyte injuries depending on the presence or absence of PPARα and expression of human or mouse PPARα. In a recent study, adult male 129/Sv WT, PPARα-KO, and humanized PPARα (hPPARα) mice were treated with 1.0 or 5.0 mg/kg of PFOA for 6 weeks (Nakagawa et al. 2012). WT and hPPARα mice demonstrated hypertrophic hepatocytes and PPARα-KO mice presented with inflammatory cell infiltrations. There was also a difference in steatosis among the 3 groups. Microvesicular steatosis was seen in PPARα-KO and hPPARα mice, macrovesicular steatosis was only observed in the PPARα-KO mice, and no changes were seen in WT mice. Mice expressing hPPARα produce PPARα at protein levels similar to WT mice, which are 10 times higher than what is found in human livers.
In this study, we examine the long-term hepatic effects following developmental exposure to PFOA in mice with and without endogenous PPARα expression. Pregnant mice were dosed with PFOA or vehicle throughout the entire pregnancy and their offspring were assessed as adults for latent liver effects. We used 2 different strains of mice; 129/Sv (both WT and PPARα-KO mice) and CD-1. Our histopathological findings suggest that low-dose gestational PFOA exposure can induce hepatic injury and cancer in a dose-dependent and PPARα-independent manner. Further studies are warranted to evaluate PPARα-independent mechanisms for liver toxicity following developmental PFOA exposures. Our initial mechanistic evaluations (Quist et al., this issue), confirm the findings here that suggest PFOA mediates its hepatotoxic effects on the liver via pathways other than PPARα.
Materials and Methods
Animals
Timed-pregnant CD-1 mice (Charles River Laboratories, Raleigh, NC) arrived on gestational day (GD) 0 (sperm positive) at the U.S. Environmental Protection Agency (EPA) where they were weighed upon arrival and distributed to make the average dam weight similar in each dose group. PPARα-KO (null) mice (129S4/SvJae-Pparatm1Gonz/J, stock #003580) and 129/Sv WT mice (129S1/SvlmJ, stock #002448) were originally purchased from the Jackson Laboratory (Bar Harbor, ME) and were maintained as an inbred colony on the 129S1/SvlmJ background at the U.S. EPA, Research Triangle Park, NC. Pregnant dams were housed individually in polypropylene cages and received chow (LabDiet 5001, PMI Nutrition International LLC, Brentwood, MO) and tap water ad libitum. Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC)-accredited animal facilities were controlled for temperature (20–24°C) and relative humidity (40–60%), and kept under a 12-hr light–dark cycle. Animals were treated humanely and with regard for alleviation of suffering, and all animal protocols were approved by the U.S. EPA National Health and Environmental Effects Research Laboratory Animal Care and Use Committee.
Dosing Solution and Procedures
PFOA, as its ammonium salt (>98% pure, linear product), was acquired from Fluka Chemical (Steinheim, Switzerland). PFOA dosing solution was prepared fresh daily in deionized water, and the dosing solution was administered at a volume of 10 µl/g. Mice received either water vehicle or PFOA, as detailed subsequently, by oral gavage once daily over the period of gestation. The highest dose (5 mg PFOA/kg/day) was shown in a previous study using CD-1 mice (Lau et al. 2006) to induce hepatomegaly, with no effect on dam body weight (BW) gain and mild postnatal mortality (˜25% increase in mortality from controls). The doses for the 129/Sv substrains were not identical because of reported differences in sensitivity to PFOA among these 2 substrains (Abbott et al. 2007)
Experimental Design
Developmental exposure
Two blocks of CD-1 mice were used in these studies. The blocks were staggered by 4 weeks, and some animals from these studies were used in other reports (Hines et al. 2009). The timed-pregnant animals were dosed with vehicle (distilled water), 0.01, 0.1, 0.3, 1, or 5 mg PFOA/kg BW resulting in a final number of 29, 29, 37, 26, 31, and 21 female offspring surviving to 18 months of age (from 12, 12, 14, 13, 12, and 6 pregnant dams), respectively, and included in this study. Some animals died before 18 months (28%, 17%, 16%, 28%, 24%, and 22% of beginning n from control, 0.01, 0.1, 0.3, 1, and 5 mg PFOA/kg BW, respectively) due to sudden, unknown causes (found on morning check, 28% of early deaths), or severe dermatitis (common to CD-1; 32%) and other health problems (40%) that required preemptive euthanasia. The percentages of early death in our study are in line with reported survival rates in 8 studies of 78-week-old CD-1 control mice, with an average death rate prior to 18 months of 21.7% (table 2 in Giknis and Clifford 2010). For the 129/Sv mice, 3 blocks of animals were used in these studies, each separated by 2 to 3 weeks. The 129/Sv WT animals were dosed with vehicle, 0.1, 0.3, 0.6, or 1 mg PFOA/kg BW resulting in a final number of 10, 10, 8, 6, and 8 female offspring surviving to 18 months of age (to be consistent with the CD-1 animals) and included in the necropsy (from 7, 7, 5, 3, and 5 pregnant dams), respectively. PPARα-KO animals were dosed with vehicle, 0.1, 0.3, 1, or 3 mg PFOA/kg BW, resulting in a final number of 6, 10, 10, 9, and 9 offspring (from 5, 9, 8, 7, and 9 pregnant dams), respectively. Details of survival in all groups are reported in Supplemental Data Table 1. Different dose ranges were used for the 3 strains due to differences in strain sensitivities to PFOA. The highest dose used per strain was selected to minimize developmental toxicities and litter loss (Abbott et al. 2007). The lower doses were selected that would result in adolescent mice with PFOA blood serum levels comparable with serum levels reported in highly exposed humans (Macon et al. 2011). All animals received PFOA by oral gavage on the mornings of GD 1 to 17. Dams were weighed daily prior to dosing to determine dose amount.
At birth, pups were individually weighed and sexed. Consistent with previous PFOA research in CD-1 mice (Lau et al. 2006; White et al. 2007; Wolf et al. 2007), pups within a treatment group were pooled and randomly redistributed among the dams of their respective treatment groups, and litters were equalized to 10 pups (both sexes represented). Among the CD-1 mice, small litters (n < 4 pups) were excluded from the remainder of the study. All 129/Sv pups were kept with their mothers to ensure survival. Litter sizes are typically smaller in 129/SV mice than CD-1, and our litter size ranged from 4 to 8 pups. Pups were weaned at 21 days of age at which point only female offspring were retained for these studies and housed 3 to 5 mice per cage. Males were either used for breeding (controls) or utilized in other studies. Growth data on these mice have been reported in Hines et al. (2009).
