
Abstract
Canagliflozin, a sodium glucose co-transporter 2 (SGLT2) inhibitor, has been developed for the treatment of adults with type 2 diabetes mellitus (T2DM). During the phase 3 program, treatment-related pheochromocytomas, renal tubular tumors, and testicular Leydig cell tumors were reported in the 2-year rat toxicology study. Treatment-related tumors were not seen in the 2-year mouse study. A cross-functional, mechanism-based approach was undertaken to determine whether the mechanisms responsible for tumorigenesis in the rat were of relevance to humans. Based on findings from nonclinical and clinical studies, the treatment-related tumors observed in rats were not deemed to be of clinical relevance. Here, we describe the scientific and regulatory journey from learning of the 2-year rat study findings to the approval of canagliflozin for the treatment of T2DM.
Keywords
Introduction
Phlorizin, an agent originally extracted from the bark of apple trees, was found in preclinical models of diabetes to reduce plasma glucose levels in an insulin-independent fashion by increasing urinary glucose excretion (UGE; Rossetti et al. 1987; Ehrenkranz et al. 2005). Subsequently, it was discovered that phlorizin increases UGE by inhibiting sodium glucose co-transporters 1 (SGLT1) and 2 (SGLT2), members of the solute carrier 5 gene family that are responsible for resorbing glucose filtered by the renal glomerulus (Ehrenkranz et al. 2005; Yao et al. 2011).
SGLT2 is a high-capacity, low-affinity glucose transporter almost exclusively expressed in the proximal renal tubule where, under euglycemic conditions, it resorbs the majority of glucose filtered at the glomerulus (Vallon et al. 2011; Abdul-Ghani, Norton, and Defronzo 2011; Defronzo, Davidson, and Del Prato 2012). Mutations reducing SGLT2 transport activity are associated with increased UGE, which is considered a benign condition (familial renal glucosuria; Santer and Calado 2010; Bailey 2011).
SGLT1 is predominantly expressed in the gut and is the transporter responsible for glucose and galactose absorption. SGLT1 is also expressed, to a lesser extent, in other tissues including the proximal renal tubule, where it functions as a high-affinity, low-capacity glucose transporter responsible for a minority of glucose resorption in nondiabetic persons (Gerich 2010; Abdul-Ghani, Norton, and Defronzo 2011). Individuals with loss-of-function SGLT1 mutations present soon after birth with severe diarrhea due to glucose and galactose malabsorption, which can be corrected by eliminating glucose from the diet. Individuals with loss-of-function SGLT1 mutations also have modest increases in UGE. Although limited numbers of these individuals have been characterized, diarrhea appears to be the only clinically relevant sequelae associated with loss-of-function SGLT1 mutations (Gorboulev et al. 2012).
SGLT2 Inhibitors as a Therapeutic Option to Treat Type 2 Diabetes Mellitus (T2DM): Development of Canagliflozin
Based on the benign phenotype associated with familial renal glucosuria and the ability of SGLT2 inhibition to reduce hyperglycemia in preclinical models of diabetes (Santer and Calado 2010; Abdul-Ghani, Norton, and Defronzo 2011), Tanabe Seiyaku Co., Ltd. (now Mitsubishi Tanabe Pharma Corporation) and Johnson & Johnson Research & Development, LLC (now Janssen Research & Development, LLC) collaborated to discover and develop SGLT2 inhibitors for the treatment of T2DM. As a result of this collaboration, canagliflozin was discovered and advanced into clinical development.
Canagliflozin, chemically known as (1S)-1,5-anhydro-1-[3-[[5-(4-fluorophenyl)-2-thienyl]methyl]-4-methylphenyl]-
In rat toxicity studies, canagliflozin was associated with increases in urinary volume due to glucosuria (i.e., osmotic diuresis) and increased food intake; both are considered to be related to the mode of action (U.S. Food and Drug Administration 2014). Marked increases in urinary calcium excretion (>10-fold), without significant changes in total serum calcium levels, were seen in rats treated with canagliflozin and were associated with reductions in parathyroid hormone (PTH) and 1,25-dihydroxyvitamin D levels. While measurement of ionized calcium, a more sensitive parameter for the assessment of calcium homeostasis, might have identified alterations with canagliflozin, this parameter was not assessed in these studies. It should be noted that modest, statistically significant increases in total serum calcium and ionized calcium have been reported in rats treated with dapagliflozin, another SGLT2 inhibitor (Tirmenstein et al. 2013). The increases in urinary calcium excretion were seen at higher canagliflozin doses than those required to maximally increase UGE (De Jonghe et al. 2014). Glucose malabsorption increases calcium absorption by stimulating colonic fermentation and reducing intestinal pH, resulting in a vitamin D–independent increase in calcium absorption (Tirmenstein et al. 2013). Histologically, bone hyperostosis was observed. Notably, hyperostosis was not seen in mice or dogs treated with canagliflozin (De Jonghe et al. 2014). Increased urinary calcium excretion and hyperostosis have also been described in rats that were orally administered poorly absorbable SGLT1 inhibitors and other SGLT2 inhibitors at doses in excess of the exposures providing maximal SGLT2 inhibition (Kissner et al. 2010; Tirmenstein et al. 2013). In a set of elegant experiments, scientists at Bristol-Myers Squibb prevented hyperostosis in rats treated with an SGLT2 inhibitor with low-potency SGLT1 inhibitory activity by feeding them a glucose-free diet to avoid carbohydrate malabsorption due to SGLT1 inhibition. Since SGLT1, which is expressed on the luminal surface of the intestinal enterocyte, is responsible for glucose absorption (Bailey 2011), it was hypothesized that SGLT1 inhibition by these compounds may lead to glucose malabsorption and, in turn, hyperostosis due to increased calcium absorption. These studies demonstrated the role of intestinal SGLT1 inhibition by high intraluminal intestinal concentrations of SGLT2 inhibitors with low-potency SGLT1 inhibitory activity in mediating hyperostosis, and increased intestinal calcium absorption (Tirmenstein et al. 2013).
