
Abstract
Vascular inflammation, infusion reactions, glomerulopathies, and other potentially adverse effects may be observed in laboratory animals, including monkeys, on toxicity studies of therapeutic monoclonal antibodies and recombinant human protein drugs. Histopathologic and immunohistochemical (IHC) evaluation suggests these effects may be mediated by deposition of immune complexes (ICs) containing the drug, endogenous immunoglobulin, and/or complement components in the affected tissues. ICs may be observed in glomerulus, blood vessels, synovium, lung, liver, skin, eye, choroid plexus, or other tissues or bound to neutrophils, monocytes/macrophages, or platelets. IC deposition may activate complement, kinin, and/or coagulation/fibrinolytic pathways and result in a systemic proinflammatory response. IC clearance is biphasic in humans and monkeys (first from plasma to liver and/or spleen, second from liver or spleen). IC deposition/clearance is affected by IC composition, immunomodulation, and/or complement activation. Case studies are presented from toxicity study monkeys or rats and indicate IHC-IC deposition patterns similar to those predicted by experimental studies of IC-mediated reactions to heterologous protein administration to monkeys and other species. The IHC-staining patterns are consistent with findings associated with generalized and localized IC-associated pathology in humans. However, manifestations of immunogenicity in preclinical species are generally not considered predictive to humans.
Keywords
Introduction
The first recombinant human (rh) proteins used in clinical practice (such as somatostatin [ProTropin™] in 1976, insulin [Humulin™] in 1982, erythropoietin [Epogen™] in 1986, interferon [Roferon] in 1986, tissue plasminogen activator [Activase] in 1987, or coagulation factors [factor VIII] in 1990) were not often pharmacologically active in the species (e.g., rats or dogs) that were traditionally used for toxicology testing. Because of the species-specific restrictions in the pharmacologic activity of these biopharmaceuticals (i.e., medical drugs derived by biotechnology including mAbs and rh proteins), preclinical testing often required the use of nonhuman primates (e.g., marmosets, monkeys, baboons, and/or chimpanzees). Typically, the human doses for these early biopharmaceuticals were in the μg/kg range, and higher doses were associated with significant super-pharmacologic (“toxic”) effects, thus limiting the multiples of the human dose (e.g., 5× human dose or 10× human dose) that were evaluated in toxicology studies to the μg/kg or low mg/kg range. In contrast, with the advent of mAb therapies (e.g., muromonab-cluster of differentiation 3 [CD3] [OKT3™] in 1986, abciximab [ReoPro™] in 1994, daclizumab [Xenepax™] in 1997, and infliximab [Remicade™], basilixumab [Simulect™], palivizumab [Synagis™], and trastuzumab [Herceptin™] in 1998), the doses of these protein drugs to be used in humans were typically in the low mg/kg range and the multiples (e.g., 5× human dose or 10× human dose) that were given to nonhuman primates were often high mg/kg range. Even at high doses, where the maximum pharmacodynamic (PD) effects were achieved, toxicity was not common. However, immunogenicity often was observed and typically resulted in neutralization and/or clearance of the therapeutic mAb (Ponce et al. 2009; Rojas et al. 2005). In some cases, the neutralizing effect could be overcome by increasing the dose or dosing frequency (Ponce et al. 2009). The requirement to determine a toxicologic effect dose level and the desire to “dose through” immunogenicity worked in concert to escalate the doses of mAbs tested in nonhuman primates (Ponce et al. 2009), so that it is not uncommon now to see doses of 50 to 300 mg/kg administered to nonhuman primates. While there was initially some concern that these high doses of “foreign” (nonmonkey) protein might lead to immune complex (IC)-related pathologic changes, this did not appear to be the case. Infusion reactions occurred infrequently and were generally not life threatening; glomerulonephritis (GN) and other IC-related pathophysiologic changes were rare.
Recently, the frequency, pathogenesis, and identification of infusion reactions and other IC-related pathologies have become topics of interest to the toxicologic pathology community as evidenced by specialty sessions at Society for Toxicologic Pathology (Leach et al. 2014) and BioSafe (BioSafe General Membership Meeting, April 2012). In the experience of three of the authors (Rojko, Price, and Raymond, unpublished), requests for immunohistochemistry (IHC)-IC studies have tripled in the interval from 2008 to 2013 compared with the time period prior to 2005. The majority of requests stem from repeated-dose toxicity studies with human mAbs or other rh therapeutic protein drugs in which one or more drug-dosed animals experience infusion reactions leading to unscheduled deaths or in which histopathology alterations potentially related to ICs are observed in dosing or recovery phase animals. Likely because monkeys are often the pharmacologically relevant species for rh therapeutic proteins/mAbs, the majority of the requests stem from monkey toxicity studies. However, during this interval, we also have evaluated rat and rabbit tissues from rh therapeutic protein toxicology studies for IC-related pathology. The increase in IHC-IC studies may indicate (1) an increasing frequency of infusion reactions or other IC-related pathologies; (2) greater numbers of monkeys in toxicology studies; (3) inclusion of longer term studies (i.e., 3–6 months or longer compared with 2–4 weeks, allowing more time for IC to develop/manifest); (4) an increasing awareness that current IHC practices allow IC evaluation on replicate paraffin sections from the toxicity studies; or (5) other unknown reasons. This article is intended to review the formation, clearance, deposition, pathogenicity, and IHC identification of biopharmaceutical-related ICs in laboratory animals with a focus on a pharmacologically relevant species (monkeys) and to provide representative case studies in which IHC provided a valuable adjunct to the pathology evaluation.
Antidrug Antibodies (ADAs) and Hypersensitivity Reactions (HSRs)
ADA in Monkeys on Preclinical Studies of Therapeutic Proteins
Following dosing with therapeutic protein drugs to animals on preclinical studies, adverse drug reactions can occur as a result of direct dose-dependent toxicity, suprapharmacologic effects, other secondary drug effects, or secondary to immunogenicity of the drug. As part of their increasing role in the biopharmaceutical industry (Ettlin 2013; van Tongeren et al. 2011), toxicologic anatomic and clinical veterinary pathologists provide critical analyses during the preclinical phase. These analyses help sort out the possible cause/effect scenarios for the adverse drug reaction, including the possibility that it is mediated by ADA and an HSR. The weight-of-evidence approach includes consideration of clinical signs, pharmacokinetic (PK)/PD and immunogenicity/ADA data, and flow cytometry and other specialized assay results as well as more traditional histopathology, hematology, and clinical chemistry findings.
Monkeys are often the most pharmacologically relevant test species for toxicity studies with rh therapeutic proteins, including mAbs. In monkeys receiving rh therapeutic protein drugs, ADA form relatively often as the rh proteins are sufficiently different from the homologous monkey proteins to be immunogenic (Brinks, Jiskoot, and Schellekens 2011; Leach et al. 2014; Ponce et al. 2009; Rojas et al. 2005). The immunogenicity can be affected by (1) the relative “foreignness” of the rh protein (e.g., chimeric mAbs may be more immunogenic than humanized or fully human mAbs in humans and/or monkeys [discussed subsequently]); (2) the cell lines used to produce the drug (e.g., different cell lines may glycosylate the rh protein differently); and/or (3) excipients or residual protein aggregates in the formulation (Brinks, Jiskoot, and Schellekens 2011; Brinks 2013), resulting in greater ADA responses. Immunogenicity can also be affected by the mechanism of action of the drug itself (Brennan et al. 2010; Srinivas et al. 1997). For example, abatacept, a human IgG constant region of immunoglobulin (Ig) molecule (Fc) fused with the extracellular domain of cytotoxic T-lymphocyte antigen 4 (CTLA-4), is highly immunosuppressive, and low doses are associated with ADA development, while higher doses do not elicit ADA, in rat preclinical studies (Fathallah, Bankert, and Balu-Iyer 2013; Keystone et al. 2012; Nash et al. 2013).
When ADA neutralize or accelerate clearance of the rh therapeutic protein, they may be associated with altered PK and/or PD properties of the therapeutic protein; however, some ADA may have no discernible effect on PK/PD levels (Leach 2013; Ponce et al. 2009; Rojas et al. 2005). An analysis of European Union mAb data registration indicated less than 60% correlation between the development of nonhuman primate ADA and human ADA to rh therapeutic proteins (van Meer et al. 2013). The measurement of monkey ADA levels is reviewed by Leach and colleagues (2014) who note that the initial ADA assays in monkeys generally measure binding ADA, while neutralizing ADA or further ADA characterization is conducted on an as-needed basis (e.g., IgE ADA may be measured if an acute infusion reaction occurs; Clark et al. 2013; Leach et al. 2014).
ADA in Humans Administered Therapeutic Proteins
The effects of human ADA responses on the PK/PD responses to rh therapeutic proteins have been reviewed recently (Chirmule, Jawa, and Meibohm 2012). In humans, relatively comprehensive information regarding ADA responses is available for multiple sclerosis patients treated with interferon β drugs as reviewed by Creeke and Farrell (2013) or tumor necrosis factor-α (TNFα) inhibitors. For interferon β drugs, human ADA responses are affected by the type of interferon β used (including the glycosylation patterns of the cell line used to produce the drug), dose, route of administration, and dosing schedule. In the first 1 to 3 months of treatment, the ADA responses usually bind but do not neutralize interferon β. Generally between 4 and 24 months, 2 to 45% of patients will make neutralizing ADA that interferes with the binding of interferon β to its cellular receptors. Escherichia coli–derived, unglycosylated interferon β-1b is more immunogenic than mammalian cell–derived, fully glycosylated interferon β-1a (Perini et al. 2001). In general, more patients receiving subcutaneous (SC) interferon β-1a drug (Rebif®) or SC interferon β-1b develop neutralizing ADA compared with those receiving an intravenous (IV) interferon β-1a drug (Avonex®), while patients receiving intramuscular interferon β-1a seldom develop neutralizing ADA (Farrell and Giovannoni 2007; Minagara and Murray 2008). To the contrary, repeated-dose rat and monkey studies have shown that intramuscular dosing with interferon β-1a (Rebif®) was associated with higher ADA titers compared with IV dosing (Ponce et al. 2009). For a different rh therapeutic protein (abatacept), SC/IV immunogenicity differences were not observed in patients (Fathallah, Bankert, and Balu-Iyer 2013; Keystone et al. 2012; Nash et al. 2013), but SC administration elicted greater ADA titers compared with IV dosing in rat preclinical studies (Srinivas et al. 1997). For adalimumab in monkeys, IV dosing was associated with higher ADA titers compared with SC dosing (Ponce et al. 2009).
Clinical studies of the chimeric anti-TNFα mAb infliximab indicate that ADA may affect serum infliximab levels. Forty-six percent of 316 patients with inflammatory bowel disease treated with infliximab developed ADA in the first few weeks after therapy. With continuing therapy, ADA declined in two-thirds of patients with good clinical responses but persisted in patients with poor clinical responses (Steenholdt et al. 2012). Furthermore, ADA levels were negatively correlated with infliximab levels. In a different study, 28% of 272 rheumatoid arthritis patients treated with the anti-TNFα human mAb adalimumab developed ADA, the ADA-positive patients had reduced adalimumab concentrations compared with ADA-negative patients, and the ADA-positive patients were more likely to discontinue treatment because of lack of sustained clinical response (38% ADA-positive patients compared with 14% of ADA-negative patients; Bartelds et al. 2011). Sustained clinical remission was observed in 24% of ADA-negative patients compared with only 4% of ADA-positive patients (Bartelds et al. 2011; Nanda, Cheifetz, and Moss 2013).
In another study, the incidences of ADA responses to mAbs were compared and sorted by molecular format (murine, chimeric, and humanized) as well as immunogenicity level (negligible, tolerable, or marked; Hwang and Foote 2005). Murine mAbs showed the highest incidence of marked immunogenicity with greater than 80% incidence of marked ADA responses, while chimeric mAbs elicited roughly 50% incidence of marked ADA responses, and humanized mAbs had only about 5% incidence of marked ADA responses. Conversely, for negligible immunogenicity, the incidences of limited ADA responses were roughly 10% for murine mAbs, 35% for chimeric mAbs, and 55% for humanized mAbs. Taken together, these data indicate that in humans, the incidence of immunologic reactions often directly correlates with the foreignness of the mAb being administered (Hwang and Foote 2005). While the Fc region differences contribute to immunogenicity, particularly for murine mAbs, residual immunogenic epitopes important for CD4+ help and ADA responses reside in the complementary determining region (CDR) of humanized and fully human antibodies (Harding et al. 2010).
Certain risk factors for developing ADA are not modeled in toxicology studies. For example, multiple sclerosis patients who smoke have increased risk for developing ADA to interferon β1-a or natalizumab (a humanized mAb directed against α4-integrin subunits) compared with their nonsmoking counterparts (Hedstrom, Alfredsson, et al. 2013; Hedstrom, Ryner, et al. 2013). In humans, ADA dcvelopment also may be affected by age, concurrent disease, exposure to immunosuppressive drugs, or other factors that may contribute to the relatively poor correlation between development of human and nonhuman primate ADAs following mAb administration (Oldenburg et al. 2004; van Meer et al. 2013).
