
Abstract
The mammalian gastrointestinal tract is home to trillions of commensal microorganisms that collectively make up the intestinal microbiota. These microbes are important environmental factors that regulate homeostasis, and alterations in the composition of the microbiota have been associated with several diseases, including inflammatory bowel disease, diabetes, and cancer. New research is beginning to uncover epigenomic pathways that may regulate this relationship with the microbiota. Epigenomic modifications alter the structure of the chromatin and therefore regulate the transcriptional program of a cell. These modifications are maintained by the dynamic activity of various modifying and demodifying enzymes, the activities of which can be influenced by metabolites and other environmental cues. Histone deacetylases (HDACs) are a class of epigenomic-modifying enzymes that are regulated by both endogenous and exogenous factors, and recent studies have suggested that host HDAC expression is important for regulating communication between the intestinal microbiota and mammalian host cells.
Introduction
An individual’s genetic makeup is highly influential with regard to the development of disease, but often the genome is not the sole factor. In addition to genetic susceptibility, there is significant evidence that the environment influences the pathogenesis of a variety of chronic diseases, such as inflammatory bowel disease (IBD), diabetes, cancer, heart disease, and allergy (Kaser, Zeissig, and Blumberg 2010; Kau et al. 2011; Mukherjee and Zhang 2011; Renz et al. 2011). Therefore, understanding the fundamental mechanisms that influence how environmental factors regulate gene expression could aid in the development of novel therapeutics to prevent or limit chronic human diseases. There is increasing evidence that the commensal microbes that normally colonize our bodies, referred to as our microbiota, can act as environmental factors that influence both health and disease (Cadwell et al. 2010; Ivanov and Honda. 2012; Kamada et al. 2013; Kaser, Zeissig, and Blumberg 2010; Kau et al. 2011).
Commensal bacteria colonize all body surfaces that are exposed to the external environment, such as the gastrointestinal and respiratory tracts as well as the skin. These bacteria have coevolved with their hosts and have formed complex ecosystems in specific anatomical locations of the mammalian body (Kamada et al. 2013; Ley et al. 2008). The number of bacteria that normally colonize the intestinal and skin surfaces greatly exceeds the number of human cells in the body. It is estimated that there are approximately 10 times more resident microbes than human cells. Further, the microbiome, which encompasses the collective genomes of these commensal microbes, makes up more than 99% of the genetic information in the human body (Backhed et al. 2005).
Recent molecular advances in high-throughput metagenomic deep sequencing analyses have greatly expanded our ability to identify bacterial communities throughout the body in human health and disease, as well as in animal models. Specifically, bacterial DNA can be isolated from distinct anatomical locations, sequenced, and compared computationally to determine microbial diversity and phylogenetic composition. These approaches have resulted in a plethora of collaborations and publications characterizing core bacterial communities in specific regions of the body under healthy and disease conditions (Caporaso et al. 2010; Cho and Blaser 2012; Huttenhower et al. 2012).
The Host–Microbiota Relationship and Intestinal Homeostasis
The majority of the commensal bacteria reside within the intestine where they directly interact with a single layer of intestinal epithelial cells (IECs) that line the lumen. It has become evident that the mammalian host has formed a symbiotic relationship with these intestinal commensal bacteria. In particular, the intestinal microbiota plays a critical role in the normal digestion of food, resulting in the release of nutrients and metabolites that are essential for the maintenance of mammalian health (Tremaroli and Backhed 2012). As the vast majority of the microbiota resides in the mammalian intestine, one strategy to decipher how the host and microbiota interact has been to focus on evaluating commensal bacteria-dependent homeostatic regulation of mammalian cells in the intestinal microenvironment.