Data collection
The exposure schematic for these studies is shown in Figure 1. At 18 months, liver and numerous other target organs (not described here) were collected from all surviving exposure groups. As denoted in Supplemental Data Table 1 and in Hines et al. (2009), some animals died prior to 18 months; these animals were not included here due to inconsistencies in age and quality of tissues that could be retrieved. This study was not designed as a liver carcinogenesis study (see Hines et al. 2009) and was the result of finding liver tumors in preterminal decedent PPARα-KO animals where we expected to find none. Although liver tissues collected from animals sacrificed before 18 months indicated the need to focus attention on the liver in these studies, those data from animals <18 months of age were not included in this analysis, as those animals were sacrificed for various reasons, including severe dermatitis in some cases. To avoid bias in liver outcomes, only those mice living until 18 months were included in analyses.
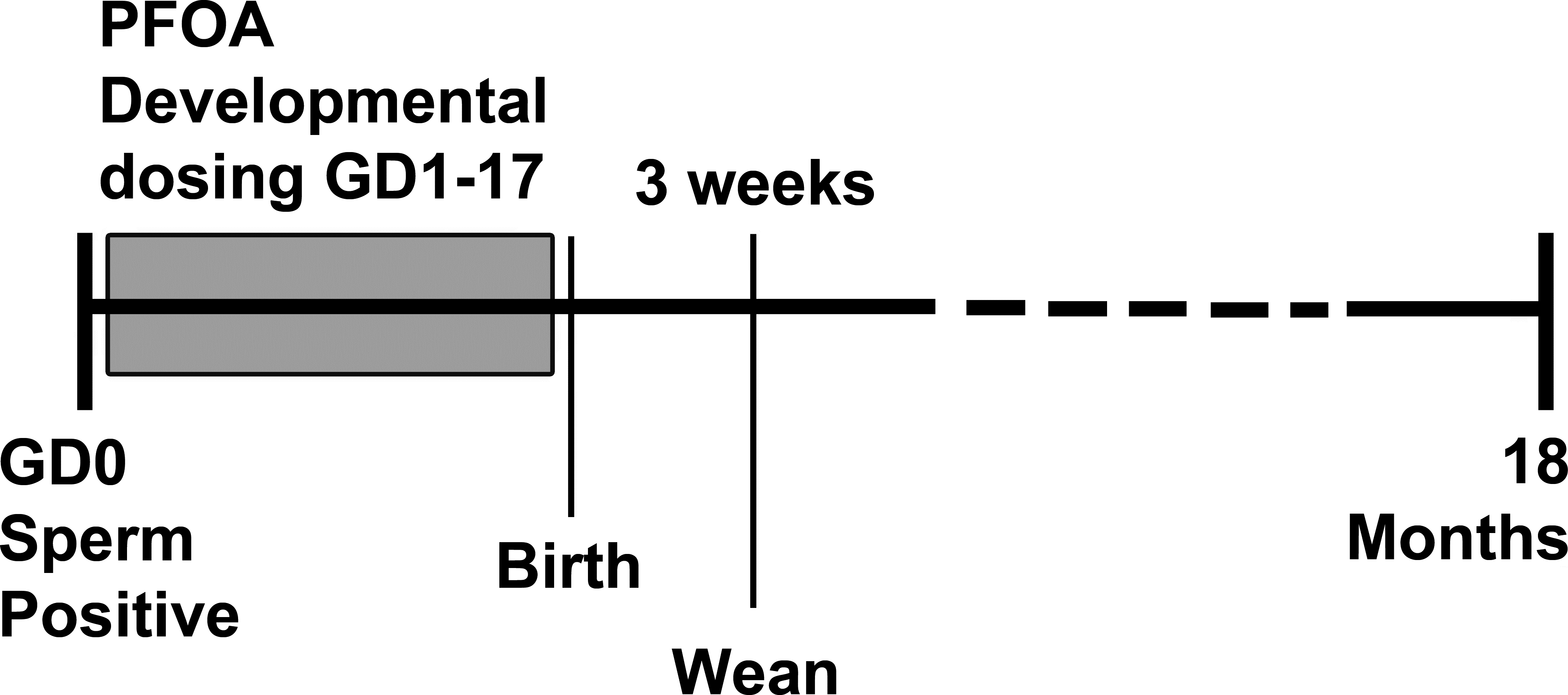
Exposure schematic for study of developmentally perfluorooctanoate acid (PFOA)-exposed female mice. Exposure was during gestation and all tissues were collected from female offspring at 18 months of age.
Pathology techniques
All 18-month-old mice underwent a full necropsy. Livers were fixed in 10% neutral buffered formalin and routinely processed for histology. Samples collected from the left lobe of the liver were cut into 5 µm-thick sections and stained with hematoxylin and eosin (H&E). Histological sections were reviewed by a team of board-certified veterinary pathologists within the National Toxicology Program (NTP) Pathology Group at the National Institute of Environmental Health Sciences (NIEHS; Pathology Working Group) in order to characterize and evaluate observed neoplastic and nonneoplastic lesions. Liver lesions were classified based on the liver nomenclature guidelines recommended by the International Harmonization of Nomenclature and Diagnostic Criteria (INHAND) documents published in Toxicological Pathology. (Thoolen et al. 2010). Neoplastic lesions were noted as presence = +1 or absence = 0, of a lesion. Nonneoplastic lesions were graded on a severity scale of 1 to 4 (1 = minimal, 2 = mild, 3 = moderate, and 4 = severe).
Statistics
Data were analyzed using Cochran–Armitage trend tests for dose-related trends in incidences for each strain/condition combination. Fisher’s exact test was used to compare each dose group to the control group and to compare strains at the same condition. Kruskal–Wallis analysis of variance (ANOVA) was used to test for differences in severities across all dose groups, and Mann–Whitney tests were used to compare each dose group to the control group within the ANOVA, as the incidence data were not normally distributed. A p value ≤ .05 was considered statistically significant.
Results
Histopathological Findings in the Liver of CD-1 Mice
Neoplastic and preneoplastic lesions were present in CD-1 mice treated with PFOA, and a single malignant lymphoma was found in untreated animals (Table 1; top panel). Lymphoma was a background lesion in 14.4% of 18-month-old historical control CD-1 females (table 6 in Giknis and Clifford 2010). The overall incidence of malignant lymphoma in our study was 2.3% (4 of 173) or 3.4% in controls only (1 of 29). Hemangiosarcomas were observed in 2 animals in the high-dose 5.0 mg/kg BW PFOA group and 1 animal in the mid-dose 0.3 mg/kg BW PFOA group, and hemangioma/hemangiosarcomas are found in only 0.4% of historical control female CD-1 mice (Giknis and Clifford 2010). Hepatocellular adenomas occurred in every dose group except 1.0 mg/kg BW PFOA and were significantly increased compared to controls at 0.3 mg/kg BW PFOA, with an overall incidence in treated animals of 4.9% (7 of 144), and specific dose responses are shown in Table 1. This differs dramatically from historical control female CD-1 mice at 18 months of age, where this lesion has not been detected (Giknis and Clifford 2010). Hepatocellular carcinomas occurred in mice dosed with 0.3 and 5.0 mg/kg BW PFOA and histiocytic sarcomas developed in mice dosed with 0.1, 1.0, and 5.0 mg/kg BW PFOA (shown in Figure 2), but neither tumor type reached statistical significance (Table 1). Basophilic or eosinophilic foci were found in 3 treated animals (0.01, 0.1, and 0.3 mg/kg BW PFOA, respectively) and were also without statistical significance.