Leach and colleagues (2014) indicate that the route of administration influence on immunogenicity observed in human clinical trials often is not observed in preclinical studies. Although ADA responses are measured in many preclinical studies, rarely is a preclinical study conducted to compare IV, SC, or IM immunogenicity as the primary end point (Ponce et al. 2009; Rojas et al. 2005). Preclinical immunogenicity usually is assessed retrospectively, comparing ADA responses from studies conducted with different product formulations at different times in different animals, often at different testing facilities. For rh therapeutic proteins, IV dosing often is conducted first. SC dosing may come later in development after efficacy has been demonstrated for IV dosing; the SC route may be included as a means to enable patient self-dosing. IV dosing may provide higher drug concentrations than SC dosing, meaning that greater amounts of ADA may be bound to drug and that the apparent immunogenicity (free ADA) may be higher than for SC dosing.
ADA-mediated HSRs
Although ADA-mediated HSRs are infrequent, they may cause significant pathophysiologic changes, even resulting in death. There are four classes of HSRs generally recognized according to the revised Gell and Coombs’ classification (Pichler 2007; reviewed in Leach et al. 2014):
Type 1 HSRs are immediate, anaphylactic reactions characterized by IgE recognition of soluble antigen (protein), mast cell activation, and histamine release. Examples include allergic asthma and food allergies. Type I HSRs have been described for monkeys exposed to human serum containing reaginic IgEs (Revenas et al. 1981) or to a rh Fcγ-Fc∊ fusion protein (Van Scott et al. 2008) and for patients retreated with the chimeric anti-interleukin 2 (IL2) receptor mAb basiliximab (Baudouin et al. 2003) or the chimeric anti-TNFα mAb infliximab (Vultaggio et al. 2010). Type Ia: Type 1 HSRs can be amplified by platelet-activating factor, release of other vasoactive amines (e.g., eosinophilic chemotactic factor of anaphylaxis), leukotrienes, and prostaglandins with attraction of neutrophils and eosinophils.
Type 2 HSRs are cytotoxic HSRs characterized by IgG (usually IgG1 or IgG3 in humans [not defined in monkeys]) and/or IgM recognition of cell surface–bound or matrix-associated antigens (proteins), complement activation and complement-dependent cytotoxicity (CDC), and natural killer (NK) cell activation and antibody-dependent cell
Type 3 HSRs are IC-mediated, anaphylactoid reactions characterized by IgG/IgM recognition of soluble antigens (proteins) and complement activation; examples include systemic lupus erythematosus (SLE) and serum sickness. A type 3 HSR has been described in a rheumatoid arthritis patient with a poor clinical response to the chimeric anti-TNFα mAb infliximab (van der Laken et al. 2007). Other acute infusion reactions to infliximab that appear to be mediated by IgG or IgM ADA in patients also may represent type 3 HSRs (Steenholdt et al. 2011; Vultaggio et al. 2010).
Type 4 HSRs are delayed-type reactions characterized by recognition of antigen by antigen-presenting cells (APCs) in the context of major histocompatibility (MHC) I or II proteins, followed by T cell activation, cytokine release, and cytotoxicity. Examples include contact dermatitis and graft versus host disease (GvHD). Type 4 reactions are divided into four subclasses based on immune reactant molecules, types of APCs, and effector cells (Leach et al. 2014; Pichler 2007).
For the purposes of this discussion, the term anaphylactic/anaphylaxis will be reserved for type 1 HSRs with a demonstrated IgE
Rh therapeutic proteins could, in principle, be associated with types 1 to 4 HSRs or CARPA in preclinical studies. In practice though, type 1 and type 3 HSRs are most likely to occur in laboratory animals, including monkeys, while type 4 HSRs are rarely observed after rh therapeutic protein drug administration (van Meerten et al. 2006; Klingbeil and Hsu 1999; Leach et al. 2014; Van Scott et al. 2008). CARPA reactions have been described only in humans and swine exposed to liposomal, micellar, or other lipid-including therapeutic drugs and have not been associated with rh therapeutic protein administration (Szebeni 2005). Type 2 HSRs may occur as part of the intended pharmacologic mechanism of action for some mAbs. For example, rituximab (Rituxan™) binding to CD20 on normal B cells in monkeys or neoplastic B cells in patients results in ADCC and B cell killing (van der Kolk et al. 2001), which causes clinical signs referred to as “infusion reactions” or “tumor lysis syndromes,” respectively. The different types of HSRs are not mutually exclusive, and some, such as type 2 and 3 HSRs might occur simultaneously.
On the basis of clinical signs, it can be difficult or impossible to distinguish the acute HSRs of types 1, 2, and 3, especially if the route of administration is IV infusion and the reactions occur during or soon after dosing. However, histopathologic and IHC evaluation can often identify type 3 HSRs, based on the demonstration of ICs, sometimes containing the rh therapeutic protein, within affected tissues. This document provides background information regarding the formation, clearance, deposition, pathogenicity, and IHC identification of ICs in laboratory animals, particularly the pharmacologically relevant monkeys that may be useful for understanding the types and consequences of generalized and localized type 3 HSRs, which may occur after administration of rh therapeutic proteins as part of preclinical drug development. References are also made to IC formation in other species, including humans. In addition, representative case studies of pathogenicity associated with immunogenicity of rh therapeutic proteins are presented.
Formation of ICs and Complement Activation
The normal physiologic biodistribution, transport, and clearance of IgG in humans and monkeys (and the implications for mAb products) has been reviewed (Rojko and Price-Schiavi 2008). That review, however, did not emphasize the formation and clearance of ICs.
Serum Levels
Healthy humans (and, by inference, healthy nonhuman primates) form and clear approximately less than 1 to 30 µg/ml of ICs each day, with IgG
One report indicates higher levels of CICs in cynomolgus monkeys from Indonesia, as compared with their counterparts from Malaya or the Philippines (Alexander, Clarkson, and Fulgham 1985). Adult macaques also typically have higher levels of CICs than juveniles (Alexander, Clarkson, and Fulgham 1985). In humans, ICs are often increased in patients with various inflammatory or neoplastic diseases (Anderson and Stillman 1980; Berkowitz et al. 1983; Brandeis et al. 1978; Carpentier et al. 1977; Doll, Wilson, and Salvaggio 1980; Hooper et al. 1983; Hubbard et al. 1981; Lavedan et al. 1991; Muratsugu 2005; Nydegger et al. 1974).
Different methodologies are currently in use for the measurement of CICs in monkeys on toxicity studies (Leach et al. 2014; Stubenrauch et al. 2010). An important consideration is the risk of false negatives due to interference by high levels of circulating drug; similar to the interference with ADA measurements by high drug levels (Leach et al. 2014; Stubenrauch et al. 2010). False positives can also occur with the formation of IC in vitro due to the close approximation of drug and ADA. For mAb drugs, Stubenrauch and colleagues (2010) have developed an enzyme-linked immunosorbent assay in which the capture reagent is a mouse mAb directed against the Fc portion of human IgG (which binds drug) and the detection reagent is a mouse anti-cynomolgus monkey mAb (which binds ADA only in the IC-complexed form). Leach and colleagues (2014) discuss the use of commercially available kits to measure ICs in monkeys on toxicity studies.
IC Composition
ICs are composed of a lattice-like network of noncovalently bound antigen and antigen-specific antibody molecules. Physicochemical characteristics of the IC lattice determine whether ICs are cleared or deposited in tissues (Mannik 1980). These include: (1) lattice size; (2) antigen concentration, valence (number of repeating units/epitopes), charge, and site/sites of production/localization; (3) antibody concentration, valence, affinity, charge, and subclass/isotype (including ability to fix complement); and (4) antigen:antibody ratio (Mannik 1980; Theofilopoulos and Dixon 1979; Wener 2007).
IC Lattice Size, Antigen and Antibody Valence
IC lattice size depends on antigen and antibody valences. Each monovalent antigen can only interact with one antibody molecule; increasing the number of repeated epitopes in the antigen allows interaction with increasing numbers of antibody molecules, thereby increasing lattice size. Antibody valence (number of antigen-binding sites) depends on subclass, multimerization, and/or fragmentation, as follows: (1) antibody valence of 0 for an Fc fragment (no antigen-binding sites); (2) antibody valence of 1 for a single chain antibody or Fab fragment; (3) antibody valence of 2 for IgG, IgD, IgE, monomeric IgA, or an antigen-binding region of immunoglobulin molecule (bivalent form; F(ab’)2) fragment; (4) antibody valence of 4 for dimeric IgA; and (5) antibody valence of 10 for pentameric IgM when formed with small haptens, and 5 for IgM when formed with large antigens (Theofilopoulos and Dixon 1979). Hence, IgM-containing ICs can form very large lattices even with monovalent antigens as long as the antigen concentration is high.
Antigen:Antibody Ratio
IC formation and lattice size also are affected by antigen:antibody ratio. In vitro experiments demonstrate that when antigen is in large excess, small IC form, while at antigen/antibody equivalence, large insoluble IC form and precipitate, and when antibody is in great excess, IC tend to be small (Mannik 1980; Wener 2007). As antigen/antibody ratios approach molar equivalence, IC lattices tend to be larger but soluble, which could deposit in tissue. In vivo, IC formation and disease are more dynamic processes, and the types of ICs formed and their sizes likely change as antigen/antibody ratios, Ig isotypes, and clearance mechanisms change over time and IC remodeling occurs in tissue deposits. The exact nature of IC formed in vivo is difficult to study, as complete mixing of antigen and antibody is complicated by interactions with complement, phagocytic cells, and other circulating cells (e.g., erythrocytes, platelets).
Figures 1 to 2 illustrate IC formation in antigen exceses, antibody excess, or at molar antigen:antibody equivalence, as well as the formation/effects of ICs of different lattice size.
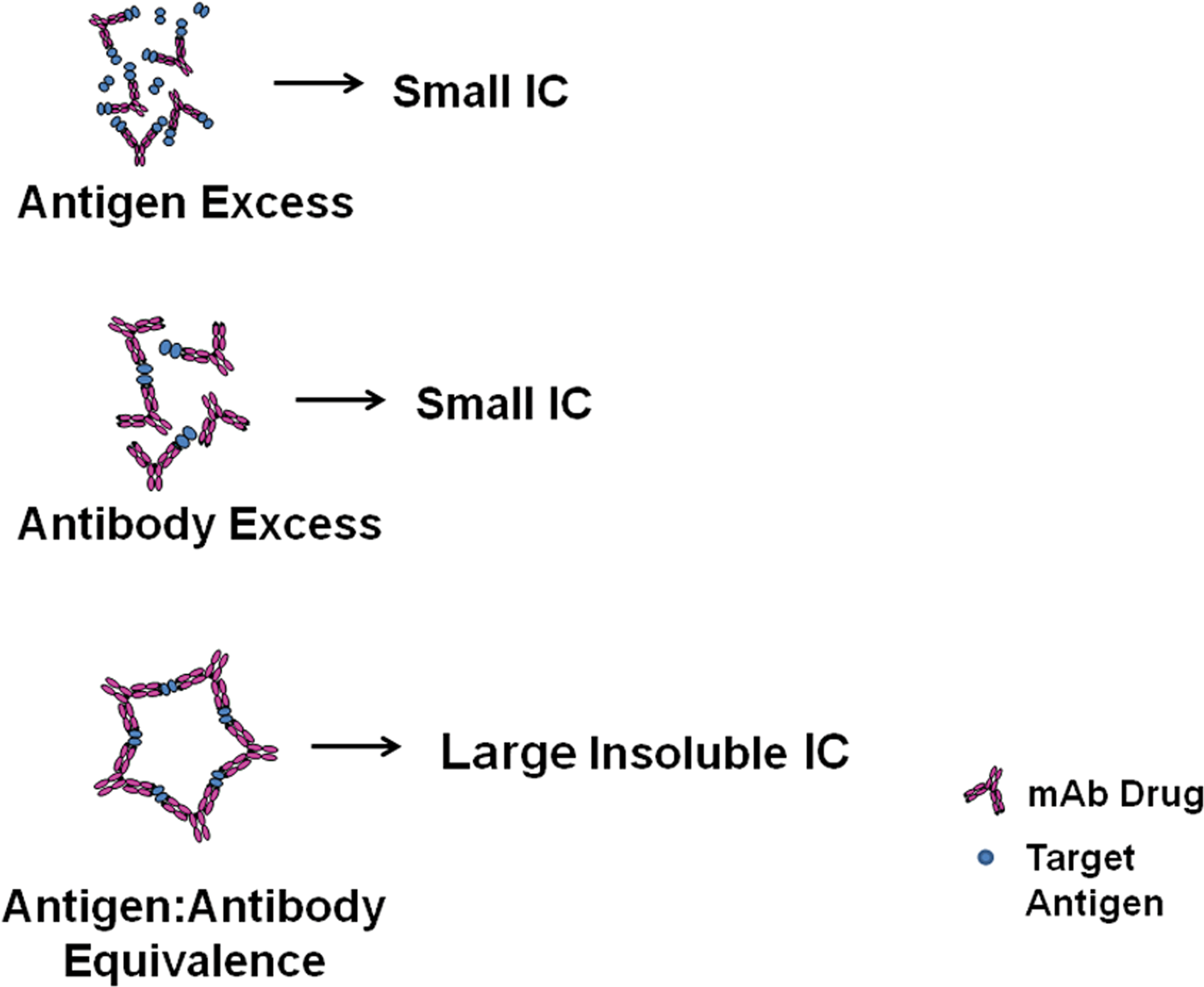
Schematic of IC lattice formation at different molar ratios of antigen and antibody. When antigen or antibody is in great excess, small soluble complexes form. When antigen and antibody are in molar equivalence, large, insoluble complexes form. As antigen/antibody ratios approach molar equivalence, ICs are larger but remain soluble. IC = immune complex.