A dynamic and complex relationship exists between the intestinal microbiota, the immune system, and the intestinal epithelium (Artis 2008; Hooper, Littman, and Macpherson 2012). IECs function as a crucial cell lineage that resides at the interface between the mammalian host and commensal bacteria. These cells form a physical barrier, sense bacteria-derived signals, and secrete antimicrobial peptides and cytokines/chemokines that, in turn, regulate the microbiota and immune cell homeostasis (Gallo and Hooper 2012; Roda et al. 2010). Further, epithelial permeability, proliferation, and expression of antimicrobial proteins can be regulated by cytokines produced by immune cells (Dahan et al. 2007). The microbiota, itself, influences immune cell homeostasis and is critical in the development/maturation of both the innate and adaptive system (Khosravi et al. 2014; Littman and Pamer 2011). In fact, recent studies have identified specific species of commensal bacteria in the intestine that drive differentiation of intestinal CD4+ T cells to distinct lineages, suggesting that there may be core species of microbes that are required for development of select immune cell populations (Atarashi et al. 2011; Littman and Pamer 2011).
In addition to intestinal homeostasis, a wide range of disease entities, including IBD, obesity, cancer, and allergy have been associated with dysregulation of the host–microbiota relationship and alterations in the diversity of intestinal commensal bacteria (dysbiosis; Arthur et al. 2012; Kamada et al. 2013; Kau et al. 2011; McLoughlin and Mills 2011). The pathogenesis of IBD, in particular, appears to be driven both by altered immune responses to commensal bacteria in addition to alterations in the composition of these commensal bacteria (Kamada et al. 2013; Kaser, Zeissig, and Blumberg 2010; Maloy and Powrie 2011). Further, commensal bacteria help to prevent pathogen infection in part by stimulating the mammalian host’s immune defenses in a manner that preserves their own survival and simultaneously limits infection (Abt and Pamer 2014). A large field of research is now dedicated toward better understanding the effects of manipulating commensal bacterial communities. Many of these studies investigate how altering the diversity of the microbiota, possibly through the use of antibiotics, could negatively impact health and disease susceptibility. Further, investigation into probiotics has significantly expanded in order to identify potential cocktails of commensal bacteria or bacteria-derived products that can beneficially regulate the immune system, confer protection against infections, or aid in digestion (Sanders et al. 2013).
Chromatin and the Epigenome
Dynamic transcriptional regulation is essential in mediating the symbiotic host–microbiota relationship. Eukaryotic cells package their DNA around histone proteins to form a higher order structure termed chromatin. The repetitive element within chromatin, called the nucleosome, is composed of 147 bp of DNA tightly wound around a histone octamer. Histone H1 functions as a linker between nucleosomes that permits further condensation of the chromatin structure. Chromatin, itself, is generally repressive by limiting access of transcriptional machinery to the genome. However, covalent nucleosomal modifications as well as ATP-dependent chromatin remodeling ATPases enable chromatin flexibility in response to specific cellular signals. The chromatin structure can, therefore, undergo local condensation or decondensation, which facilitates various processes such as DNA replication, repair, or transcription (Richmond and Davey 2003). The most well-characterized mechanisms that regulate the chromatin structure are DNA methylation and histone modifications, each of which can modify gene expression without altering the associated DNA sequence. Histone N-terminus tails extend from the nucleosomal core and provide a template for various covalent modifications such as acetylation, phosphorylation, methylation, SUMOylation, ubiquitination, and more. The pattern of modifications on these histone tails establish a “histone code” that guides changes in chromatin structure and/or directs recruitment of specific cofactors (Jenuwein and Allis 2001; Strahl and Allis 2000).
While covalent DNA and histone modifications are most commonly discussed as epigenetic phenomena, the term epigenetics often suggests heritability of the associated changes in gene expression. A broader concept of the epigenome has more recently been adopted to refer to the combination of histone and DNA modifications and associated proteins that package the genome and define the transcriptional program of a cell (Arrowsmith et al. 2012). Epigenomic modifications are maintained by the balanced activity of various enzymes. Alterations in epigenomic modifications provide a mechanism by which the environment can alter gene expression and influence disease pathogenesis. For this reason, epigenomic mechanisms have been implicated in the development of most chronic conditions with complex gene–environment etiologies, including cancer, diabetes, allergy, atherosclerosis, and IBD (Begin and Nadeau 2014; Feil and Fraga 2011; Handel et al. 2010; Renz et al. 2011).