In phase 1 studies in patients with T2DM, canagliflozin increased UGE and lowered plasma glucose levels, but was not associated with symptoms of carbohydrate malabsorption (Rothenberg et al. 2010). At the highest clinical dose used in phase 3 studies (300 mg once daily), canagliflozin did not increase hydrogen breath content following a standardized meal (a marker of carbohydrate malabsorption; Schwartz et al. 2010) and did not alter urinary calcium excretion or serum PTH and 1,25-dihydroxyvitamin D levels (Rosenstock et al. 2012; Bode et al. 2013). Thus, in contrast to studies in rats, canagliflozin treatment was not associated with carbohydrate malabsorption or its sequelae in patients with T2DM.
In a 12-week phase 2 study in patients with T2DM, canagliflozin increased UGE and lowered HbA1c (Rosenstock et al. 2012). Canagliflozin was generally well tolerated; an increased incidence of genital mycotic infections was noted in women treated with canagliflozin (Nyirjesy et al. 2012). Based on the phase 2 study findings, canagliflozin at doses of 100 and 300 mg given once daily was advanced into phase 3 development. The phase 3 development program was composed of 9 trials conducted globally at approximately 1,500 sites in over 10,000 patients (Janssen Research & Development, LLC 2013).
Genotoxicity Assessment and Tumor Findings in 2-Year Canagliflozin Rodent Studies
Based on results from a battery of in vitro and in vivo studies, canagliflozin was not found to be genotoxic. Tumors or preneoplastic lesions were not observed in chronic toxicology studies, including a 6-month study in rats and a 12-month study in dogs (De Jonghe et al. 2014). Initial results from the 2-year rodent carcinogenicity studies became available at a time when all clinical trials of canagliflozin in the phase 3 program were ongoing and over 90% of patients had been enrolled. No treatment-related tumors were found in Swiss Albino mice treated with canagliflozin 10, 30, and 100 mg/kg/day, but in Sprague-Dawley rats administered the same doses, tumors were detected in the kidneys (renal tubular tumors [RTTs]), adrenal glands (pheochromocytomas), and testes (Leydig cell tumors [LCTs]; Table 1; De Jonghe et al. 2014). Basophilic RTTs (adenomas and carcinomas) were noted in both sexes; these were localized in the cortex and outer stripe of the outer medulla (OSOM). Two RTTs in the mid-dose group were of the amphophilic-vacuolar (AV) phenotype characteristic of spontaneously occurring tumors in rats (Hard et al. 2008; Crabbs et al. 2013) and were deemed by an independent expert panel masked to treatment allocation to be incidental and not treatment related. Pheochromocytomas (benign and malignant) were observed in both sexes accompanied by medullary hyperplasia. Relative to the highest canagliflozin dose being studied in the phase 3 program, the safety margin based on the no observed effect level (30 mg/kg/day) for basophilic RTTs and pheochromocytomas was ≥4.5×. LCTs were associated with interstitial cell hyperplasia and atrophy of the accessory sex glands (seminal vesicles, prostate, and coagulating gland). As LCTs were detected at the lowest canagliflozin dose studied, a safety margin could not be calculated for these tumors (De Jonghe et al. 2014).
Treatment-related tumors in the 2-year rat study.

Note. Bold indicates a treatment-related effect based on comparison with incidence rate in vehicle and historical controls. AV = amphophilic-vacuolar (a spontaneous tumor type); HCD = historical control data; NA = not available.
aAnimal no. 333 had an adenoma and a carcinoma.
bAnimal no. 740 had an adenoma and a carcinoma.