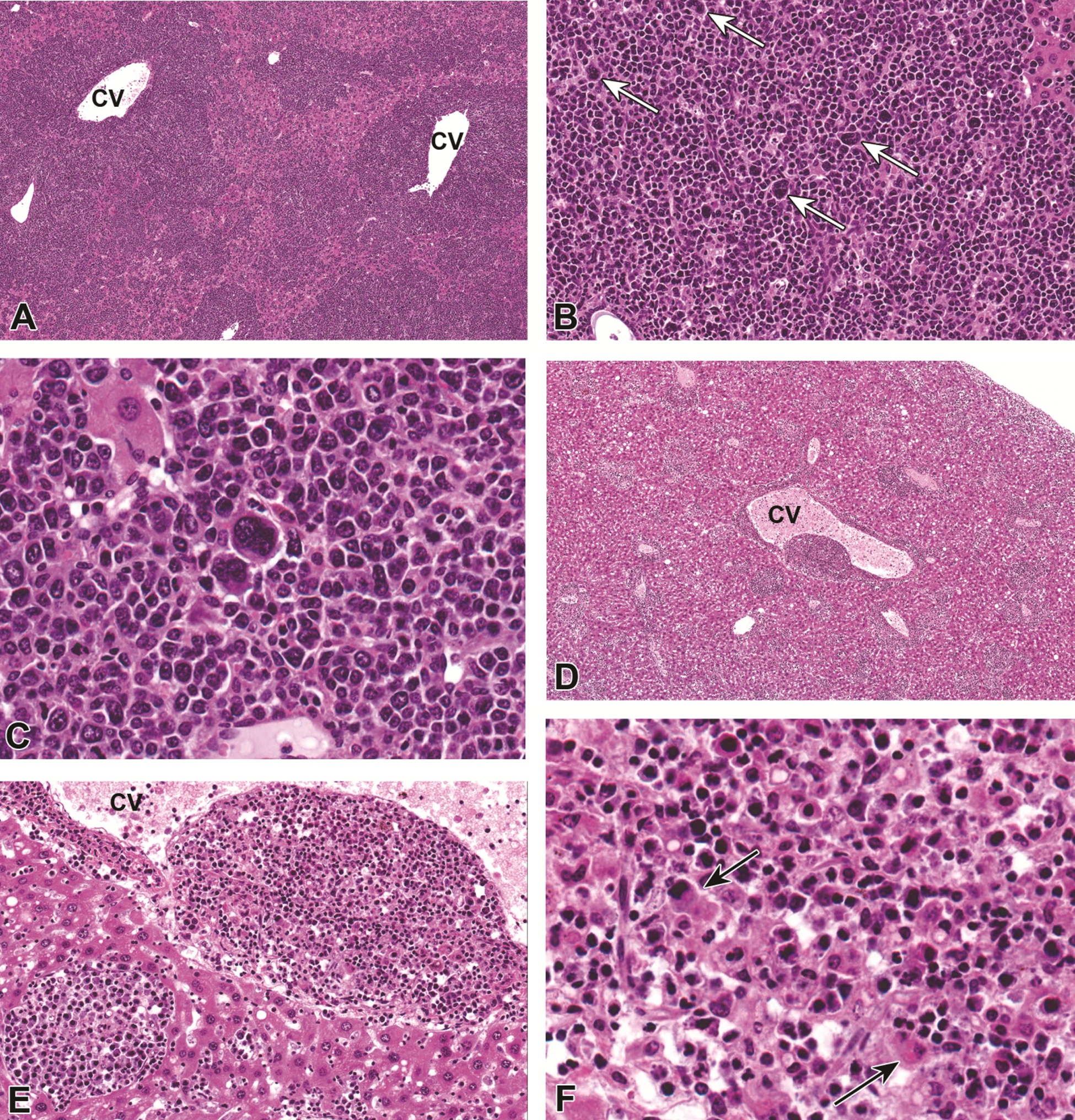
Histiocytic sarcoma in perfluorooctanoate acid (PFOA)-treated CD-1 mice. (A–C) Liver, CD-1 mouse, 0.1 mg/kg PFOA. A densely cellular, poorly demarcated, infiltrative neoplasm is diffusely expanding centrilobular to midzonal areas, compressing and replacing the existing hepatic parenchyma, CV = central vein. 4×. Hematoxylin and eosin (H&E). (B) The neoplasm is composed of sheets of a homogeneous round cell population that is histiocytic in appearance and contains numerous multinucleated giant cells (arrows). 20×. H&E. (C) Higher magnification of multinucleated giant cells. 40× magnification. (D–F) Liver, CD-1 mouse, 5.0 mg/kg PFOA. (D) A similar population of histiocytic round cells are multifocally infiltrating the hepatic parenchyma, surrounding both central and portal veins, CV = central vein. 4×. H&E. (E) Neoplastic cells are often distending vascular structures and partially obstructing vascular lumina, CV = central vein. 20×. H&E. (F) Neoplastic round cells occasionally exhibit erythrophagocytosis (arrows). 40×. H&E.
Neoplastic and nonneoplastic incidences (and severity scores) in livers after PFOA treatment (mg/kg) in CD-1 mouse strain.