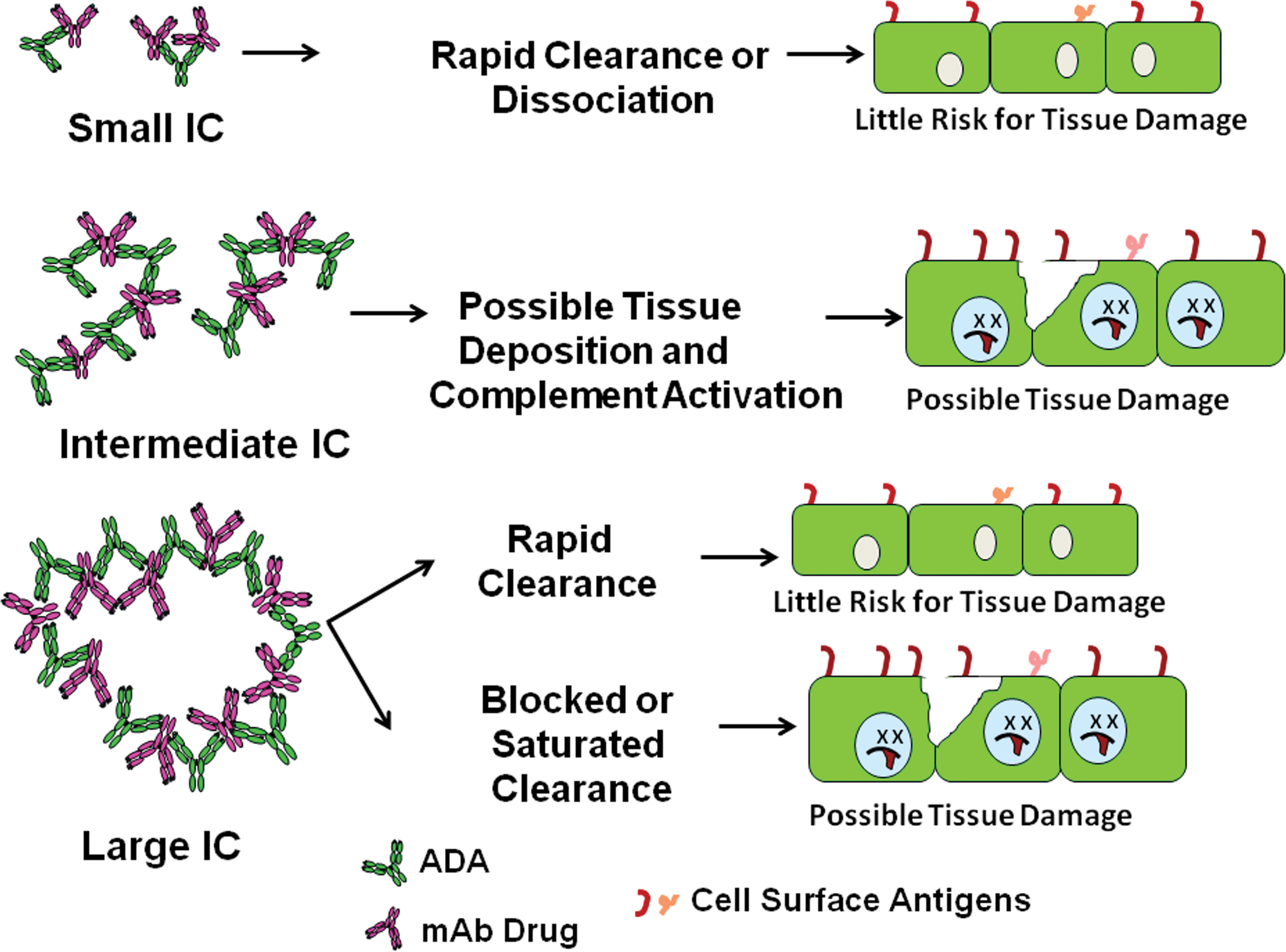
Schematic of effect of IC lattice formation on IC tissue deposition, tissue deposition, and pathogenicity. Small ICs, which may form under great antigen or antibody excess, generally remain in circulation or are rapidly cleared or dissociated and pose little risk for tissue damage. Intermediate-sized IC can deposit in tissue and activate complement. Large ICs are generally rapidly cleared through the phagocytic system, but when clearance mechanisms are blocked or saturated, large ICs can deposit in tissue, activate complement, and cause tissue damage. IC = immune complex.
Large versus Small IC Lattices
ICs with large lattices activate the classical complement pathway (CCP) efficiently, resulting in the attachment of C3b to the Fc region of the IC antibody component. In primates, C3b can then mediate the binding of IC to the complement receptor 1 (CR) on erythrocytes. Physiologic levels of erythrocyte-bound ICs are readily cleared by liver Kupffer cells or spleen macrophages via FcR and/or CR1. In rodents, the C3b immune adherence receptor for ICs is not CR1 or CR2 but appears to be membrane-bound factor H on rat platelets, neutrophils, and splenocytes and mouse neutrophils and platelets (Alexander et al. 2001; Quigg et al. 1997; Quigg and Holers 1995). Rodent platelets do express Fcγ receptors, but these do not appear to bind or transport ICs (Fernandez-Gallardo et al. 1989). In contrast, human and cynomolgus monkey platelets do not express C3b receptors but do express cellular receptors for the Fc region of the Ig molecule; γ indicates receptor for IgG Fc (FcγRII), which can participate in IC-mediated clearance by liver and spleen macrophages, particularly if erythrocyte CR1 mechanisms are saturated (Mahan et al. 1993).
ICs with large lattices can deposit in tissues or bind to FcγR on circulating or tissue neutrophils when FcR- or complement
Antigen and Antibody Concentration
The higher the antigen and antibody concentration/density, the greater the likelihood of antigen/antibody interaction, lattice initiation, IC formation, and potential for IC (-mediated) disease (ICD). IC formation can occur in circulation or locally (Mannik 1980). Thyroglobulin (a low
Charge
Antigen and antibody charge also affect IC lattice formation, complement activation, and tissue deposition (Gauthier and Mannik 1990; Michelin et al. 2002; Theofilopoulos and Dixon 1979; Wener 2007). Cationic antigens bind to anionic sites in glomerular basement membrane, knee joint, or dermoepidermal junction as shown by injection of antigen or preformed ICs into mice, rats, or rabbits (Adler et al. 1983; Border et al. 1981; Gallo, Caulin-Glaser, and Lamm 1981; Gauthier and Mannik 1990; Isaacs and Miller 1982; Joselow, Gown, and Mannik 1985; Theofilopoulos and Dixon 1979; van den Berg and van de Putte 1985; Wener 2007). Preformed ICs containing cationic antibodies may deposit in glomerular and/or small myocardial blood vessel walls with ICs containing neutrally charged antibodies remaining in circulation (Krant, Gauthier, and Mannik 1989).
Complement Activation
Lattice size (reviewed previously), antibody subclass, isotype, and other functional characteristics affect complement activation by IC as well as plasma and/or tissue IC clearance and tissue IC deposition (Lucisano Valim and Lachmann 1991). Antibodies may be precipitating or nonprecipitating and can compete among themselves for antigen to form ICs. In vitro analysis of precipitating and nonprecipitating mouse anti
In humans, IgG1- and IgG3-containing ICs readily activate the CCP (via C1q binding) over a wide range of antigen densities and antigen:antibody ratios, but do not activate the complement alternative pathway (CAP; Lucisano Valim and Lachmann 1991). IgG2-containing ICs can activate either the CCP or CAP, but only do so when formed under conditions of high antigen density and antigen:antibody equivalence or antibody excess (Lucisano Valim and Lachmann 1991). IgG4-containing ICs do not activate either the CCP or CAP (Lucisano Valim and Lachmann 1991). IgM-containing ICs do not activate CAP and activate the CCP only when formed under conditions of high antigen density and antigen:antibody equivalence or antibody excess. IgA-containing ICs activate the CAP over a wide range of antigen densities and antigen:antibody ratios, but do not activate the CCP (Lucisano Valim and Lachmann 1991). In other species, including nonhuman primates, the contribution of the antibody class and isotype to complement activation and IC formation may vary from that in humans.
After complement activation, C3b molecules attach to the Fc regions of ICs formed by all IgG isotypes, IgM and IgA, which facilitates binding by CR1 and subsequent clearance (see IC Clearance section subsequently), with one exception. IgG1- or IgG3-containing ICs formed in antigen excess are poorly cleared by CR1 (see IC Clearance section subsequently) and thus are available for tissue deposition and pathogenicity (Lucisano Valim and Lachmann 1991). All other factors being equal, ICs formed from high
In addition to C3b, C4b may deposit on IC via CR1-like adherence receptors. Small differences in Ig variable regions may affect the formation of ICs, complement activation, C3 and C4 cleavage, and C3b and/or C4b deposition on ICs (Yokoyama and Waxman 1993).
Antigen:antibody complexes that have not fixed complement are still reversible and can dissociate (Theofilopoulos and Dixon 1979). Complement fixation can break bonds in the IC lattice, thereby making the ICs smaller, more soluble, and more easily cleared (Frank and Hester 2009). Complement activation is greatest when ICs form in conditions of antibody excess and/or at equivalence and when antigen (epitope) density is high.
Relevant Human Clinical Experience
In a study of 71 patients treated with the anti-TNFα chimeric mAb infliximab for various chronic inflammatory diseases, 11 developed severe HSRs, and an additional 11 failed to respond to therapy (Vultaggio et al. 2010). Three of the 11 patients with severe HSRs developed IgE ADA and were skin prick positive to infliximab; a different 3 out of 11 patients with severe HSRs were IgE negative and skin prick negative but developed IgM ADA. Other ADA, characterized as nonisotype-specific, were evident in 8 out of 11 patients with severe HSRs and 2 11 patients who failed to respond to infliximab therapy. Evaluation of serial serum samples indicated that the ADA were demonstrable beginning at 2 to 3 weeks after the initial infusion and that ADA were present prior to clinical signs in the majority of the patients experiencing HSRs.
In a different study, 8% of 315 patients with inflammatory bowel disease experienced acute severe infusion reactions; 95% of reactors had IgG ADA but none had IgE ADA. Reactors were younger than nonreactors at the time of first infliximab treatment and more likely to have undergone episodic therapy (Steenholdt et al. 2011). Notably, infliximab-IgG anti-infliximab ICs were isolated from two rheumatoid arthritis patients with poor clinical responses; higher levels and larger sized complexes were isolated from the patient who also experienced a type 3 HSR postdosing (van der Laken et al. 2007).
In humans administered rh interferon β, ADA are primarily IgG1 and IgG4 and not IgM; however, most patient sera are tested after chronic administration of the drug. These ADA have been shown to form ICs with endogenous interferon β in vitro and activate complement (Sethu et al. 2013). The optimal antigen:antibody ratio for complex formation was 5:1, suggesting that the complexes formed best under antigen excess.
Nonimmune Aggregation of Ig
Nonimmune IgG aggregates can enhance immunogenicity or simulate ICs. Igs, including mAbs, can aggregate during production, formulation, compounding, or by exposure to silicone oil in plastic syringes (Kahook et al. 2010; Liu et al. 2011; Thirumangalathu et al. 2009). Aggregation is increased by heat, shear stress, and/or agitation and may be reduced by filtration (e.g., 0.2-µm filters remove insoluble but not soluble aggregates), inclusion of excipients and surfactants (e.g., polysorbate) and choice of vehicle (e.g., glycine may reduce aggregation; Lahlou et al. 2009; Vazquez-Rey and Lang 2011). While highly concentrated solutions generally are more likely to foster aggregation during dosing (Turner et al. 2011), some aggregates can form in less concentrated solutions (Vazquez-Rey and Lang 2011). Repackaging also may increase the risk for mAb aggregation by exposure to silicone oil particulate surfaces as has been shown for bevacizumb and ranibizumab (Kahook et al. 2010; Liu et al. 2011; Thirumangalathu et al. 2009). In mice, aggregation also can affect human IgG1 mAb biodistribution and immunogenicity with SC administration leading to persistence of aggregates in the injection site, faster clearance of aggregates after plasma uptake, and greater increases in cytokine levels compared with IV administration (Filipe et al. 2014). Similarly, aggregates in rh interferon β drugs were associated with increased induction of neutralizing ADA in both humans and transgenic mice with lessening of immunogenicity by inclusion of a surfactant (Rifkin et al. 2011). Finally, it is plausible that residual aggregates in mAb or IgG Fc fusion drug formulation could act as IC even without an ADA component. In dextran-treated mice with impaired Kupffer cell clearance, heat-aggregated human IgG is deposited in kidney glomerulus (Kimura et al. 1985). In rats, cocaine and morphine but not alcohol foster accumulation of human IgG aggregates in kidney glomerulus (Pan and Singhal 1994). Large, heat-aggregated human IgG aggregates have been demonstrated to activate human neutrophils in vitro (Gale et al. 1984).
IC Clearance
IC Clearance Mechanisms
IC clearance is biphasic in humans and monkeys. First, ICs are transferred from blood CR1 (or CR1-like) erythrocytes to FcγRII, FcγRIII, CR1, CR2 or CR4 (receptor for iC3b), or mannose receptors on liver and/or spleen phagocytes (Hepburn et al. 2006; Kavai and Szegedi 2007). Next, ICs are cleared from liver or spleen (Davies et al. 1990). Transfer of the ICs to liver or spleen phagocytes leads to IC destruction but generally occurs without harm to the erythrocytes that are recycled (Birmingham and Hebert 2001; Cosio et al. 1981; Cosio, Hebert, et al. 1987; Cosio, Sedmak, and Nahman 1990; Hebert, Birmingham, Mahan, et al. 1994; Hebert, Birmingham, Shen, et al. 1994; Reist et al. 1993).