Non–tumor-related findings from the rat carcinogenicity study included a dose-related increase in hyperostosis of the sternum and/or stifle (distal femur and proximal tibia) apparent in both sexes with canagliflozin 30 and 100 mg/kg/day. Atrophy of male accessory sex glands was found in rats starting with 30 mg/kg/day of canagliflozin. Histologic findings in the kidney with canagliflozin treatment included increased mineralization, a marginal increase in simple tubule hyperplasia (only in high-dose males), tubular and pelvic dilation, and an increase in chronic progressive nephropathy (CPN; predominantly grades 2 and 3; De Jonghe et al. 2014). Although CPN is considered to be involved in the pathogenesis of RTT, the severity of CPN associated with RTT formation is grade 4 or 5 (Hard and Khan 2004).
A cross-functional, preclinical, and clinical team was established to assess the clinical relevance of these findings and to determine whether the phase 3 program could continue. Based on review of extant literature, associations were found between RTTs, pheochromocytomas, and LCTs and carbohydrate malabsorption in rats; these associations were caused by a variety of foodstuffs and agents approved for human use. None of the agents causing carbohydrate malabsorption and associated with RTT formation in rats were known to cause the corresponding tumors in humans. For example, acarbose, an α-glucosidase inhibitor approved for use to treat T2DM, delays glucose absorption in the intestinal lumen and causes carbohydrate malabsorption in rats. Evidence of carbohydrate malabsorption in rats treated with acarbose includes cecal enlargement (Creutzfeldt et al. 1985; Lembcke et al. 1987) and increases in cecal carbohydrate content (Lembcke et al. 1987). In a rat carcinogenicity study, acarbose, at doses similar to those recommended for clinical use, induced RTTs in Sprague-Dawley rats; acarbose did not cause RTTs in Wistar rats, mice, or hamsters (Schulter 1988; PRECOSE 2011). Acarbose has not been associated with an increase of RTTs in humans (Roe 1989; Hollander 1992).
In rats, treatment-related pheochromocytomas are caused by a variety of nongenotoxic agents (Tischler and Delellis 1988) that may act through a variety of pharmacodynamic mechanisms including dysregulation of calcium homeostasis, hypoxia, neurogenic or hormonal signals (growth hormone and prolactin), and stress (Tischler et al. 1996; Greim et al. 2009). Treatments causing carbohydrate malabsorption in rats—such as lactose, sorbitol, xylitol, mannitol, and lactitol—are associated with abnormalities in calcium handling and in treatment-related adrenal medullary hyperplasia and pheochromocytomas (Roe and Bar 1985; Baer 1988; Sinkeldam et al. 1992; de Groot et al. 1995; Tischler et al. 1996). Unlike in rats, lactose and xylitol do not induce pheochromocytomas in mice (Lynch et al. 1996). Agents causing carbohydrate malabsorption in humans have not been linked to an increased risk of pheochromocytomas (Baer 1988).
In rats, a diverse set of agents cause LCT formation (Clegg et al. 1997). However, agents causing LCTs in rats have not been linked to LCTs in humans; this discrepancy is likely related to well-known physiological differences (Cook et al. 1999; Cohen and Arnold 2011), including differences in the number of Leydig cell luteinizing hormone (LH) and luteinizing hormone-releasing hormone (LHRH) receptors (Prentice and Meikle 1995). The number of reported LH receptors per Leydig cell is 14 times greater in the rat compared with humans (Huhtaniemi 1983). Furthermore, the rat appears to be unique in possessing the LHRH receptor as it is not present in humans (Clayton and Huhtaniemi 1982), monkeys (Mann et al. 1989), or mice (Wang et al. 1983). Rats, but not humans, lack sex hormone–binding globulin (SHBG), resulting in enhanced susceptibility to alterations in hormone levels (Cook et al. 1999). Thus, treatment-related LCTs in rats have a poor predictive value for human risk. An increase in the incidence of Leydig cell hyperplasia and LCT is observed when animals are fed poorly digestible sugars (such as lactose) and sugar alcohols (such as lactitol; Woutersen 1986; Sinkeldam et al. 1992; de Groot et al. 1995). This phenomenon is not seen in mice (Bär 1992). Acarbose also induces LCT formation in Sprague-Dawley rats. There does not appear to be an association between lactose consumption and increased incidence of LCT in humans (Roe 1989; Ursin et al. 1990; Bär 1992). Although the mechanism for carbohydrate malabsorption-induced LCT formation in rats is not known, alteration of the hypothalamic–pituitary–gonadal axis resulting in increased secretion of LH, a Leydig cell mitogen, is an established mechanism shared by a number of nongenotoxic agents that induce LCTs in rats but not in humans (Clegg et al. 1997).