Do Epigenomic Pathways Regulate the Host–Microbiota Relationship?
As discussed earlier, the immune system is a key component that shapes the microbiota and the epigenome clearly mediates development and differentiation of immune cells (Cedar and Bergman 2011; Cuddapah, Barski, and Zhao 2010; Natoli 2010; O’Shea et al. 2011). Recent work demonstrates more direct links between commensal bacteria-derived signals and host epigenomic pathways, particularly through histone acetylation. Acetylation of histone tails is believed to disrupt the DNA–histone interaction, causing local relaxation of the chromatin and permitting access for transcription machinery (Chen, Tini, and Evans 2001). Furthermore, histone acetylated-lysines are the preferred substrate for proteins that propagate increased acetylation and promote transcriptional activation (Eberharter and Becker 2002).
Conversely, removal of the acetyl groups by histone deacetylases (HDACs) generally promotes tighter DNA–histone associations and inhibits transcriptional activity. There are 18 known HDACs that are classified into 4 groups based on their homology to yeast HDACs and subcellular location. The class I, II, and IV HDACs require zinc for their enzymatic deacetylase activities, whereas class III HDACs (sirtuins) depend on nicotine adenine dinucleotide as a cofactor (Sauve et al. 2006). These enzymes are often found in large complexes that are recruited to the chromatin through interactions with transcription factors. Further, their activity and recruitment to the genome can be altered by hormones, dietary compounds, and bacteria-derived products (Chen et al. 2012; Dashwood and Ho 2007; Donohoe and Bultman 2012; Haberland et al. 2009; Kim et al. 2010; Perissi and Rosenfeld 2005). In vivo studies suggest that the specificity of HDACs in regulating distinct gene programs differs with cell identity, available associating proteins, and the cell signaling environment (Haberland et al. 2009).
Importantly, the zinc-dependent HDACs can be targeted by a large class of inhibitors that are currently being utilized or examined for the treatment of various types of cancer, as well as inflammatory and degenerative conditions (Haberland et al. 2009; Huang. 2006). The therapeutic potential of HDAC inhibitors (HDACi) is promising; however, the mechanisms underlying their clinical effects are not fully understood. Studies directed toward better understanding their specificity and mode of action are ongoing (Ververis et al. 2013).
While studies have indicated that the microbiota regulates DNA and histone methylation–dependent pathways in the host (Ganal et al. 2012; Kellermayer et al. 2011; Obata et al. 2014; Olszak et al. 2012; Takahashi et al. 2011), recent work over the last year has brought histone acetylation and HDACs to the forefront as a critical level of epigenomic regulation that mediates the interplay between mammalian host cells and the intestinal microbiota (Figure 1). The class I HDAC, HDAC3, regulates transcription through histone deacetylation, but has also been suggested to deacetylate nonhistone targets and possess enzyme-independent effects (Choudhary et al. 2009; Sun et al. 2013; You et al. 2010). As discussed earlier, IECs are central to integrating signals from the intestinal microenvironment to regulate intestinal homeostasis. Recent work revealed that loss of IEC-specific HDAC3 expression led to extensive alterations in gene expression, changes in histone acetylation, impaired Paneth cell survival, and decreased intestinal barrier function. Lack of HDAC3-dependent regulation in IECs also resulted in significant alterations in the composition of the intestinal microbiota as well as increased susceptibility to intestinal damage and inflammation. Interestingly, elimination of the microbiota by generating a germ-free HDAC3-deficient mouse strain restored Paneth cell homeostasis and the functionality of the intestinal barrier, suggesting that HDAC3 in IECs mediates functional cross talk between commensal bacteria and host cells (Alenghat et al. 2013). However, the underlying microbiota-dependent mechanisms that orchestrate HDAC3 function in this pathway remain to be defined.