IC clearance by erythrocytes is unique to primates, as described for humans, monkeys, chimpanzees, and baboons (Kimberly et al. 1989; Nielsen et al. 1994, 1997). In humans, the mean number of CR1 molecules is approximately 500 on erythrocytes, 20,000 on neutrophils, 21,000 on B lymphocytes, and 30,000 on monocytes (Birmingham and Hebert 2001). However, as there are many more erythrocytes in blood compared with leukocytes, ∼85% of the CR1 in primate blood are on erythrocytes. Nonprimate erythrocytes do not serve a similar IC shuttle function; however, platelets appear to provide a partial IC buffering function in nonprimates (Taylor et al. 1985). In mice, soluble ICs are removed by liver while particulate ICs may localize to spleen (Finbloom and Plotz 1979a, 1979b).
These clearance mechanisms apply to ICs formed by endogenous antibodies against self- (e.g., antithyroid) or nonself- (e.g., antistreptococcal) antigens as well as to antibodies formed after administration of a heterologous protein (e.g., bovine serum albumin [BSA], human mAb drug). Experimentally, cross
In humans and monkeys, more recently formed erythrocytes, including reticulocytes, have higher CR1 expression compared with mature or aged erythrocytes (Hebert et al. 1992; Lach-Trifilieff et al. 1999). Thus, stimulating erythropoiesis leads to increases in CR1 expression in cynomolgus monkeys, presumably facilitating increased IC clearance (Hebert et al. 1992). Decreased erythrocyte CR1 can also be demonstrated in humans with human immunodeficiency virus (HIV) infection, SLE, and increased CICs (Inada et al. 1986; Lach-Trifilieff et al. 1999). The transfer of ICs from erythrocytes to liver or spleen may be defective in SLE patients who have reduced FcγRII, FcγRIII, and mannose receptor expression on phagocytes (Kavai and Szegedi 2007).
Three Examples of IC Clearance Kinetics
IC clearance may be affected by IC composition, particularly when the antigen is also an antibody (thereby increasing lattice size and speed of clearance). In examples 1 and 2 (presented subsequently), a mAb drug was administered to humans (example 1) or monkeys (example 2), followed by a clearing antibody (simulating an ADA-like response). In example 3, the ICs were formed by the mAb drug complexing to its antigen in vivo (IgE).
In example 1, an 131I
In example 2, the kinetics of IC formation, distribution, and clearance were determined in monkeys by sequentially administering radiolabeled chimeric antihuman TNFα mAb infliximab followed by radiolabeled, purified monkey anti
Example 3 involves the therapeutic mAb omalizumab (Xolair™, rhuMAb-E25) whose intended mechanism is to form intravascular IC with endogenous Igs. Omalizumab is a humanized anti
Other Factors Affecting Clearance
IC clearance may be affected by the size of the ICs, and by alterations in immune function and/or complement activation. Human and/or monkey studies with preformed ICs indicate that very small ICs do not fix complement or bind to erythrocyte CR1. More than 90% of these soluble ICs are cleared in the liver; approximately 2 to 6% are cleared in the spleen. SLE patients have elevated levels of autoimmune antibodies and ICs and may have decreased C3/C4 concentrations due to persistent complement activation and consumption of these factors. In fact, innate complement deficiencies are associated with the development of SLE (Sturfelt and Truedsson 2005). Phase 1 (plasma to liver) clearance rates are comparable in healthy volunteers and SLE patients but phase 2 (clearance from liver) rates may be reduced in the latter (Davies et al. 1990, 2002; van der Woude et al. 1984). Serum IgG levels are inversely correlated with the rate of IC clearance, suggesting that FcR saturation might contribute to decreased clearance and decreased liver or splenic uptake, and increased tissue deposition in SLE, rheumatoid arthritis, or other IC
Figure 3 illustrates Fc-mediated clearance of IC by liver Kupffer cells; Figure 4 illustrates primate erythrocyte CR1-mediated clearance of IC by phagocytic cells, and Figure 5 illustrates tissue IC deposition and/or tissue destruction (detailed subsequently) which occurs when IC clearance mechanisms are saturated.
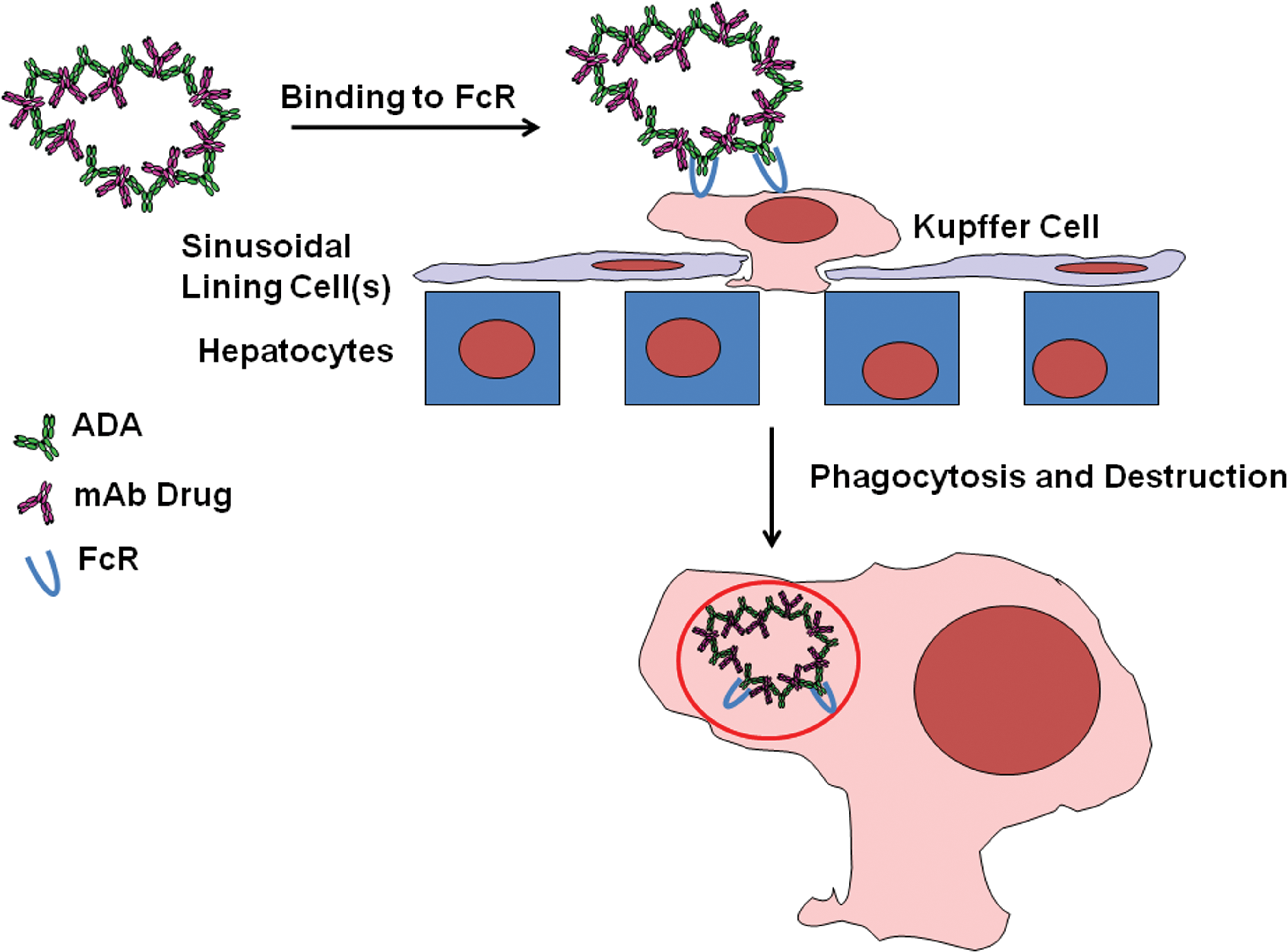
Schematic of Fc receptor-mediated IC clearance by liver Kupffer cells. IC = immune complex; Fc = constant region of immunoglobin molecule.
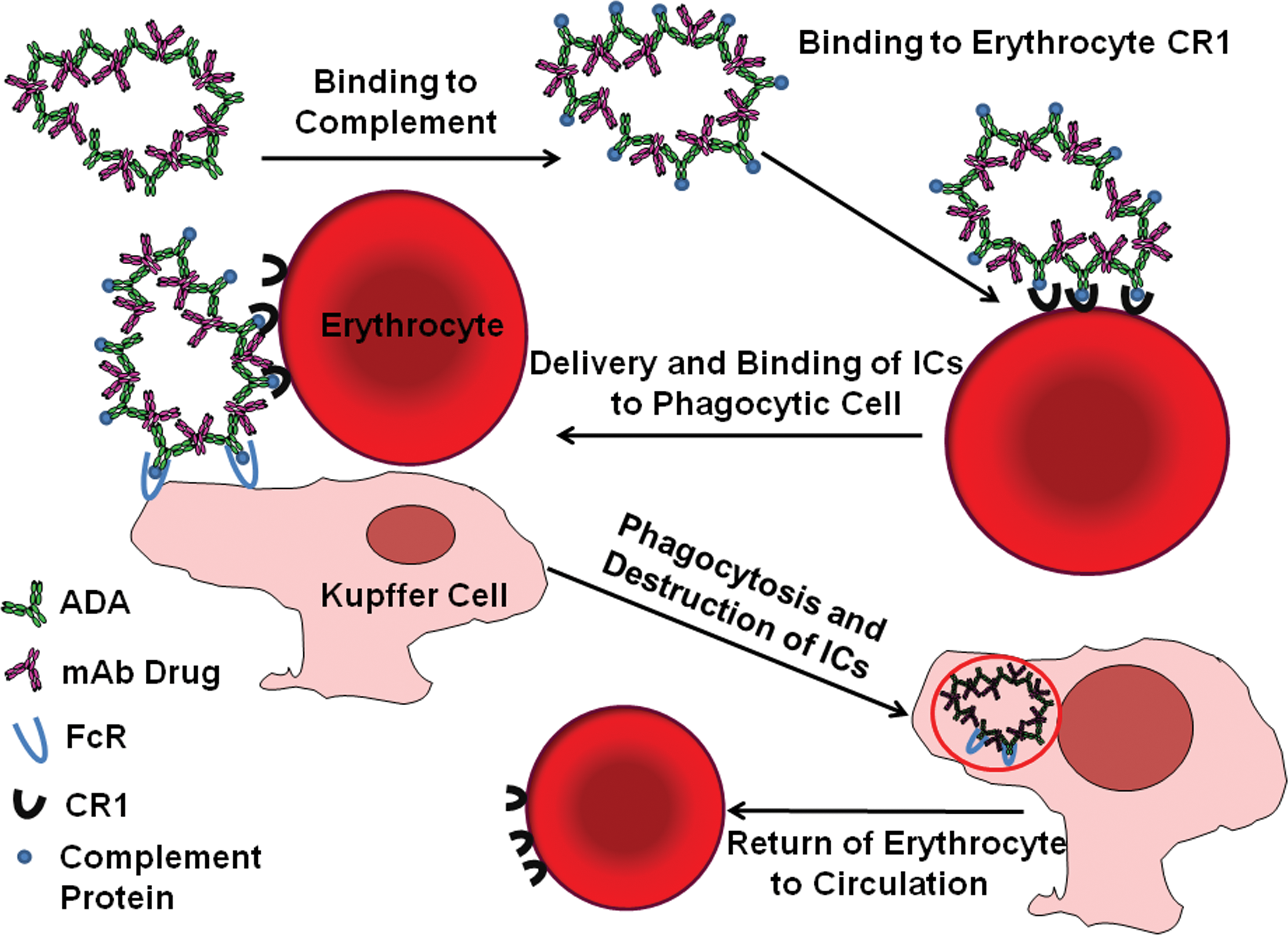
Schematic of primate erythrocyte CR1 (complement)-mediated IC clearance. IC = immune complex; CR1 = complement receptor 1, binds C3b component.