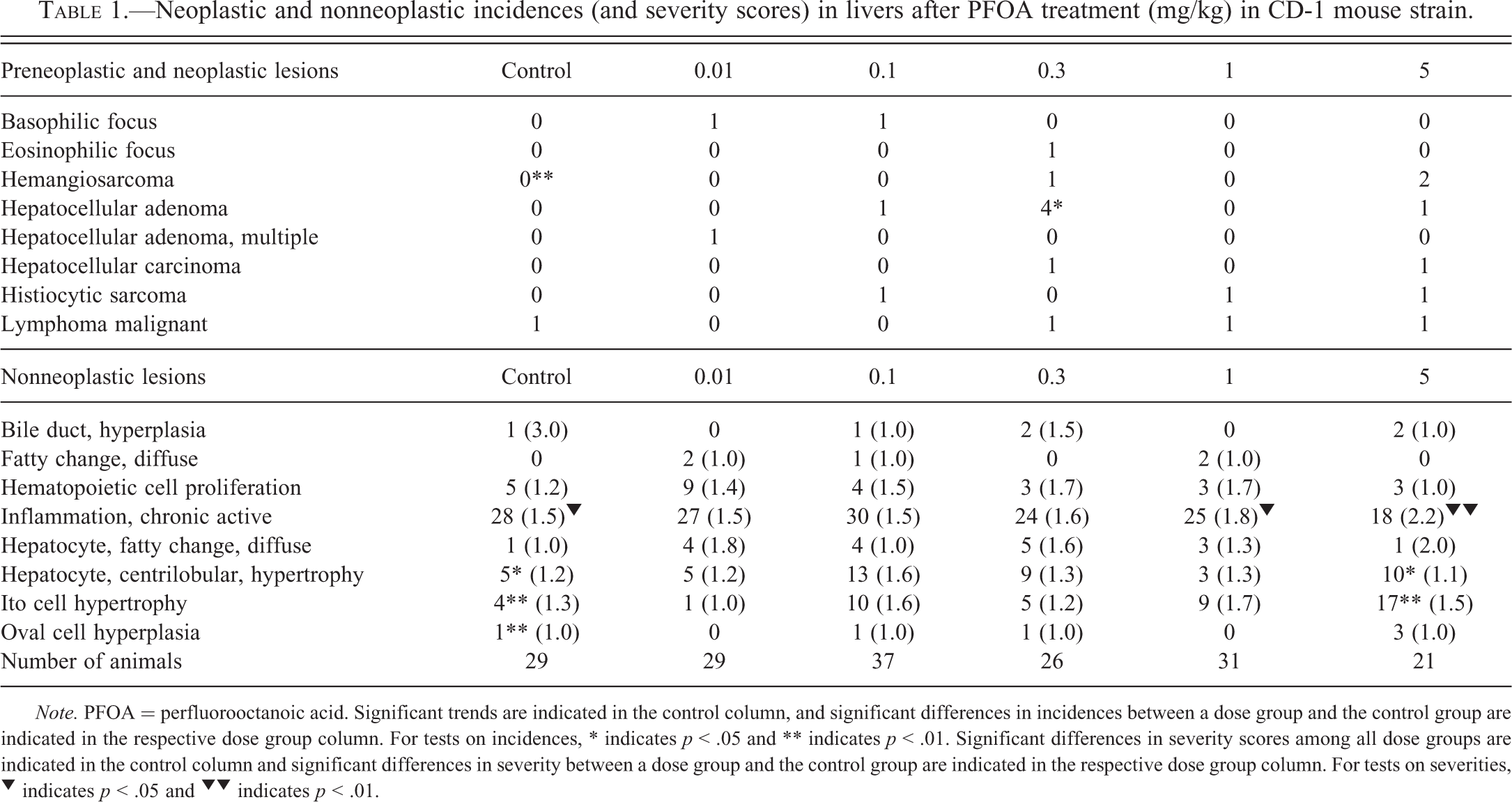
Note. PFOA = perfluorooctanoic acid. Significant trends are indicated in the control column, and significant differences in incidences between a dose group and the control group are indicated in the respective dose group column. For tests on incidences, * indicates p < .05 and ** indicates p < .01. Significant differences in severity scores among all dose groups are indicated in the control column and significant differences in severity between a dose group and the control group are indicated in the respective dose group column. For tests on severities, ▾ indicates p < .05 and ▾ ▾ indicates p < .01.
Numerous nonneoplastic liver lesions were noted in CD-1 mice. Developmental PFOA exposure caused a significant dose-related increase in oval cell hyperplasia (Table 1; bottom panel). There was also a dose-dependent increase in Ito cell and centrilobular hepatocyte hypertrophy following prenatal PFOA exposure. In several dosed groups, these incidences were 2- to 4-fold the incidence in the control group, and reached a significant increase above controls at 5.0 mg/kg BW PFOA. Chronic inflammation was common in CD-1 mice, and there was a dose-related increase in severity scores in PFOA-exposed livers; mean severity in the 2 highest dose groups was significantly higher than controls.
Histopathological Findings in the Liver of WT and PPARα-KO Mice
In the vehicle-treated 129/Sv WT mice, no tumors were found. A hemangiosarcoma developed in a PPARα-KO control mouse and was the only incidence of this tumor type in the 129/Sv strain (Table 2; top panel). The only tumor found in treated 129/Sv WT mice was a histiocytic sarcoma in the 0.1 mg/kg PFOA dose group. Hepatocellular adenomas developed in 5 PFOA-treated PPARα-KO mice, with 1 affected animal in each dose group and 2 animals in the highest dose group, leading to an overall incidence of 13.2% in treated animals (Table 2 and Figure 3). However, this occurrence did not reach statistical significance (p = .11). An Ito cell tumor developed in 1 PPARα-KO mouse treated with 0.3 mg/kg PFOA. Focal hepatocyte changes occurred in 3 PPARα-KO mice. Clear cell focus developed in 1 animal at 0.1 mg/kg PFOA and eosinophilic foci developed in 1 animal at each of 0 and 0.3 mg/kg PFOA. In 129/Sv WT mice, eosinophilic foci developed in 1 animal at each of 0.3 and 0.6 mg/kg PFOA. The lesion closest to reaching statistical significance was hepatocellular adenomas (p = .11), and the 2- to 3-fold increase in incidence over that seen in CD-1 likely failed to reach significance due to the smaller n in this strain (difficult to breed and raise litters).