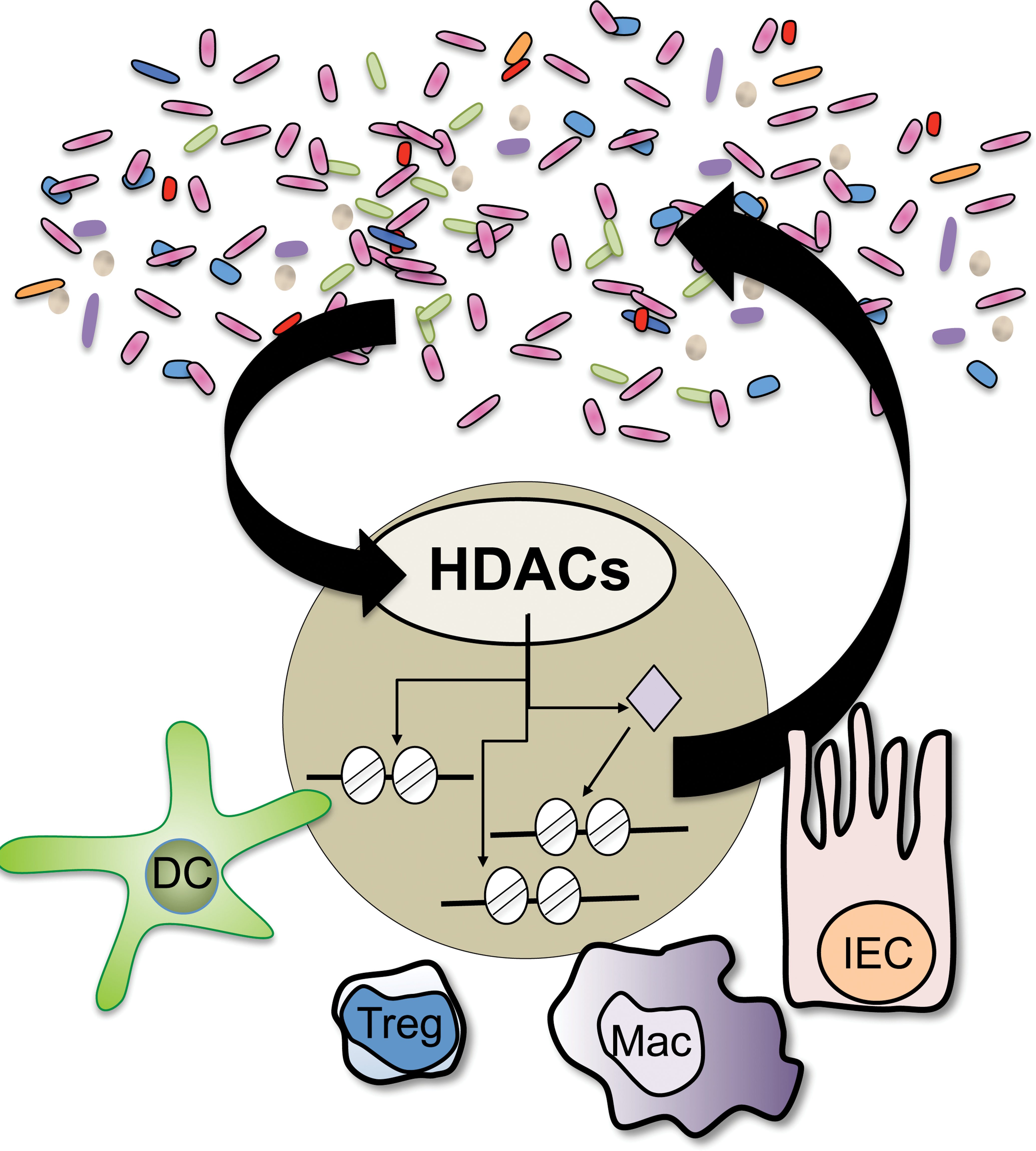
Recent studies indicate that histone deacetylases (HDACs), a family of epigenomic-modifying enzymes, mediate dynamic regulation between the microbiota and multiple cell lineages in the mammalian intestine. Manipulation of these pathways may result in altered intestinal homeostasis and susceptibility to inflammation, as well as changes in the diversity of the microbiota.