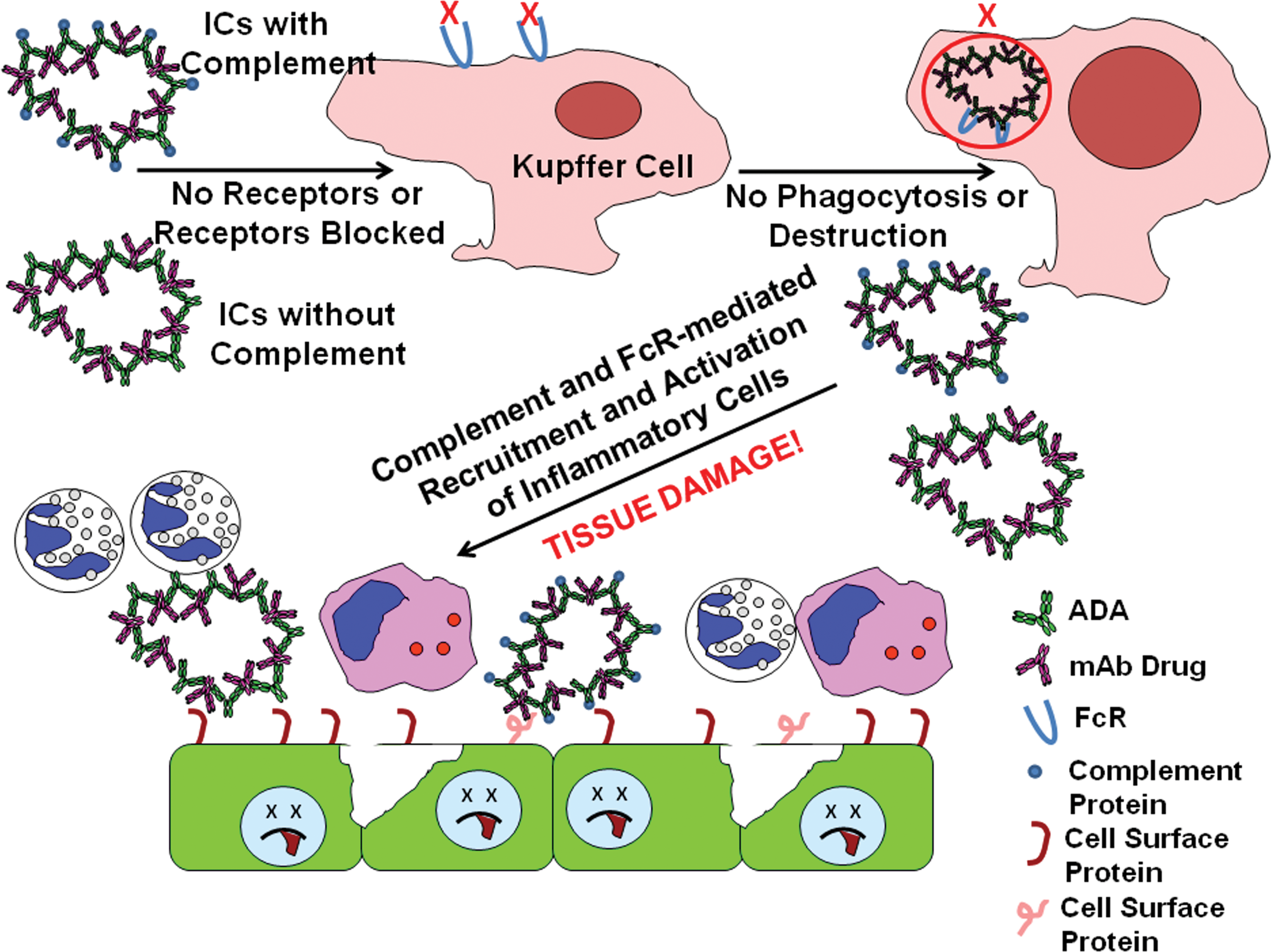
Schematic of saturation of IC clearance mechanisms, leading to tissue IC deposition and/or tissue damage/destruction. IC = immune complex.
Deposition and Pathogenicity of ICs: Complement Activation, Coagulation, and Release of Inflammatory Mediators
Deposition on Leukocytes (Neutrophils, Monocyte/Macrophages) and/or Platelets
ICs typically deposit on neutrophils, monocyte/macrophages, and/or platelets in experimental ICD (e.g., GN induced by repeated BSA or bovine gamma
ICs bind to FcγRIIa (CD32) on human neutrophils,monocytes/macrophages, and/or platelets, but the receptors are in greatest number on neutrophils (Cosio et al. 1981; Hart, Alexander, and Dransfield 2004). IC activation of neutrophils may require binding of the ICs to both FcγRII and FcγRIII (CD16; Brennan et al. 1991) and may be different for circulating (not yet primed) and migrating or tissue neutrophils (Wright et al. 2010). Binding of ICs to neutrophils and monocytes is inhibited by heat
Sequelae to Leukocyte Phagocytosis of ICs
Repeated infusion of heterologous BGG to cynomolgus monkeys leads to IC formation, phagocytosis by leukocytes, and classical pathway complement activation, with decreases (∼25%) in plasma C3 and C4 levels, increases in plasma C3a levels, marked (>50–75%) reductions in neutrophils and monocyte/macrophages and limited (∼13%) reductions in platelets, with sequestration/uptake of IC
In rabbits, neutrophil phagocytosis of ICs is a feature of ferritin–anti
Infusion Reactions and Generalized Type 3 HSRs
Experimental studies have shown that IC
The ratio of antigen to antibody also affects the rate of IC formation and the severity of the associated reactions. In one series of studies (Birmingham and Hebert 2001; Camussi et al. 1980; Cats, Lafeber, and Klein 1975; Davies et al. 1990), monkeys were dosed for 8 weeks on Monday through Friday (5 days) with an amount of BGG calculated to be stoichiometrically equivalent to the anti
Studies of repeated
One report indicates that mAb-associated infusion reactions in monkeys are mediated by complement recognition of and binding to key amino acids in the mAb Fc region, thus contributing to CDC, although these amino acids are not needed to maintain ADCC functions (Tawara et al. 2008). Alterations in mAb Fc sequence (i.e., substitution of serine for proline at position 331 and substitution of alanine for lysine at position 322) that reduce CDC also reduce the risk for mAb
In humans, the role of ICs is not well understood for mAb infusion reactions but IgG ADA responses, ICs, and type 3 HSRs may contribute to serum sickness
The term “first
Other mAb or drug infusion reactions do not appear to be mediated by ICs/type 3 HSRs and may be caused by IgE
Tissue Deposition
In monkeys, glomerular disease with proteinuria generally develops within 5 to 8 weeks of daily BGG infusion at a dose of 5 mg/kg or greater and coincides with reduced CR1 expression by erythrocytes and reduced IC clearance (Cosio, Hebert, et al. 1987; Cosio, Birmingham, et al. 1987; Hebert et al. 1991; Hebert, Birmingham, Shen, et al. 1994). Glomerular disease and/or coronary arterial lesions also are described in BSA
Studies in mice have included single- or multiple-dose infusion of preformed complexes into naive mice or infusion of antigen into sensitized mice. After a single dose, the ICs initially localize to neutrophils, Kupffer cells, and sinusoidal lining cells in liver; neutrophils, macrophages, and sinusoidal lining cells in spleen; and neutrophils, macrophages, and endothelium in alveolar capillaries or larger vessels in lung. In kidney, ICs were found in glomerulus (capillary walls, intercapillary spaces, and basement membrane) but also were observed in walls of afferent arterioles, peritubular capillaries, tubular epithelium, and tubular lumens (Mellors and Brzosko 1962). After 48 hr, the ICs were found primarily associated with germinal center cells in spleen with limited glomerular deposition in kidney (Mellors and Brzosko 1962). If multiple doses of preformed ICs were infused and followed by hyperimmune serum, anaphylaxis was evident and IC deposits were observed to occlude glomerular or alveolar capillary lumens. Microthrombi were present and associated with ICs phagocytosed by neutrophils and macrophages (Mellors and Brzosko 1962). Notably, studies in Fc receptor
IHC Correlates of IC Deposition and Pathogenicity in Monkeys
Over the past 6 years, several of the current authors (Rojko, Price, and Raymond, unpublished) have performed retrospective IHC evaluation of paraffin tissues from monkeys that experienced IC
The IHC correlate of IC formation and deposition is the presence of granular deposits containing antibody and/or complement components as well as antigen. The IHC for the case studies presented subsequently was conducted using immunoperoxidase stains on paraffin sections because of their improved morphology compared with cryosections and excellent sensitivity for the Ig and complement stains performed (Chowdhury et al. 2005; Molne, Breimer, and Svalander 2005; Yamashina et al. 1989). In paraffin sections, granular deposits typically range from approximately 0.5 to 3 µm in diameter; the lower size limit is estimated based on the limit of resolution of light microscopy, while the higher size limit is estimated by comparison to platelets (2–3 µm in diameter). The upper size limit is consistent with the formation of aggregates of ICs. Granular deposits are distinguished from leakage and precipitation of proteinaceous material (presumably due to formalin fixation and/or desiccation during processing) by the following criteria: Leaked/precipitated protein is evident as variably stained, sometimes smudgy material centered around vessels, or associated with inflammation/hemorrhage with decreasing concentration gradients and alignment along connective tissue fibers; Granular deposits are discrete, sometimes aggregated, usually densely stained material either intravascular or dispersed extracellularly in a nonconcentration gradient in blood vessel walls, granular deposits are observed in intima and/or media, often at the internal elastic lamina (boundary between intima and media) or vessel branch points; in kidney glomeruli, granular deposits may be observed in mesangial, subepithelial, and/or subendothelial locations. Figure 6 illustrates glomerular granular deposits containing monkey IgM from a monkey with spontaneous (nondrug-associated) glomerulopathy; granular deposits also are deposited in other sites, including interstitium; granular deposits may be associated with erythrocytes (as part of IC clearance) or platelets and/or demonstrated at the surface of, or phagocytosed by neutrophils, monocytes/macrophages, and/or Kupffer cells; other intravascular granular deposits may appear as globular intravascular microthrombi. Granular deposits contain Ig and/or complement components. When a staining method is available, the granular deposits often can be shown to contain antigen (e.g., administered drug).
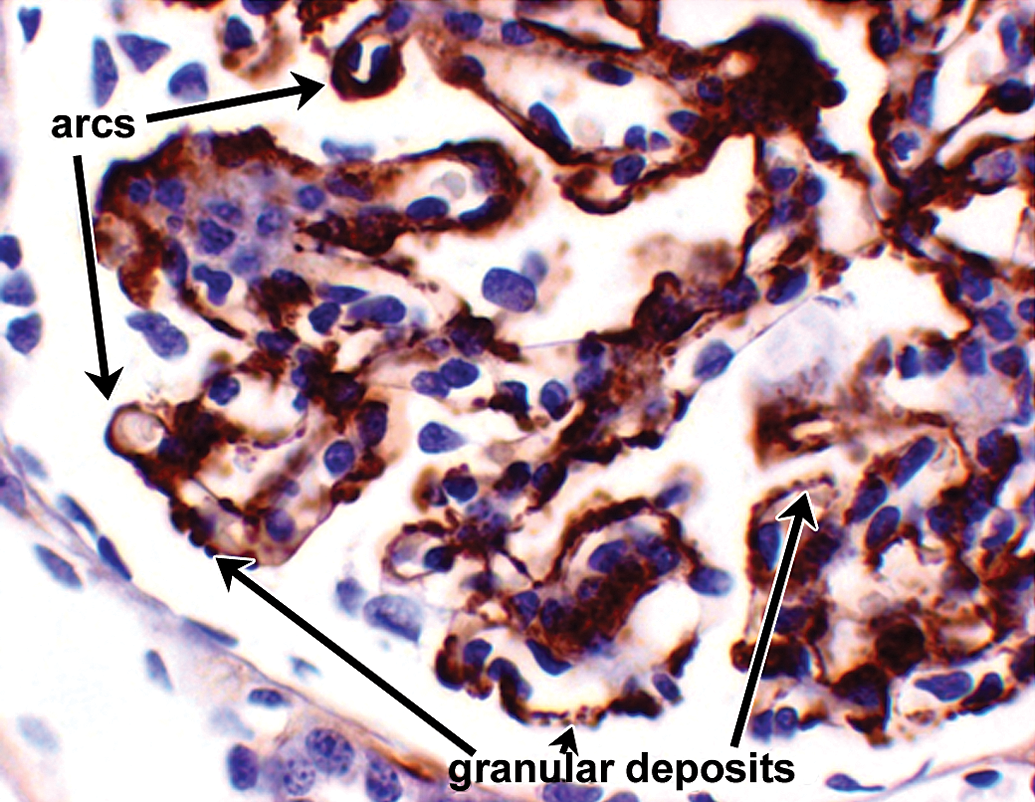
Example of granular deposits in kidney glomerulus revealed by IHC staining for monkey IgM. Extensive granular deposits observed in subepithelial, subendothelial, and mesangial locations. Some of the granular deposits are identified by arrows, a double row of granular deposits (indicative of subepithelial and subendothelial deposits) lies above the short arrow. When granular deposits become confluent, they form arc shapes (arrows). 60×. This was an example of spontaneous glomerulopathy in a control cynomolgus monkey. Other photographs from this same example are presented in Figures 15 to 19. IHC = immunohistochemistry.