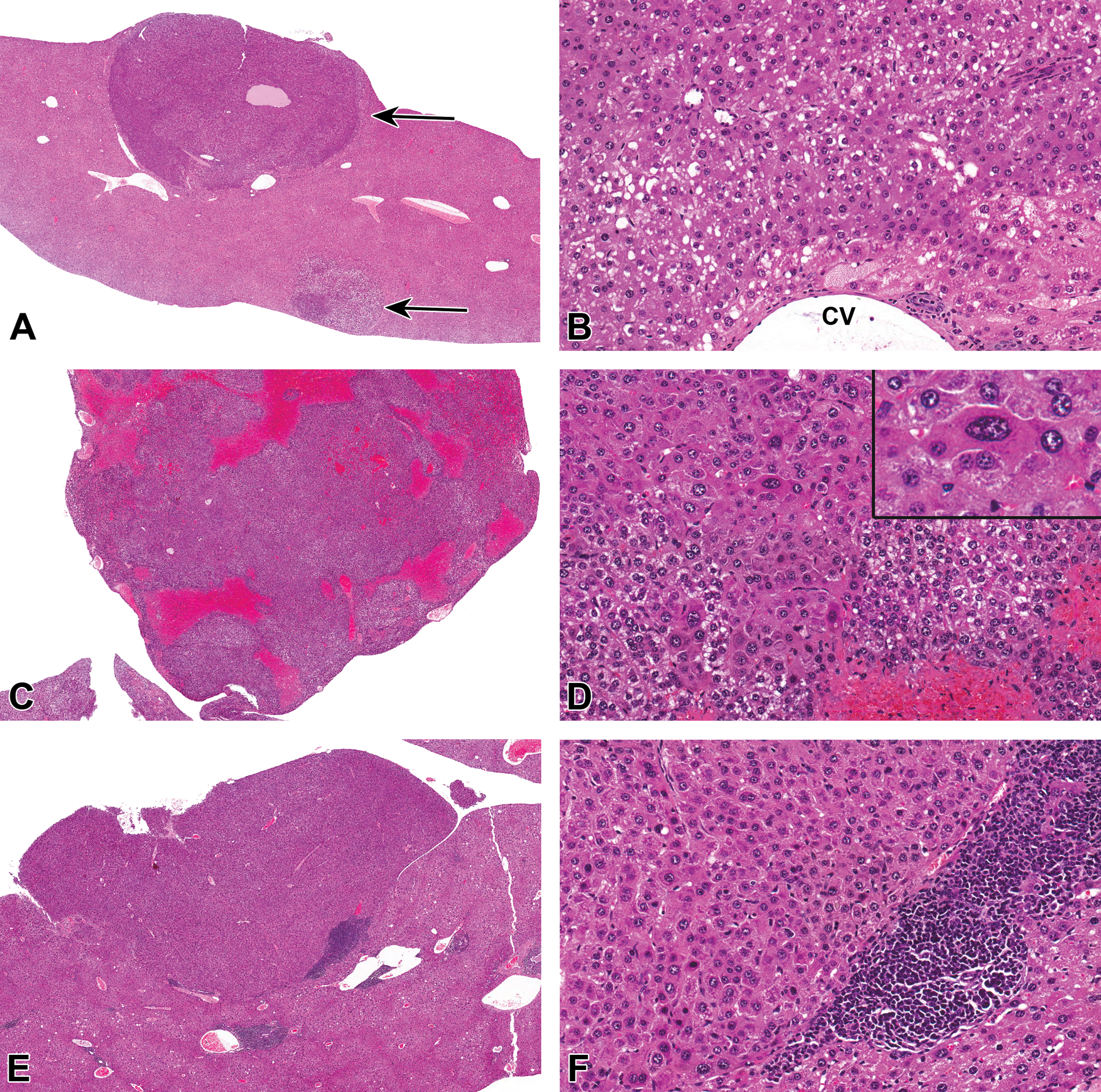
Hepatocellular adenomas in perfluorooctanoic acid (PFOA)-treated CD-1 and peroxisome proliferator–activated receptor-alpha-knockout (PPARα-KO) mice. (A–B) Liver, CD-1 mouse, 0.3 mg/kg PFOA. (A) Two, discrete, unencapsulated, and expansile adenomas are arising from the subcapsular hepatic parenchyma (arrows). 2×. Hematoxylin and eosin (H&E). (B) Adenomas are composed of compact lobules of neoplastic hepatocytes exhibiting increased cytoplasmic basophilia with hyperchromatic nuclei. Hepatocytes are often distended with variable amounts of discrete, clear, and intracytoplasmic vacuoles (lipid), CV = central vein. 20×. H&E. (C and D) Liver, PPARα-KO mouse, 3.0 mg/kg PFOA. (C) A well-demarcated and unencapsulated, polypoid mass is arising from the capsular surface of the liver. 2×. H&E. (D) Hepatocytes are often enlarged and contain 1 or more enlarged hyperchromatic nuclei (karyomegaly). 20×. H&E. Inset: higher magnification of neoplastic hepatocytes. 40×. H&E. (E and F) Liver, PPARα-KO mouse, 3.0 mg/kg PFOA. (E) A similar, discrete, hepatic nodule (adenoma) is expanding the subcapsular region and compressing the underlying hepatic parenchyma. 2×. H&E. (F) Neoplastic cells exhibit increased basophilia and hyperchromatic nuclei with loss of the normal radial architecture and are sometimes bordered by large numbers of inflammatory cells. 20×. H&E.
Neoplastic and nonneoplastic incidences (and severity scores) in livers after PFOA treatment (mg/kg) in 129/Sv mouse strain.
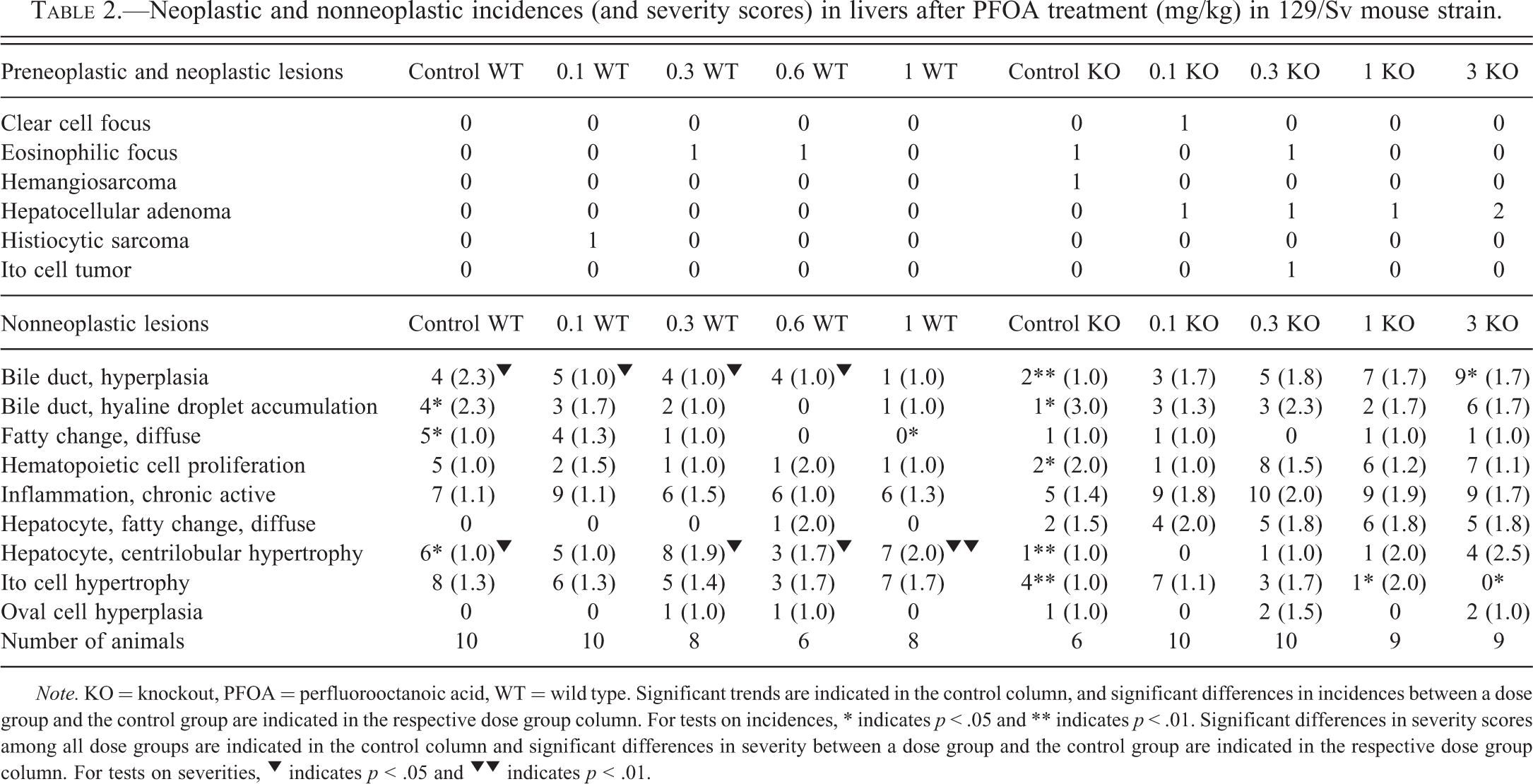
Note. KO = knockout, PFOA = perfluorooctanoic acid, WT = wild type. Significant trends are indicated in the control column, and significant differences in incidences between a dose group and the control group are indicated in the respective dose group column. For tests on incidences, * indicates p < .05 and ** indicates p < .01. Significant differences in severity scores among all dose groups are indicated in the control column and significant differences in severity between a dose group and the control group are indicated in the respective dose group column. For tests on severities, ▾ indicates p < .05 and ▾ ▾ indicates p < .01.