The microbiota can produce by-products that modify the epigenome of host cells and in turn alter the cell’s function and the intestinal environment. HDACs in immune cells have been highlighted as targets of microbiota-derived short chain fatty acids (SCFAs). SCFAs that are produced by commensal bacteria through fermentation of dietary carbohydrates in the colon can function as HDAC inhibitors (Macfarlane and Macfarlane 2003) and have been found to regulate the development and function of several immune cell lineages (Brestoff & Artis. 2013). A series of recent high-profile publications demonstrated that SCFAs derived from commensal bacteria in the large intestine exert anti-inflammatory effects in the colon by stimulating histone acetylation of the FoxP3 locus and driving differentiation of regulatory T cells (Tregs; Arpaia et al. 2013; Furusawa et al. 2013; Smith et al. 2013). Smith et al. suggested a mechanism by which SCFAs decreased expression of HDAC6 and/or HDAC9 in Tregs in a G-protein-coupled receptor-dependent manner. Consistent with these SCFA findings, synthetic pan-HDACi were previously found to limit colitis through expansion of Foxp3+ Tregs and associated differences in HDAC9 expression (de Zoeten et al. 2010; Glauben et al. 2008; Tao et al. 2007).
The effects of butyrate, a microbiota-derived SCFA, on histone acetylation in myeloid cell lineages have also been examined. Arpaia et al. found that butyrate increased histone H3 acetylation in dendritic cells, enhancing their ability to facilitate Treg differentiation, and Chang et al. recently demonstrated that treatment of macrophages with butyrate increased histone acetylation and decreased expression of pro-inflammatory cytokines (Arpaia et al. 2013; Chang et al. 2014). While butyrate clearly increases levels of histone acetylation in immune cells, it remains unclear how this relates to physiologic concentrations of SCFAs in the colon and whether commensal-derived SCFAs increase histone acetylation primarily through direct or indirect regulation of HDAC expression versus inhibition of HDAC enzymatic activity. Further, multiple outcomes and mechanisms have been suggested for butyrate treatment during colitis, which warrants a more thorough examination of the differential effects of SCFAs on specific HDACs in different host cells, in the context of protective and pathologic immunity (Berndt et al. 2012; Chang et al. 2014; Hamer et al. 2010; Kim et al. 2013; Tarrerias et al. 2002).
Collectively, it is becoming evident that proper innate and adaptive immune system development and function supports the maintenance of a healthy intestinal microbiota and that epigenomic pathways in host epithelial and immune cells mediate cross talk between the microbiota and the mammalian host. Although deciphering the role of epigenomic regulation in mediating the mammalian relationship with commensal microbes is still in its infancy, these recent advances provoke several questions regarding the potential relevance of evaluating epigenomic end points and the microbiota in toxicologic pathology. Can we evaluate epigenomic modifications in the gut to assess intestinal health? How does clinical inhibition of epigenomic pathways affect the intestinal microbiota? What is the impact of drug-dependent alterations of the microbiota on epigenomic regulation? How does variability in the microbiota alter drug metabolism, efficacy, and toxicity? Continued basic mechanistic and translational studies will dictate how best to focus our efforts toward addressing these issues.
Footnotes
Acknowledgments
The author thanks T. Fung, J. Brestoff, and M. Abt for discussions and reading of the article. This work is supported by the National Institutes of Health (DK093784) and the Burroughs Wellcome Fund Career Award for Medical Scientists.
Author Contribution
T. Alenghat substantially contributed to conception or design, acquisition and interpretation of data, drafted the article, critically revised the article content, gave final approval, and agrees to be accountable.
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The author(s) received no financial support for the research, authorship, and/or publication of this article.