Key IHC findings are categorized by the types of IC Systemic or generalized drug–antidrug IC formation associated with type 3 HSRs involving systemic complement activation may have granular deposits in the following locations: intravascular proteinaceous material (CICs) or in perivascular interstitium; intravascular (sometimes sequestrated) and tissue monocyte/macrophages and/or neutrophils; may include drug- or IC-coated platelets; liver Kupffer cells and/or sinusoidal lining cells; spleen or lymph node macrophages and/or sinusoidal lining cells; dendritic cells in various tissues. Systemic or localized type 3 HSRs, drug–antidrug IC formation, and localized deposition in kidney may have granular deposits in the following locations: mesangium, subepithelial, and/or subendothelial locations in kidney glomerulus; tubular epithelium and/or basement membrane; interstitium, either extracellular or in macrophages. Localized drug–antidrug IC formation and localized deposition in blood vessel walls, and localized (Arthus intima and/or media of small to medium perivascular interstitium, either extracellular or in macrophages. Localized type 3 HSRs, drug–antidrug IC formation, and localized deposition in interstitium, joints, heart valves, choroid plexus, or skin may have granular deposits in interstitium, macrophages, dendritic cells, or other cell types. Limited localized drug–antidrug IC formation without HSR occurrence/clearance by liver Kupffer cells, spleen, or other tissue macrophages and/or dendritic cells. IC do not form or are below the limits of IHC detection and no IC
Thus, our observations of IHC staining patterns in tissues of monkeys administered heterologous therapeutic proteins are consistent with the patterns predicted by experimental studies of IC
Case Studies
In order to better understand the range of findings associated with IC formation in toxicity testing of therapeutic proteins or mAbs, a variety of case studies are presented in which IHC was used to demonstrate IC. These case studies are not comprehensive in scope but rather are intended to illustrate various findings associated with generalized or localized IC formation during toxicity testing with therapeutic proteins or mAbs in primates and rodents. Other studies are presented in which IC were not evident. Taken together, these case studies are intended to illustrate the challenges associated with demonstrating IC
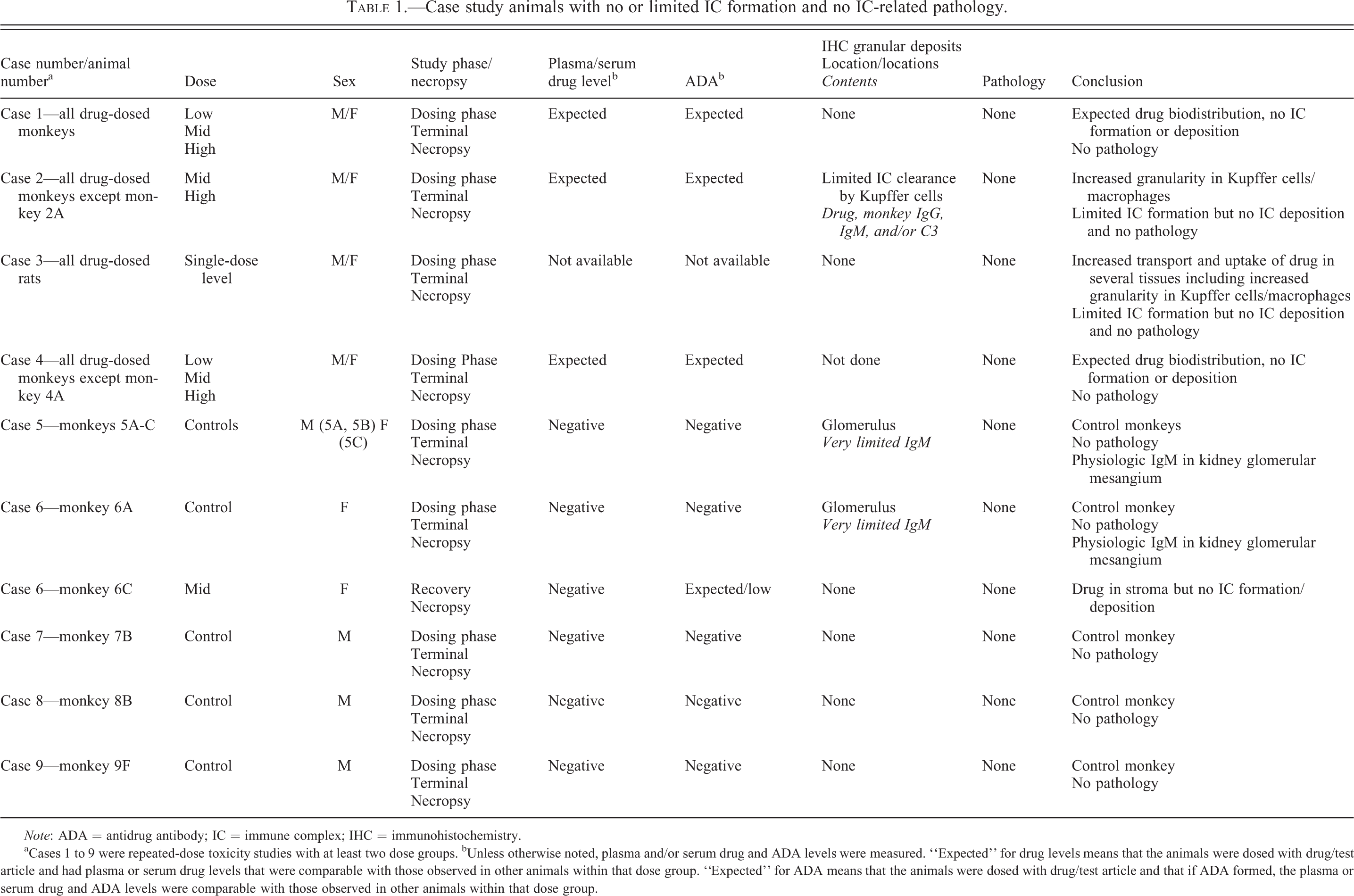
Case study animals with no or limited IC formation and no IC-related pathology.
Note: ADA = antidrug antibody; IC = immune complex; IHC = immunohistochemistry.
aCases 1 to 9 were repeated-dose toxicity studies with at least two dose groups. bUnless otherwise noted, plasma and/or serum drug and ADA levels were measured. “Expected” for drug levels means that the animals were dosed with drug/test article and had plasma or serum drug levels that were comparable with those observed in other animals within that dose group. “Expected” for ADA means that the animals were dosed with drug/test article and that if ADA formed, the plasma or serum drug and ADA levels were comparable with those observed in other animals within that dose group.
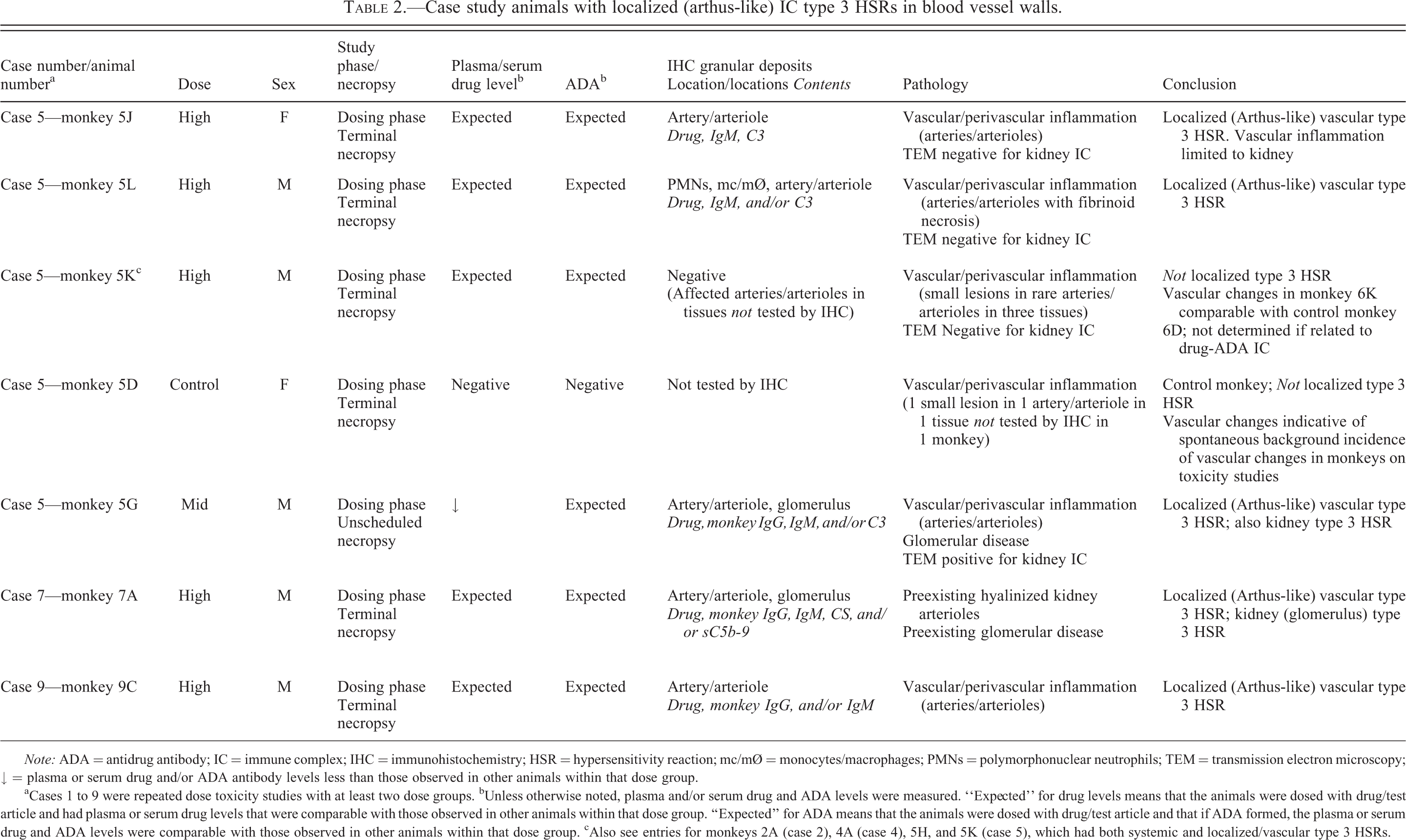
Case study animals with localized (arthus-like) IC type 3 HSRs in blood vessel walls.
Note: ADA = antidrug antibody; IC = immune complex; IHC = immunohistochemistry; HSR = hypersensitivity reaction; mc/mØ = monocytes/macrophages; PMNs = polymorphonuclear neutrophils; TEM = transmission electron microscopy; ↓ = plasma or serum drug and/or ADA antibody levels less than those observed in other animals within that dose group.
aCases 1 to 9 were repeated dose toxicity studies with at least two dose groups. bUnless otherwise noted, plasma and/or serum drug and ADA levels were measured. “Expected” for drug levels means that the animals were dosed with drug/test article and had plasma or serum drug levels that were comparable with those observed in other animals within that dose group. “Expected” for ADA means that the animals were dosed with drug/test article and that if ADA formed, the plasma or serum drug and ADA levels were comparable with those observed in other animals within that dose group. cAlso see entries for monkeys 2A (case 2), 4A (case 4), 5H, and 5K (case 5), which had both systemic and localized/vascular type 3 HSRs.
Case study animals with systemic or localized (Arthus-like) IC type 3 HSRs and IC deposition in kidneys.

Note: ADA = antidrug antibody; IC = immune complex; IHC = immunohistochemistry; HSR = hypersensitivity reaction; TEM = transmission electron microscopy; ↓ = plasma or serum drug and/or ADA antibody levels less than those observed in other animals within that dose group.
aCases 1 to 9 were repeated dose toxicity studies with at least two dose groups. bUnless otherwise noted, plasma and/or serum drug and ADA levels were measured. “Expected” for drug levels means that the animals were dosed with drug/test article and had plasma or serum drug levels that were comparable with those observed in other animals within that dose group. “Expected” for ADA means that the animals were dosed with drug/test article and that if ADA formed, the plasma or serum drug and ADA levels were comparable with those observed in other animals within that dose group. cAlso see entries for monkeys 4A (case 4), 5E, 5F, 5H, and 5I (case 5), 6B, and 6E (case 6), which had both systemic and kidney type 3 HSRs. In case 6, overall incidence of glomerular pathology was 5/10 at low dose, 7/10 at high dose, and 9/10 at high dose.
Case study animals with variable drug exposure and/or biodistribution and systemic IC type 3 HSRs/infusion reactions.
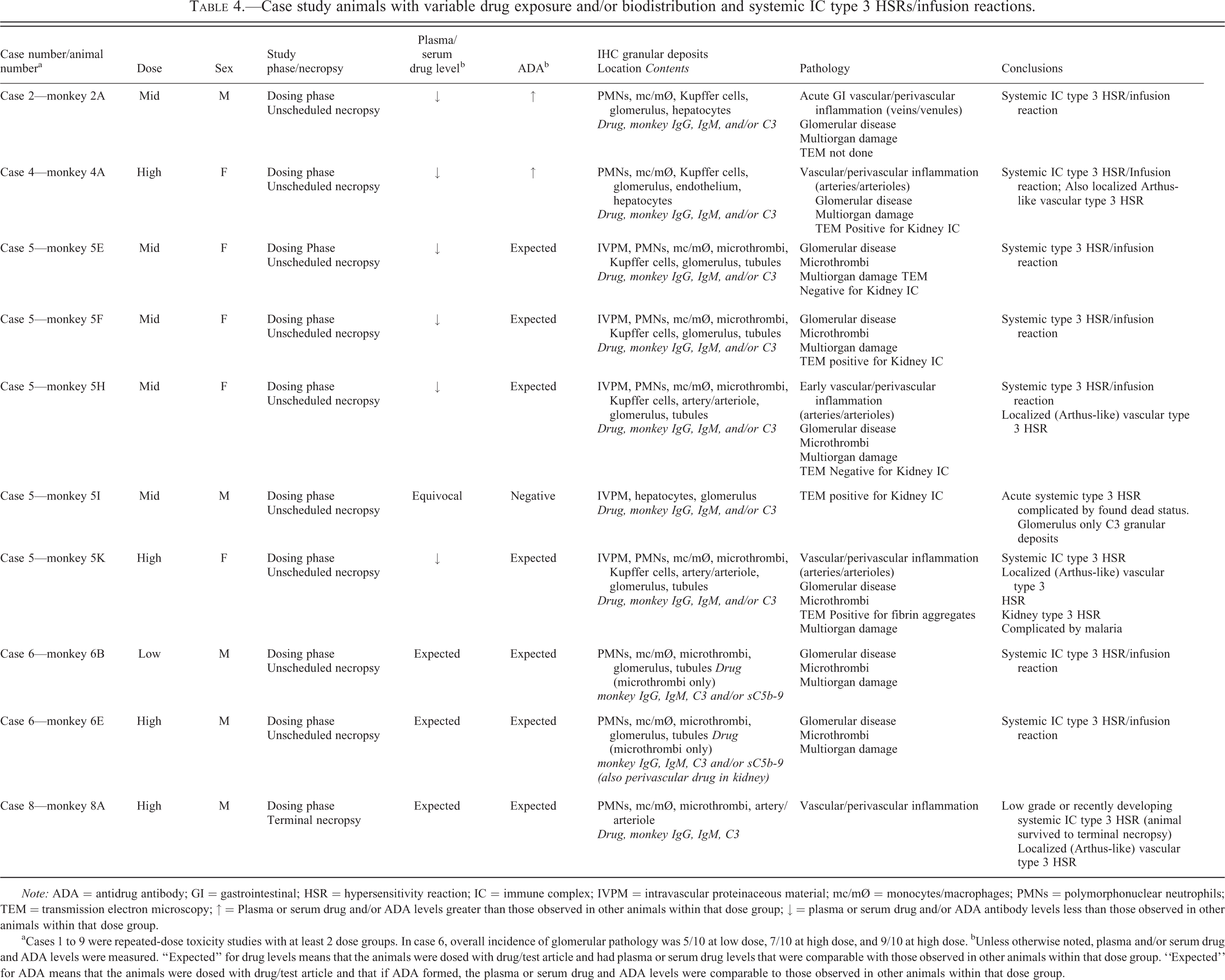
Note: ADA = antidrug antibody; GI = gastrointestinal; HSR = hypersensitivity reaction; IC = immune complex; IVPM = intravascular proteinaceous material; mc/mØ = monocytes/macrophages; PMNs = polymorphonuclear neutrophils; TEM = transmission electron microscopy; ↑ = Plasma or serum drug and/or ADA levels greater than those observed in other animals within that dose group; ↓ = plasma or serum drug and/or ADA antibody levels less than those observed in other animals within that dose group.