The nonneoplastic changes in the 129/Sv strain were also numerous. A significant PFOA dose-related increase was evident for incidences of both bile duct hyperplasia and bile duct inclusion bodies in PPARα-KO mice (Table 2, bottom panel). Conversely, the 129/Sv WT mice showed no increase in incidences of either bile duct hyperplasia (but a decreasing trend for severity with dose) or hyaline droplet accumulation (but a decreasing trend for incidence) following PFOA exposures. Incidences of Ito cell hypertrophy decreased with PFOA treatment in PPARα-KO mice (Figure 4). Hematopoietic cell proliferation significantly increased with PFOA dose in PPARα-KO mice but was not significantly related to dose in the 129/Sv WT mice. The incidence of centrilobular hepatocyte hypertrophy significantly increased with PFOA dose in the PPAR KOs, and while the incidence in 129/Sv WT mice did not significantly change with dose, severity increased significantly with PFOA dose in 129/Sv WT mice. A similar increase in mean severity was noted in PPARα-KO mice, but that effect did not reach significance. These latter findings are novel and inconsistent with previous reports of adult PFOA exposure in male PPARα-KO and WT mice that suggested minimal or absence of PFOA-induced hepatic microscopic abnormalities in animals null for mouse PPARα (Cheung et al. 2004; Nakamura et al. 2009).
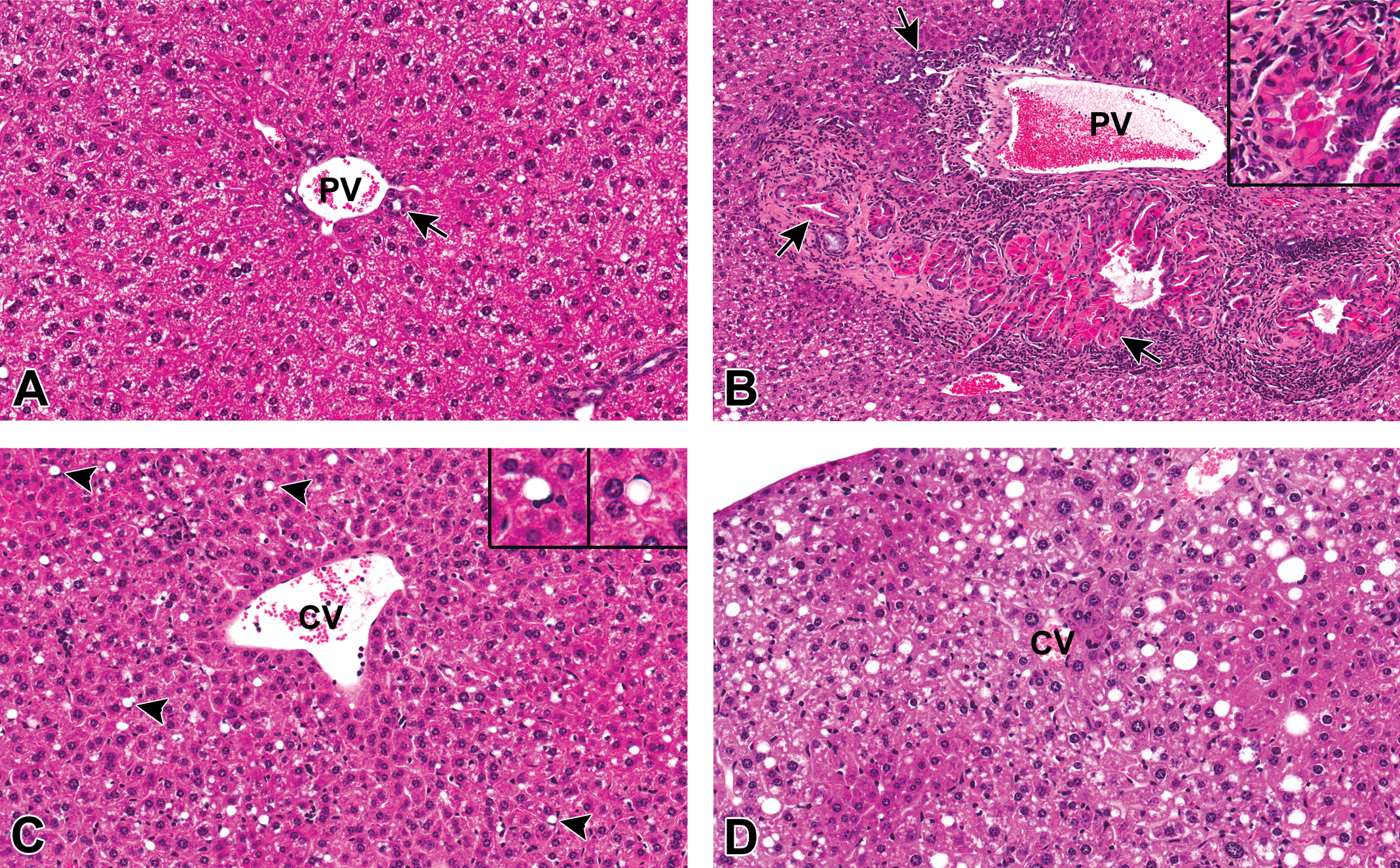
Nonneoplastic hepatic lesions in peroxisome proliferator–activated receptor-alpha-knockout (PPARα-KO) mice. (A) Liver, PPARα-KO mouse, 0 mg/kg perfluorooctanoate acid (PFOA). Control liver from an untreated PPARα-KO mouse. PV = portal vein, arrow = bile duct. 40×. Hematoxylin and eosin (H&E). (B) Liver, PPARα-KO, 3.0 mg/kg PFOA. There is marked bile duct proliferation relative to the control mouse A (bile duct hyperplasia) as well as marked accumulations of homogenous, intracytoplasmic, brightly eosinophilic, globular material within biliary epithelial cells (hyaline droplet accumulation). Note the accumulation of primarily lymphocytic inflammatory infiltrates bordering the portal area, as well as increased deposition of fibrous connective tissue. PV = portal vein, arrow = bile duct. 20×. H&E. Inset: higher magnification of intracytoplasmic hyaline droplet accumulation within biliary epithelial cells and bile ducts. 40×. H&E. (C) Liver, PPARα-KO mouse, 0.3 mg/kg PFOA. Numerous hypertrophied Ito (satellite) cells contain a single, discrete vacuole that peripheralizes the nucleus (arrowheads). CV = central vein. 40×. H&E. Insets: enlarged images of hypertrophied Ito cells. (D) Liver, PPARα-KO mouse, 3.0 mg/kg PFOA. Hepatocytes within the centrilobular and midzonal areas are slightly enlarged relative to the mice without hypertrophy in A and C. Hypertrophied hepatocytes are characterized by finely granular, eosinophilic cytoplasm with a “ground glass” appearance containing hyperchromatic and often enlarged (karyomegaly) nuclei (centrilobular to midzonal hepatocellular hypertrophy). Ito (satellite) cell hypertrophy and mixed fatty change are also present. CV = central vein. 40×. H&E.