The relationship between carbohydrate malabsorption and formation of RTTs, pheochromocytomas, and LCTs suggested that these tumors could be due to glucose malabsorption mediated by canagliflozin-induced intestinal SGLT1 inhibition. The discrepancy between rats and humans in their susceptibility to develop these tumors in response to carbohydrate malabsorption supported the contention that carbohydrate-induced tumor formation in rats was not relevant to human safety. Given that canagliflozin caused carbohydrate malabsorption in rats but not in humans, it was deemed that the rat findings were not clinically relevant and a mechanistic program to further bolster this hypothesis was initiated.
Regulatory bodies, ethics review boards (ERBs), and investigative sites were promptly made aware of the findings from the 2-year rat study and the proposed mechanistic basis for why these findings were not deemed to be relevant to human safety. Patients were reconsented using a revised informed consent document (ICD) that described the tumor findings from the rat study. Two central and 2 local ERBs did not approve the revised ICD, and patients from sites under the control of these ERBs were withdrawn from the studies. Approximately 150 of the 10,301 patients (1.5%) either withdrew from the phase 3 program due to lack of ERB approval for the revised ICD or elected to withdraw consent due to the tumor findings. Given the limited loss of patients from the studies, sufficient statistical power was maintained, and the trials were not compromised due to the excessive loss of patients.
Mechanistic Program to Assess Human Relevance of Treatment-related Rat Tumors
Carbohydrate Malabsorption in Rats Treated with Canagliflozin
While it was inferred, based on findings with another SGLT2 inhibitor with low-potency SGLT1 inhibitory activity, that canagliflozin caused glucose malabsorption in rats, studies were undertaken to directly assess this assumption (Mamidi et al. 2014). Studies were conducted under treatment conditions associated with adrenal and renal tumor formation in the 2-year rat study (i.e., Sprague-Dawley rats administered canagliflozin 100 mg/kg/day). Glucose content and pH of the distal gastrointestinal tract were examined as indices of glucose malabsorption. Absorption of orally administered 3-O-methyl glucose (3-OMG), a nonmetabolizable sugar whose absorption from the gastrointestinal tract is mediated by SGLT1 (Moriya et al. 2009), was used to assess whether the degree of intestinal SGLT1 inhibition caused by canagliflozin led to glucose malabsorption. 3-OMG was administered to fasted rats 1 hr postdose at the end of the 2-week canagliflozin dosing period, and 3-OMG plasma levels were measured at various times. In addition, approximately 24 hr after the last dose, pH in the colon was measured, and glucose levels in the duodenum, jejunum/ileum, and cecum were determined by physically clamping the sections, flushing the contents from the lumen, and then analyzing each sample for glucose concentrations. Carbohydrate malabsorption leading to an increase in the distal intestinal tract of sugars results in bacterial fermentation and reduces intestinal pH (Mamidi et al. 2014). A significant reduction in 3-OMG plasma levels was seen in rats treated with canagliflozin (Figure 1). An increase in mean ± standard error (SE) cecal glucose content was seen after a 2-week treatment with canagliflozin 100 mg/kg/day relative to vehicle control (15.5 mg ± 2.3 vs. 9.1 mg ± 1.3; p < .05). A decrease in jejunal/ileal pH with canagliflozin treatment was observed (5.93 ± 0.10 vs. 6.5 ± 0.05, mean ± SE, in canagliflozin- and vehicle-treated rats, respectively; p < .01; Mamidi et al. 2014). Results from these studies confirm that in rats, under the conditions of canagliflozin treatment, glucose malabsorption occurs presumably through inhibition of intestinal SGLT1.
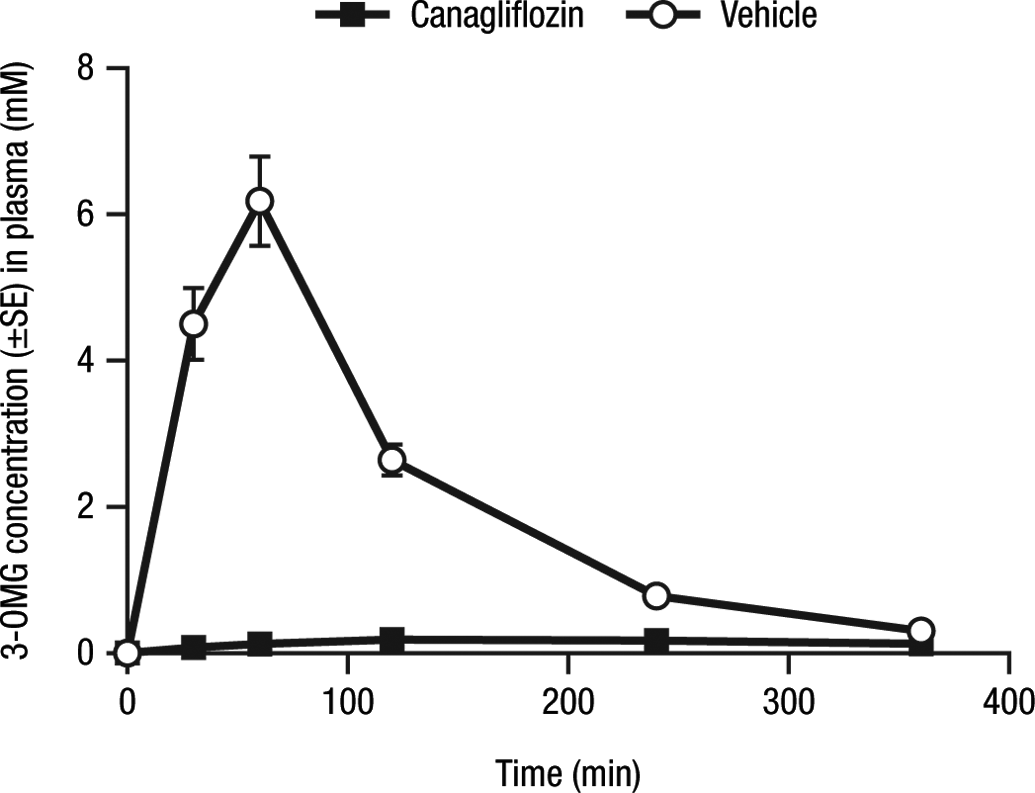
Effects of canagliflozin on 3-OMG absorption in rats. 3-OMG = 3-O-methyl-glucose; SE = standard error. Reprinted from Chemico-Biological Interactions; Vol. 221; Mamidi et al. (2014), with permission from Elsevier.