aCases 1 to 9 were repeated-dose toxicity studies with at least 2 dose groups. In case 6, overall incidence of glomerular pathology was 5/10 at low dose, 7/10 at high dose, and 9/10 at high dose. bUnless otherwise noted, plasma and/or serum drug and ADA levels were measured. “Expected” for drug levels means that the animals were dosed with drug/test article and had plasma or serum drug levels that were comparable with those observed in other animals within that dose group. “Expected” for ADA means that the animals were dosed with drug/test article and that if ADA formed, the plasma or serum drug and ADA levels were comparable to those observed in other animals within that dose group.
Case 1: IC Formation or Deposition Does Not Always Occur or Is Not Demonstrated
A therapeutic mAb drug was administered to cynomolgus monkeys at various doses using a repeated dose schedule. There were no clinical, clinical pathology, or histopathology findings. Low ADA levels were evident in dosed monkeys. IHC staining for drug (human IgG) and endogenous monkey IgG was performed on multiple tissues from both control and drug-dosed animals. Within these tissues, both endogenous monkey IgG and drug were localized in cell types and regions consistent with expected physiologic distribution, uptake, transport, and clearance for IgG (e.g., intravascular and interstitial locations). As expected, the therapeutic mAb drug localization was a subset of the endogenous monkey IgG in each tissue. There were no drug or monkey IgG
Case 2: IC-mediated Postdosing Reaction, Acute Venular Inflammation, and Multiorgan Damage in a Single Study Monkey; Limited IC Clearance in Other Dosed Study Monkeys
A therapeutic mAb drug was administered to cynomolgus monkeys IV at various doses using a once-weekly, repeated dose schedule. In one mid
In the remaining dosed animals at terminal necropsy (designated as all dosed monkeys except 2A in Table 1), there was evidence of limited IC clearance in Kupffer cells (increased IHC granularity with human [mAb] IgG, monkey IgG, IgM, and/or C3 stains) but no clinical signs, no histopathology, and no IHC evidence of IC deposition in tissues or bound to/phagocytosed by circulating/sequestered neutrophils. Thus, the IHC results supported the drug findings from the tissue cross
Case 3: Transient Exaggerated Physiologic Response to Human IgG in Rats without Demonstrated IC Formation
Rats were administered two doses of vehicle, therapeutic human mAb drug, or polyclonal human IgG at one dose level. IHC staining for human IgG (to detect drug or polyclonal human IgG) and endogenous rat IgG was performed on multiple tissues. There were no histopathology findings and no evidence of IC deposition in either treatment group. Administration of either drug or polyclonal human IgG was associated with increased IgG transport and uptake in several tissues, including kidney, liver, and choroid plexus, but IC-related granular tissue deposits were not observed. These findings were expected physiologic findings given the dose level and schedule. The increased IgG transport and uptake was judged to reflect a transient exaggerated physiologic response to the exogenously administered heterologous (human) IgG (either drug or polyclonal human IgG). These data indicated that IgG transport and clearance mechanisms can be saturated but that saturation of IgG clearance mechanisms did not, in this case, result in pathogenic IC formation or deposition.
Case 4: IC-mediated Postdosing Reaction, Thrombocytopenia, Tissue Leukocytosis, and Fibrin Thrombi in a Single Study Monkey
A therapeutic mAb (drug) was administered by IV injection twice weekly to cynomolgus monkeys. One dosed female (animal 4A in Table 4) became moribund on day 29 of the dosing phase approximately 6 hr after the 8th dose and was euthanized. Blood samples collected on day 25 (prior to the 7th dose) were below the limit of quantitation for drug but had increased ADA levels compared with other high-dose animals. Hematology and clinical pathology parameters evaluated in day 22 predose blood samples were comparable with control study animals; however, a diagnostic sample collected postdosing revealed decreases in platelets, total protein, albumin, albumin:globulin ratio, and serum electrolytes (Ca, P, Na, K, Cl) as well as increases in serum triglycerides.
In kidney, histopathology revealed eosinophilic globular to fibrillar deposits in glomerular capillaries and small medullary vessels. Phosphotungstic acid hematoxylin (PTAH) staining confirmed the presence of fibrin thrombi in some glomerular capillaries. Other microscopic findings in kidney included slight perivascular neutrophilic infiltrates, slight tubular dilation, and fine cytoplasmic vacuolation of proximal tubules. In lung, slight leukocytosis, minimal acute inflammation, and minimal hemorrhage were evident. In liver, changes included moderate leukocytosis and marked vascular inflammation in a single blood vessel near the liver hilus.
Kidney, lung, spleen, and liver tissues were stained for drug (human IgG) as well as endogenous monkey IgG, IgM, IgA, and complement (C3, membrane attack complex [terminal components of complement] sC5b-9) components. IC-related granular deposits consistent with a type 3 HSR were observed in multiple tissue elements, including: Granular deposits primarily containing drug with lesser amounts of monkey IgG, IgM, and/or C3; intravascular and tissue neutrophils, monocyte/macrophages, and liver Kupffer cells; endothelium of small vessels (e.g., liver sinusoids, kidney glomerular and peritubular capillaries, spleen marginal zone); intima and media at branch points of the inflamed vessel in the liver hilus (IgM predominated with lesser amounts of drug), associated with endothelial vacuolation; rare mesangial, subepithelial, and/or subendothelial locations within kidney glomerular tuft); Granular deposits containing IgM only; intima/media of a single artery with vascular/perivascular inflammation; hepatocytes in liver.
By IHC, hepatocytes also had increased cytoplasmic globules containing monkey IgG, IgM, IgA, and C3 but not drug. IHC also revealed increases in monkey IgG and C3 staining of the proximal tubular vacuoles, consistent with increased resorption, likely following glomerular loss.
These findings were interpreted to represent systemic formation of IC in circulation with adherence to, and uptake by, phagocytic cells (neutrophils, monocyte/macrophages, liver Kupffer cells) and deposition on endothelium of small vessels and intima and/or media of a few larger vessels as well as deposition on kidney glomerulus and hepatocytes. The drug predominance likely indicates that the IC formed under conditions of antigen excess (with monkey ADA forming the antibody component and drug forming the antigen component). However, since the drug is itself an antibody, this might have contributed to formation of the large (insoluble) complexes that were more typical of those formed under antibody excess conditions.
Case 5: Systemic and/or Localized Type 3 HSRs with Vascular/Perivascular Inflammation in Multiple Study Monkeys
Cynomolgus monkeys were administered various IV doses of a therapeutic mAb drug on a repeating dose schedule in a six-month dosing/two-month recovery study. Several unscheduled death (animals 5E–5I, 5K in Tables 2–4) and terminal necropsy (animals 5J, 5L) dosing phase monkeys developed evidence of possible type 3 HSRs; there was no clear relationship to dose level. Clinical pathology for the early sacrifice animals (including animal 5I who was found dead) included mild to moderate decreases in albumin and lower platelet counts and red cell mass. Histopathology revealed the following alterations:
Minimal to moderate vascular perivascular/inflammation in small arteries or large arterioles, often with fibrinoid necrosis of the vessel wall, in kidney (5E, 5G, 5H, 5L), liver (5G, 5K, 5L); recanalized thrombus in lung (5L). Periodic acid–Schiff stain (PAS)-positive thickening of the glomerular basement membrane in kidney (5E, 5F, 5G, 5H, 5I). Minimal congestion in lung (5E). Moderate diffuse hepatocytic vacuolation (5I). Slight medial hypertrophy in blood vessels in lung (5I) and spleen (5G).
IHC staining was performed for drug (human IgG), monkey IgG, IgM, IgA, C3, and SC5b
Case 6: IC-mediated Disease in Dosing Phase and Recovery Animals
Cynomolgus monkeys were administered various doses of an rh protein drug in a repeated dose once
Glomerular disease without thrombosis was observed in a few other dosing or recovery phase animals; the incidence was dose dependent, but the severity was dose independent. Animal 6D was a recovery mid
Case 7: IC-mediated Preexisting Glomerular Disease Unrelated to Dose or Treatment
The drug was a human mAb administered SC once weekly to cynomolgus monkeys for a total of five doses. A single-dosed animal (7A in Tables 2–3) had preexisting renal disease as evidenced by increased blood urea nitrogen and serum creatinine levels and proteinuria prior to dose. The animal survived to terminal sacrifice. Histopathology and IHC revealed hyalinized cortical and afferent/efferent arterioles with corresponding IgM
Case 8: Primary IC-mediated Vascular/Perivascular Inflammation and Secondary Renal Cortical Necrosis; IC-mediated Choroid Plexus Inflammation
The drug was a human mAb administered IV once weekly to cynomolgus monkeys for a total of five doses. In a single high-dose terminal necropsy monkey (8A in Table 4), histopathology revealed vascular/perivascular inflammation in kidney and choroid plexus (brain) as well as thrombosis/cortical necrosis in kidney. IC
Other histologic changes (e.g., renal cortical necrosis, basophilic tubules, proteinaceous cast formation) and IHC findings (e.g., granular deposits in tubular epithelial cells/lumens) in kidney were considered secondary to the primary IC-mediated vascular/perivascular inflammation and subsequent ischemia or glomerular protein loss and reuptake by proximal tubules.
Case 9: IC-associated Macrophage Sequestration in Multiple Tissues
The drug was a human mAb administered SC to cynomolgus monkeys for a total of five doses. Histopathology revealed increased large mononuclear cells sequestered in blood vessel lumens or perivascular interstitium in lung, liver, spleen, and/or kidney in five dosed animals (low-dose monkey 9A, mid-dose monkey 9B, high-dose monkeys 9C, 9D, 9E). IHC staining identified the sequestered cells as CD68-positive macrophage lineage cells with increased drug and monkey IgG staining and granularity at the membrane and prominent membrane/cytoplasmic processes consistent with activation. Although well-defined granular deposits were not demonstrated, it was likely that drug-ADA ICs were involved in the clustering of the leukocytes in blood vessel lumens. This interpretation was based on (1) colocalization of drug with monkey IgG, (2) increases in staining at cell margins/membrane, (3) increases in staining in aggregated macrophages, (4) increases in staining associated with platelet and erythrocyte fragments, and (5) lack of similar histopathology and IHC findings in a control animal (monkey 9F).
Two high-dose animals (monkeys 9C, 9D) each had perivascular and/or vascular inflammation in one or more tissues. IHC evaluation revealed IC deposition in the wall of the inflamed vessels, with IC composed of drug, monkey IgG, and/or IgM. Monkey 9C also had sinusoidal cell hypertrophy in liver. IHC staining revealed increased drug, monkey IgG, and IgM granularity but no well-formed granular deposits in enlarged Kupffer cells. This staining pattern was likely indicative of increased IC turnover. Thus, the constellation of histopathology and IHC findings in these dosed animals was consistent with an IC-mediated pathogenesis rather than a direct drug effect.
Discussion
The case studies illustrate that a weight of evidence approach should be taken to understand the role of IC Plasma and/or serum drug and ADA levels; Histopathology characteristic of IC electron microscopic evaluation of selected tissues (e.g., kidney for glomerular disease); Hematology including erythrocyte numbers and morphology, platelet numbers; Other information (CIC, CH50, or complement component levels) may also be useful but often is not available; Expected (target) and unexpected (offtarget) epitope/protein receptor distribution; tissue cross IHC evaluation for granular deposits or atypical distribution of drug, endogenous IgG, IgM, C3, sC5b
In preclinical studies, IHC evaluation of tissue IC is particularly useful, as staining can be performed retrospectively on paraffin sections from the toxicity study. IHC staining may demonstrate that drug does or does not colocalize with granular deposits of endogenous IgG, IgM, IgA, C3, and sC5b
The case studies present a variety of situations in which IHC staining was performed to evaluate IC deposition in monkey or rat tissues following administration of rh protein or mAb drugs. Different information was available from each case, and different animals within each case had different outcomes, based on their propensity to form/deposit pathogenic ICs in tissue. Furthermore, the IC
The information from cases 1 to 9 mentioned previously can be analyzed on an individual as well as study group basis (Tables 1–4).