Discussion
In this study, we describe several neoplastic and nonneoplastic signs of liver toxicity in 2 strains of 18-month-old mice following prenatal PFOA exposures. These lesions demonstrate that low-dose gestational PFOA exposure can impart long-term and persistent liver injuries. Importantly, our previous data predict that 28-day-old mice exposed to full gestational PFOA exposure at 0.3 mg PFOA/kg exhibited serum levels comparable with highly exposed humans (Emmett et al. 2006), and by 12 weeks of age the serum PFOA levels in those same mice had returned to background levels (Macon et al. 2011). There are still important differences between the mouse model and human exposure. The half-life of PFOA in the mouse is significantly shorter than that reported in humans. Therefore, mice require a higher in utero exposure to achieve similar serum concentrations as those reported in peripubertal children or adolescents in highly exposed regions of the United States (Emmett et al. 2006). While human exposure to PFOA is reportedly higher in infants compared with their mothers (8.0 µg/L in serum of 6-month-olds and 1.7 µg/L in their mothers; Fromme et al. 2010), it is not as high as the concentrations just after birth in gestationally exposed mice. Humans are also continuously exposed to PFOA at low levels throughout life.
Centrilobular hepatocyte hypertrophy significantly increased in incidence with PFOA dose in CD-1 and 129/Sv PPARα-KO mice, and increased in severity in the 129/Sv WT mice. Hepatocellular adenomas were significantly increased in CD-1 mice, although a dose–response relationship was not found. Hepatocellular adenomas were also evident in over 13% of PFOA-exposed PPARα-KO mice, demonstrating that these lesions are stimulated by prenatal PFOA exposure, a process that is seemingly independent of PPARα activation. Importantly, hepatocellular adenomas are not found in historical CD-1 control female mice (485 mice examined; Giknis and Clifford 2010). Additionally, in the only other long-term study of the 129/Sv mice available, with similar power as this study (n = 6–9/group), Peters, Cattley, and Gonzalez (1997) reported no hepatocellular adenomas in PPARα-KO and induction of those tumors in adult PPARα-WT mice fed Wyeth-14,643, a PPARα-inducer, after 11 months. These results indicate that Wyeth-14,643 induces hepatocellular adenomas via PPARα activation and since Wyeth-14,643 wasn’t able to induce tumors in PPARα-KO animals and PFOA-exposed PPARα-KO animals developed tumors in this study, this may indicate that low-level exposures to PFOA activate pathways other than PPARα in the mouse liver. Further, in this study, bile duct hyperplasia and hematopoietic cell proliferation were liver lesions that were only significantly increased with increasing dose of PFOA in the PPARα-KO mice. These long-term liver injuries in PPARα-KO mice suggest that potentially human relevant PPARα-independent pathway/pathways of hepatic injury and cancer exist following prenatal PFOA exposures.
Our findings here support the majority opinion of the U.S. EPA Scientific Advisory Board (2006) during their review of EPA’s Draft Risk Assessment of Potential Human Health Effects Associated with PFOA and Its Salts, which concluded that “based on current evidence, that it is possible that PPARα agonism may not be the sole MOA for PFOA, that not all steps in the pathway of PPARα activation-induced liver tumors have been demonstrated, that other hepato-proliferative lesions require clarification, and that extrapolation of this MOA across the age range in humans is not supported.” (quote begins on pg 2 of SAB document).
While there was not a significant increase in incidences of any 1 type of liver tumor in PPARα-KO mice, there was a higher incidence of tumors overall in the PFOA-exposed PPARα-KO mice compared with the PFOA-exposed 129/Sv WT mice, which may be due to some protective properties of PPARα (Ito et al. 2007). The 129/Sv WT mice had a single incidence of histiocytic sarcoma while the PPARα-KO mice had incidences of hemangiosarcoma (1 animal), hepatocellular adenoma (5 animals), and Ito cell tumor (1 animal). The original design of these studies was not intended for tumor burden or liver toxicity evaluations, but livers were collected and evaluated at necropsy and became of interest when liver tumors were discovered in the PPARα-KO mice. Additional experiments using more animals are needed to validate our observations that PPARα-KO mice are susceptible to PFOA-induced liver injuries and tumor formation through PPARα-independent and potentially human relevant pathways.
CD-1 mice and 129/Sv WT mice exposed to PFOA differed in that the CD-1 mice had increased incidences of hemangiosarcoma, Ito cell hypertrophy, and oval cell hyperplasia and an increased severity of chronic active inflammation. The PPARα-KO mice had significantly increased incidences of bile duct hyperplasia and hematopoietic cell proliferation compared with the 129/Sv WT mice. Within the 129/Sv strain, PFOA results in an increase in bile duct hyperplasia and hematopoietic cell proliferation in the PPARα-KO mice. Consistent in both the 129/Sv stains and CD-1 mice was that PFOA exposure increased centrilobular hepatocyte hypertrophy. Since all mice exposed to PFOA developed hepatocyte hypertrophy, regardless of the presence of PPARα, it is reasonable to conclude that a PPARα-independent mechanism may be responsible for this change in hepatocytes. PFOA exposure resulted in hepatocellular adenoma in about 5% of CD-1 mice, and those tumors were also evident in nearly each dose group, as well as 13% of PFOA-exposed PPARα-KO mice. Perhaps in a larger animal study, hepatocellular adenoma may have reached significance in the PPARα-KO mice, as well.