Effects of a Glucose-free Diet on Nontumorigenic Effects Elicited by Canagliflozin in Rats
Fructose transport, which is mediated by GLUT5, is not inhibited by canagliflozin (Barone et al. 2009). By substituting fructose for glucose, glucose malabsorption due to canagliflozin-induced SGLT1 inhibition can be prevented, thus allowing the effects of canagliflozin to be assessed without confounding by the effects of glucose malabsorption. An isocaloric diet with 40% fructose substituted for glucose as the primary carbohydrate source or the standard glucose-containing diet, along with a daily oral gavage of canagliflozin at doses of 0 and 100 mg/kg, were fed to male Sprague-Dawley rats (30/group) for 6 months with interim necropsies at 1 and 3 months (10/group; Mamidi et al. 2014). In rats fed a standard diet and treated with canagliflozin, urinary calcium excretion was substantially increased (∼9- to 20-fold), while in canagliflozin-treated rats fed the glucose-free diet, only a modest increase was seen (∼2- to 3-fold). Decreases in PTH and 1,25-dihydroxyvitamin D caused by canagliflozin were largely prevented by substituting the standard diet with the glucose-free diet. Rats dosed with canagliflozin and fed the standard diet exhibited distention and soft contents of the cecum and/or colon at necropsy, consistent with signs of carbohydrate malabsorption and hyperostosis; these findings were not observed in rats treated with canagliflozin and fed the glucose-free diet. These data demonstrate the ability of a glucose-free diet to largely prevent canagliflozin-induced carbohydrate malabsorption and, importantly, to abrogate the sequelae of glucose malabsorption (i.e., increased urinary calcium excretion, alterations in PTH and 1,25-dihydroxyvitamin D, and hyperostosis).
Effects of a Glucose-free Diet on Canagliflozin-induced Adrenal Medullary and Renal Tubule Cell Proliferation and Renal Tubule Injury
In the earlier study, the effects of carbohydrate malabsorption in rats induced by canagliflozin on adrenal medullary and renal tubule cell proliferation, a necessary step in the formation of pheochromocytomas and RTTs, respectively, were assessed (Mamidi et al. 2014). To assess cell proliferation, bromodeoxyuridine (BrdU) was administered continuously for 7 consecutive days via a subcutaneously implanted osmotic minipump. Paraffin sections of kidneys and adrenal glands were labeled immunohistochemically for BrdU. In canagliflozin-treated rats, BrdU labeling in the kidney cortex and OSOM was increased at 1 month, with further increases at 3 and 6 months, in rats fed the standard diet compared with rats fed the glucose-free diet. Canagliflozin increased cell proliferation in the adrenal medulla at 1, 3, and 6 months in rats fed the standard diet but not in rats fed the glucose-free diet (Table 2; Mamidi et al. 2014).
Effects of dietary glucose on adrenal medullary and renal cell proliferation in rats treated with canagliflozin.
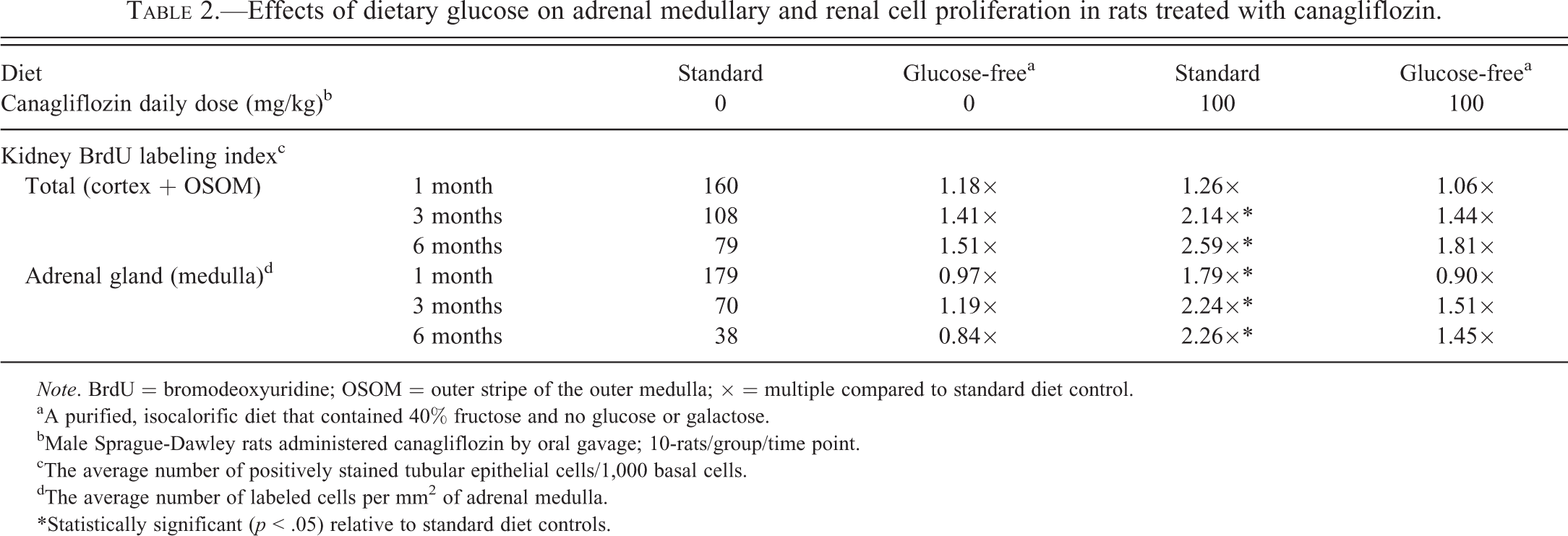
Note. BrdU = bromodeoxyuridine; OSOM = outer stripe of the outer medulla; × = multiple compared to standard diet control.
aA purified, isocalorific diet that contained 40% fructose and no glucose or galactose.
bMale Sprague-Dawley rats administered canagliflozin by oral gavage; 10-rats/group/time point.
cThe average number of positively stained tubular epithelial cells/1,000 basal cells.
dThe average number of labeled cells per mm2 of adrenal medulla.
*Statistically significant (p < .05) relative to standard diet controls.
One common mechanism by which nongenotoxic agents cause tumors in rodents is by inducing cellular injury, which leads to a regenerative, proliferative response (Cohen 1998; Hard, Boorman, and Wolf 2000; Lock and Hard 2004; Doi et al. 2006). Since proximal tubule kidney injury molecule-1 (KIM-1) expression is increased in rats following kidney injury, expression of this marker was examined (Mamidi et al. 2014). Canagliflozin treatment increased KIM-1 staining in the renal cortex and OSOM in rats fed the standard diet, but not in rats fed the glucose-free diet (Table 3; Mamidi et al. 2014). Double labeling with BrdU and KIM-1 demonstrated that increased proliferation and proximal tubule cell injury occurred in the same anatomic location (Figure 2).