PFOA has been linked to hepatic injury and the formation of the tumor triad of liver, Leydig cell, and pancreatic acinar-cell tumors in adult exposed male rats (Biegel, Hurtt, and Frame 2001). The mechanism for formation of those tumors is thought to be through PPARα-dependent pathways, yet the direct mechanism is still incompletely characterized (Klaunig, Hocevar, and Kamendulis 2012). Of the few studies that have evaluated low gestational exposure to PFOA (Lau et al. 2006; Wolf et al. 2007; Macon et al. 2011), this is the first to document persistent or long-term liver effects of this chemical in CD-1 or 129/Sv PPARα-KO mice. One study has reported that 3 mg/kg BW gestational PFOA exposure in murine PPARα-KO mice and human- or murine-PPARα-expressing 129/Sv mice caused hepatocellular hypertrophy only in the murine-PPARα-expressing mice at 20 days of age (Albrecht et al. 2013). The 129/Sv mouse strain used by those authors (obtained from the National Institutes of Health [NIH]) is reportedly less sensitive to PFOA than 129/Sv WT mice in this study (obtained from the Jackson Laboratories), as their studies failed to recapitulate postnatal lethality at PFOA exposures of 0.6 or 1.0 mg/kg as was previously shown in these 129/Sv WT mice (Abbott et al. 2007), but caused a modest increase at 3 mg/kg (Albrecht et al. 2013). The authors further justified these differences in the background strain sensitivity by observing the maternal serum PFOA observed in mice treated with 3 mg PFOA/kg during gestation on PND 20 in the Albrecht et al. (2013) study, which ranged from 2,066 to 6,812 ng/ml, compared with the maternal serum PFOA observed in the Abbott et al. (2007) study mice at PND 22 that had been exposed to one-tenth that dose (0.3 mg PFOA/kg) during gestation and generated a similar serum concentration (means at weaning: 2,840 ng/ml, dams; 2,150 ng/ml, pups). Another study using 3 strains of 8-week-old adult male 129/Sv mice (WT, PPARα-KO, and mice with hPPARα) orally gavaged with 1.0 to 5.0 mg/kg BW PFOA for 6 weeks reported liver injuries in all 3 strains, albeit they all looked different compared with each other (Nakagawa et al. 2012). These high-dose effects are not surprising, given that developmental PFOA exposures at 1 mg/kg in our previous studies had already been shown to induce transient hepatocellular hypertrophy in CD-1 mice (Macon et al. 2011).
Our findings suggest that 129/Sv PPARα-KO mice are susceptible to PFOA-induced liver injury at relatively low exposures, and these effects can be seen at 18 months in most dose groups, leading us to believe that the early lesions are permanent and potentially progress over time. The injuries to the bile ducts and hepatocytes in PFOA-exposed mice may deteriorate further with age to manifest long after the chemical has been eliminated from the animals by PND 84 (Macon et al. 2011). This is further addressed in an accompanying article in which PFOA-induced liver injury is manifested in CD-1 mice by PND21 (See Quist et al., this issue). PFOA liver injury in 129/Sv PPARα-KO mice (inflammatory cell infiltration, micro- and macrovesicular steatosis, and hydropic degeneration) may be representative of disease pathways that are relevant to humans. While PPARα is present in human livers, it is detected at one-tenth of the levels found in mice, and human PPARα does not appear to activate the cellular proliferation pathways as has been reported in mouse livers (Klaunig, Hocevar, and Kamendulis 2012). An hPPARα mouse model would still express PPARα at levels many times greater than that found in human liver. PFOA is easily excreted into the bile duct in WT mice but is thought to accumulate in PPARα-KO mice, posing a potential risk for mitochondrial dysfunction (Nakagawa et al. 2012). This accumulation in PPARα-KO mice may also be a reason for the significantly increased bile duct hyperplasia and inclusion bodies noted in PFOA-exposed animals in this study, a finding inconsistent with that in the WT 129/Sv in these studies.
Other peroxisome proliferating chemicals have been shown to cause liver injury in PPARα-KO mice. Male 129/Sv mice (PPARα-KO and WT of the NIH origin) were fed di(2-ethylhexyl)phthalate (DEHP) in their diet from 3 weeks until 22 months. On the 23rd month, they were assessed for liver toxicity. Surprisingly, and similar to what was found for PFOA in our study, the incidence of liver tumors was higher in PPARα-KO mice exposed to DEHP than in similarly exposed WT mice (Ito et al. 2007). One MOA suggested by those authors was an increase in oxidative stress and the lack of the protective properties of PPARα, resulting in the PPARα-KO mice being more susceptible to tumor formation. We may have also seen this phenomenon in the PPARα-KO mice exposed to PFOA in this study. As previously stated, these PFOA-dosed PPARα-KO animals exhibited more tumors than their 129/Sv WT PFOA-dosed counterparts.
Another possible mechanism for PFOA-induced hepatic toxicity is mitochondrial disruption. One study (Berthiaume and Wallace 2002) used adult male Sprague-Dawley rats, treated with several peroxisome proliferators, including PFOA, at a very high dose (100 mg/kg; orders of magnitude higher than body burdens in our studies) by single intraperitoneal injection to determine liver effects. On the third day after injection, livers were assessed for peroxisome proliferation and mitochondrial disruption. PFOA and perfluorooctanesulfonate (PFOS) were both shown to significantly increase peroxisomes compared with DMSO-treated animals. However, only PFOA was shown to induce mitochondrial biogenesis. In their opinion, mitochondria disruption was not associated with the biological response to peroxisome proliferators.
Liver injuries in 2 strains of mice, significantly increased hepatocellular adenomas in CD-1 mice and potentially tumorigenesis in PPARα-KO mice, suggest that PFOA can cause liver toxicity through PPARα-independent pathways following prenatal exposures that dissipate by about 12 weeks of age. Careful evaluation of the timing and doses of PFOA exposure, in addition to the background strain sensitivity, should be part of the health evaluation of PFOA going forward. Further evaluation of the health risks of PFOA is needed to determine the human relevance of PFOA’s non-PPARα pathways that result in liver toxicity. Novel PFOA modes of action are suggested in the accompanying article (Quist et al., 2014) which demonstrates that PFOA exposure is associated with hepatic mitochondrial alteration in CD-1 mice.
Footnotes
Acknowledgment
The authors would like to thank Ms. Beth Mahler in the Cellular and Molecular Pathology Branch, NIEHS, for her assistance in preparing the figures for this article and Dr. Barbara Abbott, U.S. EPA, for supplying the pregnant 129/Sv mice for these studies.
Author Contributions
Suzanne E. Fenton contributed to conception or design; Erin M. Quist, Adam J. Filgo, Amy E. Brix, Grace E. Kissling, Mark J. Hoenerhoff, and Suzanne E. Fenton contributed to data acquisition, analysis, or interpretation; Erin M. Quist, Adam J. Filgo, and Suzanne E. Fenton drafted the manuscript; and Erin M. Quist, Adam J. Filgo, Grace E. Kissling, and Suzanne E. Fenton critically revised the manuscript. All authors gave final approval and agreed to be accountable for all aspects of work in ensuring that questions relating to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Abbreviations
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.