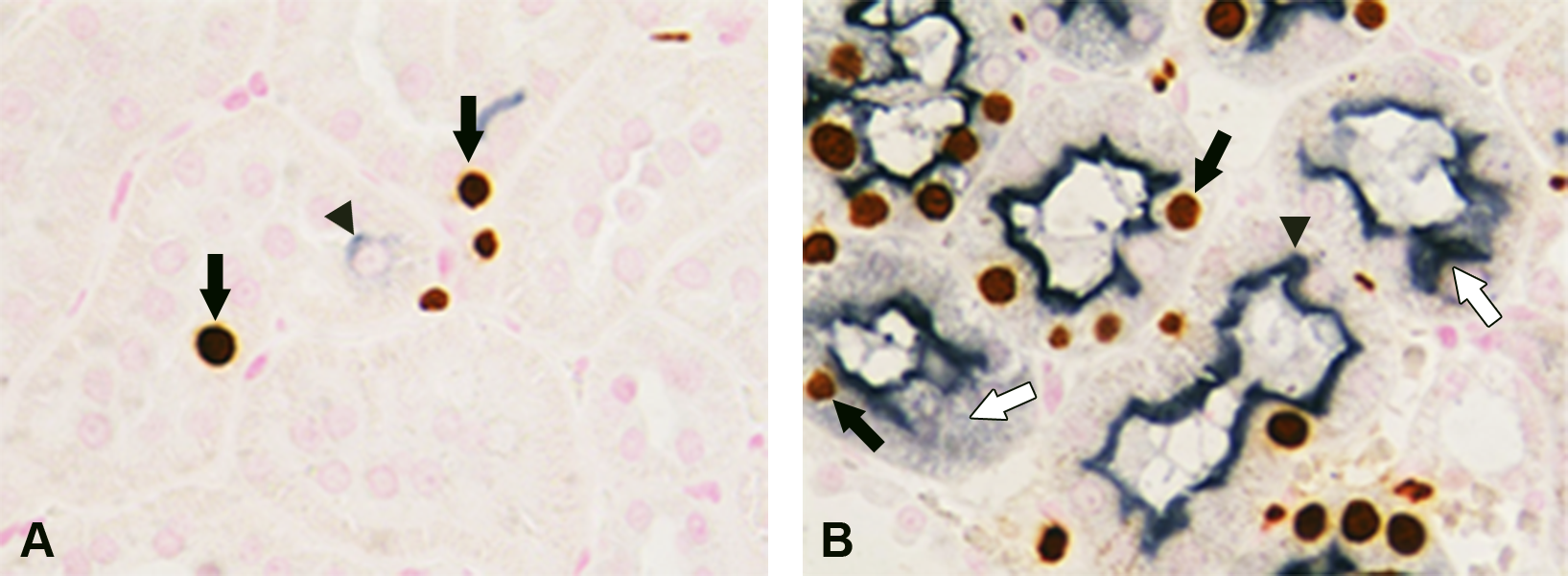
Colocalization of renal cell proliferation and injury in rats treated with canagliflozin. (A) Infrequent bromodeoxyuridine (BrdU)-labeled nuclei (black arrows) and nonspecific kidney injury molecule-1 (KIM-1)-stained cytoplasm (arrowhead) observed in the cortical tubules of the renal cortex in control rats. (B) In a male rat dosed with canagliflozin 100 mg/kg/day for 6 months and fed a glucose-containing diet, numerous brown-stained BrdU-labeled nuclei (black arrows) are present in cortical tubules with diffuse KIM-1 staining (dark blue) of the brush border (arrowhead) and cytoplasm (white arrows). BrdU was detected using avidin-biotin complex (ABC) and diaminobenzidine (DAB) peroxidase substrate; KIM-1 was detected using ABC and DAB-nickel; nuclear fast red was used to counterstain. Reprinted from Chemico-Biological Interactions; Vol. 221; Mamidi et al. (2014), with permission from Elsevier.
Effects of dietary glucose on kidney cell injury in rats treated with canagliflozin.

Note. KIM-1 = kidney injury molecule 1; OSOM = outer stripe of the outer medulla; × = multiple compared to standard diet control.
aA purified, isocalorific diet that contained 40% fructose and no glucose or galactose.
bMale Sprague-Dawley rats administered canagliflozin by oral gavage; 10 rats/group.
cAverage total areas of positively stained renal tubules, defined as mm2/section.
*Statistically significant (p < .05) relative to the standard diet control.
These data suggest that renal proximal tubule cell injury could be the inciting event that leads to a regenerative proliferative response and, ultimately, to RTT formation. The ability of a glucose-free diet to block canagliflozin-induced renal tubule proliferation (a proximal step in tumor formation) supports the hypothesis that RTTs seen in rats treated with canagliflozin are secondary to carbohydrate malabsorption and are not due to a direct effect of canagliflozin on these organs. In the clinical development program, renal injury was monitored in long-term studies by examining urinary albumin excretion, a marker of glomerular and renal tubule injury. In a subset of individuals who underwent a 24-hr urine collection in a 52-week study of canagliflozin in patients with T2DM and moderate renal impairment (estimated glomerular filtration rate ≥30 and <50 ml/min/1.73 m2), median percentage reductions in urine albumin excretion were seen with canagliflozin 100 and 300 mg (–34.4% and –49.0%), while an increase was seen with placebo (14.3%; Yale et al. 2014). Thus, long-term treatment with canagliflozin in clinical studies was not associated with evidence for glomerular or renal tubule injury as assessed by urinary albumin excretion.
Effects of Canagliflozin on LH and Testosterone (T) in Rats and Humans Treated with Canagliflozin
LCTs were associated with atrophy of the accessory sex glands (i.e., seminal vesicles, prostate, and coagulating gland), suggesting that canagliflozin treatment may have altered the hypothalamic–pituitary–testicular (HPT) axis. To assess the effects of canagliflozin on the HPT axis, LH and T were measured in a study conducted under the same conditions as those utilized in the 2-year rat study (De Jonghe et al. 2014). Male Sprague-Dawley rats (30/group) were administered canagliflozin 0 or 100 mg/kg/day orally for 7 months. Blood samples for LH and T measurements were collected monthly and at the same time each day to minimize effects associated with diurnal variation in hormone concentrations. Validated radioimmunoassay methods were used to determine serum concentrations of LH (as described by Niswender et al. 1968) and T.
Relative to vehicle-treated male rats, canagliflozin treatment was associated with sustained, statistically significant increases in LH levels (1.46- to 1.84-fold), with increases at month 1 (earliest time point examined) through month 5, while T levels were not significantly altered by canagliflozin treatment (De Jonghe et al. 2014). The increase in LH, a known Leydig cell mitogen, with canagliflozin treatment provided a plausible mechanism for LCT formation in rats.
To determine whether canagliflozin treatment alters the HPT axis in humans, LH and T were measured in blood samples archived from a 12-week phase 2 study conducted in patients with T2DM treated with placebo or canagliflozin (Rosenstock et al. 2012; Janssen Research & Development, LLC 2012). In the placebo (n = 24) and canagliflozin 300 mg (n = 30) groups, baseline LH (IU/L, mean [SD]) were 4.90 (2.52) and 5.18 (4.20), respectively. The least squares (LS) mean (SE) changes from baseline to week 12 were 0.60 (0.45) and –0.28 (0.40) with placebo and canagliflozin 300 mg, respectively; LS mean differences (95% confidence interval) between the groups were –0.88 (–2.06, 0.31). T levels were not affected by canagliflozin treatment. Since LH levels were not affected in human patients treated with canagliflozin, the LCTs induced by elevated LH in canagliflozin-treated rats were not deemed to be of clinical relevance.
Treatment-related Tumors in the 2-Year Rat Study and Relevance to Humans: Conclusions
While canagliflozin is nongenotoxic and is not associated with tumor formation in mice, in the 2-year rat study, treatment-related increases were seen in RTTs, pheochromocytomas, and LCTs. By inhibiting intestinal SGLT1-dependent glucose absorption, canagliflozin treatment in rats fed a standard diet resulted in glucose malabsorption and its sequelae (e.g., marked increases in urinary calcium excretion, decreases in PTH and 1,25-dihydroxyvitamin D, and hyperostosis). By substituting fructose, whose intestinal absorption is not dependent on SGLT1, the sequelae of canagliflozin-induced carbohydrate malabsorption were prevented. Feeding a glucose-free diet prevented canagliflozin-induced increases in adrenal and renal tubule cell proliferation, a key step in tumorigenesis. Additionally, the glucose-free diet also prevented canagliflozin-induced renal tubule injury seen in rats fed a standard diet and treated with canagliflozin. Canagliflozin treatment of male rats was also associated with sustained elevations in LH, a Leydig cell mitogen causing LCT formation in rats.
In contrast to the studies in rats, canagliflozin treatment in humans was not associated with evidence of carbohydrate malabsorption or its sequelae. No alterations in LH or T were evident in men with T2DM treated for 12 weeks with canagliflozin. Moreover, there was no evidence for renal tubule injury in patients treated long-term with canagliflozin, as determined by urinary albumin excretion.
Given the predicate examples of agents causing carbohydrate malabsorption being associated with RTT, LCT, and pheochromocytoma formation in rats, but not in humans, and the lack of evidence for carbohydrate malabsorption or renal tubule injury in patients treated with canagliflozin in the clinical program, the tumor findings in the 2-year rat study were not deemed to have clinical relevance.
Footnotes
Acknowledgments
The authors are grateful to Dr. Richard Sharpe (University of Edinburgh) for advice on the mechanistic studies related to Leydig cell tumors, Dr. Terry Nett (Colorado State University) for assistance with the mechanistic studies and for the conduct of assays for testosterone and luteinizing hormone, and Dr. Sam Cohen for his input on developing the strategy for the mechanistic program. The authors are grateful to Dr. Gordon Hard and Dr. Jerry Hardisty (Experimental Pathology Laboratories) for assistance with microscopic evaluation of the kidneys.
Author Contributions
Kirk Ways, Mark D. Johnson, Calvert Louden, James Proctor, Sandra De Jonghe, and Rao N. V. S. Mamidi contributed to conception or design; Kirk Ways, Mark D. Johnson, Calvert Louden, James Proctor, Sandra De Jonghe, and Rao N. V. S. Mamidi contributed to data acquisition, analysis, or interpretation; Kirk Ways, James Proctor, and Rao N. V. S. Mamidi drafted the manuscript; Kirk Ways, Mark D. Johnson, Calvert Louden, James Proctor, Sandra De Jonghe, and Rao N. V. S. Mamidi critically revised the manuscript. All authors gave final approval and agreed to be accountable for all aspects of work in ensuring that questions relating to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: These studies were supported by Janssen Research & Development, LLC. Editorial support was provided by Kimberly Fuller, PhD, of MedErgy, and was funded by Janssen Global Services, LLC.