IC Do Not Form or Are below the Limits of IHC Detection and No IC-related Pathology Occurs
Case 1 (all mAb
Control (undosed) monkeys also have limited IC formation/deposition although very small amounts of granular IgM or sC5b
Limited Localized Drug-ADA IC Formation/Clearance by Liver Kupffer Cells, Spleen or Other Tissue Macrophages, and/or Dendritic Cells
The formation and deposition of IC depend on the balance between drug and ADA response (Table 1). The mAb
In other instances (not included in the above-listed cases), some of us (Rojko, Price, and Raymond, unpublished) have observed limited granular deposits containing drug and other IC components in small numbers of macrophage or dendritic cells in spleen, lymph node, joint, aorta, or various subepithelial sites, sometimes in association with phagocytosed erythrocytes or platelets. As target epitope/receptor was not evident in the same cells in undosed monkey tissues, these findings were interpreted as clearance of drug via IC. Drug-ADA IC clearance also likely contributed to the sinusoidal (Kupffer) cell hypertrophy in monkey 9C from case 9. Limited IC deposition in blood vessel walls is described at the end of the next section.
Localized Drug-ADA IC Formation and Localized Deposition in Blood Vessel Walls, and Localized (Arthus-like) Type 3 HSRs
In monkeys with vascular/perivascular inflammation, IHC staining often demonstrates granular deposits associated with intima and/or media of small arteries and/or arterioles (or less often small veins/venules), primarily in affected vessels (Table 2; Figures 7
–14). Often these IC
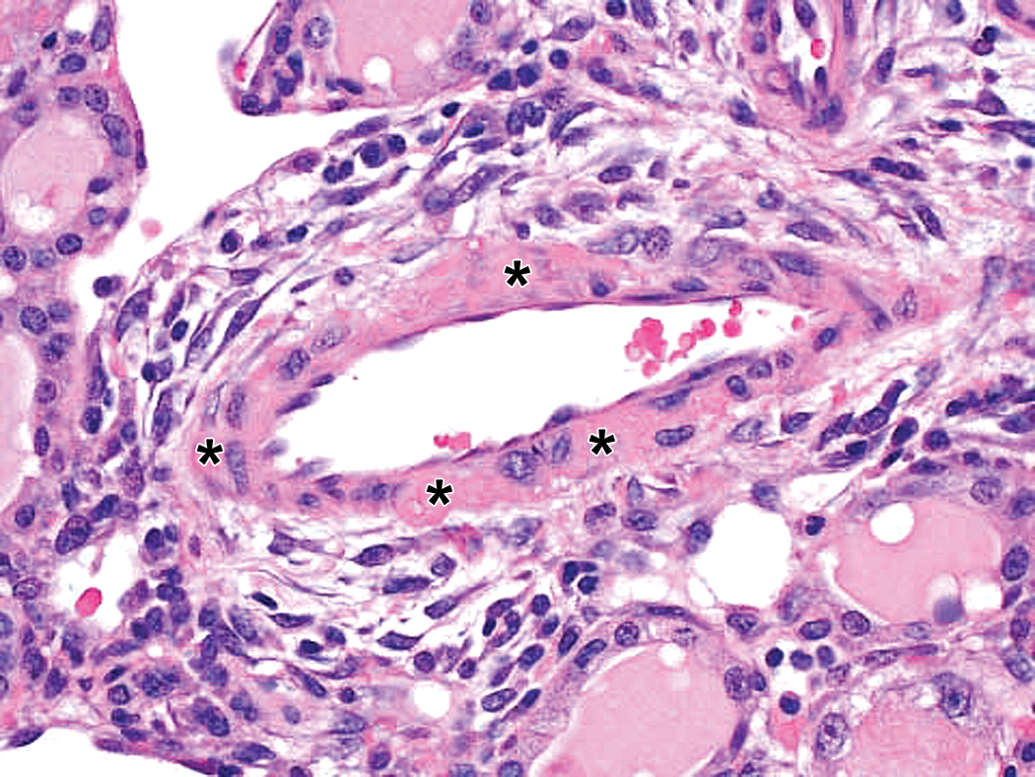
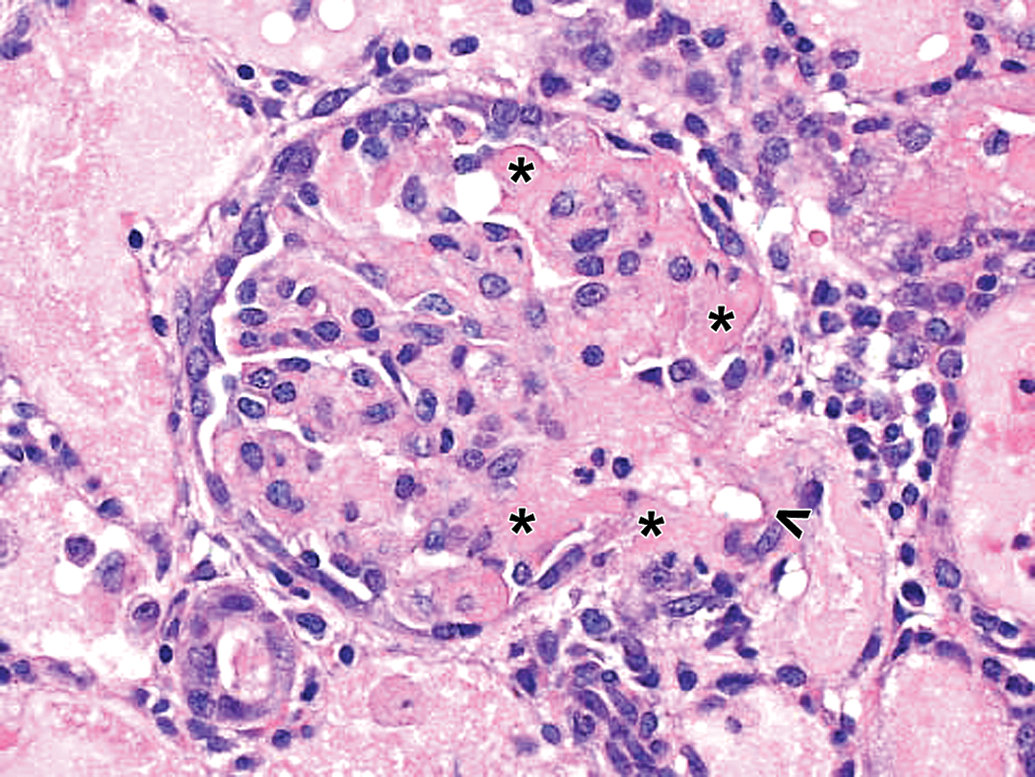
Drug-ADA-associated vascular/perivascular inflammation in kidney in a cynomolgus monkey. Hematoxylin and eosin (H&E) stain reveals fibrinoid/hyaline change (asterisks) in a small artery/large arteriole with mononuclear cells in outer media and adventitia. 40×. ADA = antidrug antibody.
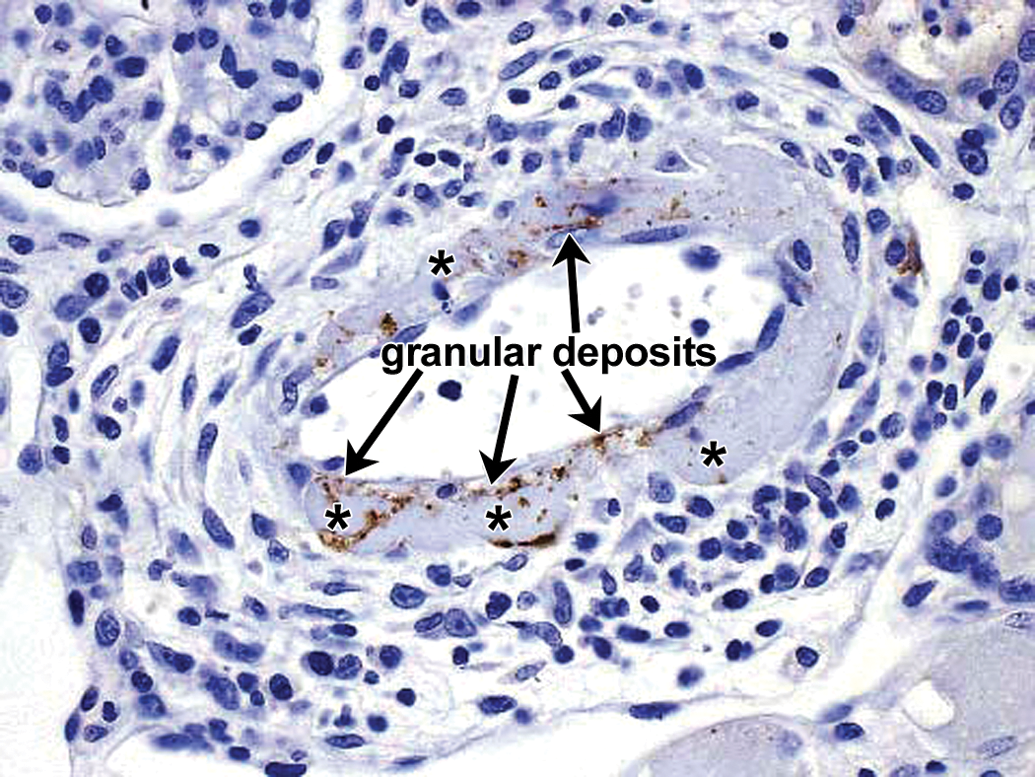
Drug-ADA-associated vascular/perivascular inflammation in kidney in a cynomolgus monkey. IHC staining for drug reveals fine-bore granular deposits containing drug and located in intima and media. Arrows indicate granular deposits, many at the internal elastic lamina. Asterisks indicate areas of smudginess in the vessel wall which correspond to areas of fibrinoid/hyaline change demonstrated in the hematoxylin and eosin (H&E) section (Figure 7). A linear array at the lower left of the vessel is suggestive of a branch point (located adjacent to the lower left asterisk). This location was confirmed to represent a branch point in serial sections. 40×. ADA = antidrug antibody; IHC = immunohistochemistry.
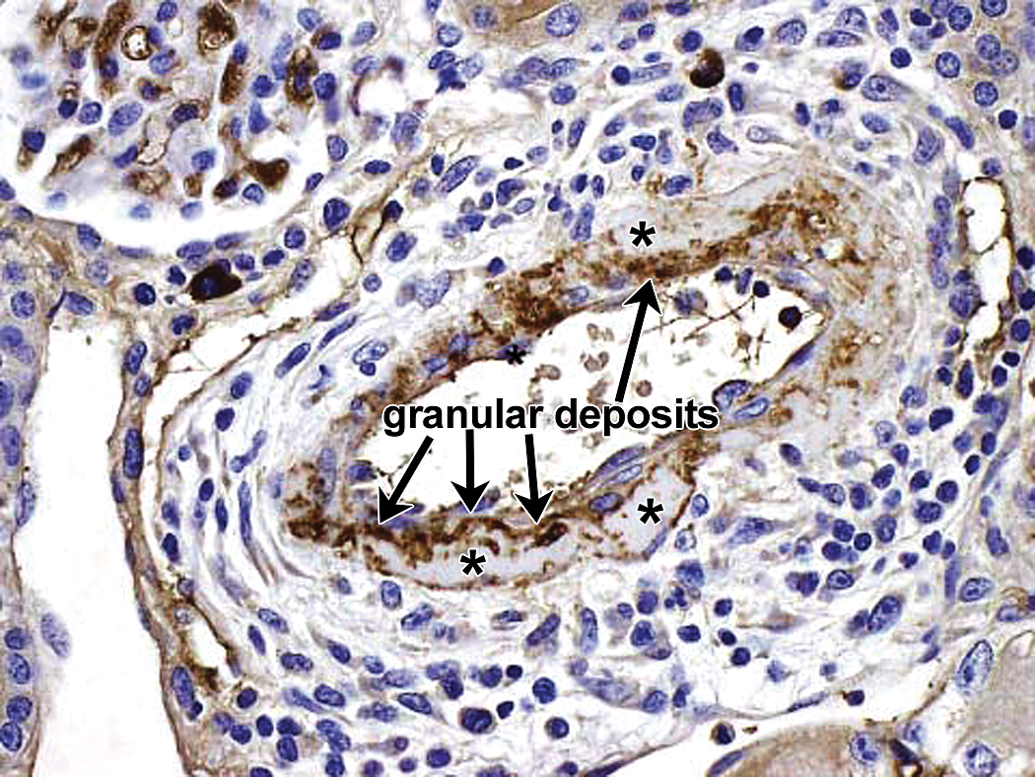
Drug-ADA-associated vascular/perivascular inflammation in kidney in a cynomolgus monkey. IHC staining for monkey IgM reveals fine-bore granular deposits containing drug and located in intima and media which colocalize with the drug-containing granular deposits in Figure 8. Arrows indicate granular deposits at the internal elastic lamina. Asterisks indicate areas of smudginess in the vessel wall, which correspond to areas of fibrinoid/hyaline change demonstrated in the hematoxylin and eosin (H&E) section (Figure 7). 40×. ADA = antidrug antibody; IHC = immunohistochemistry.
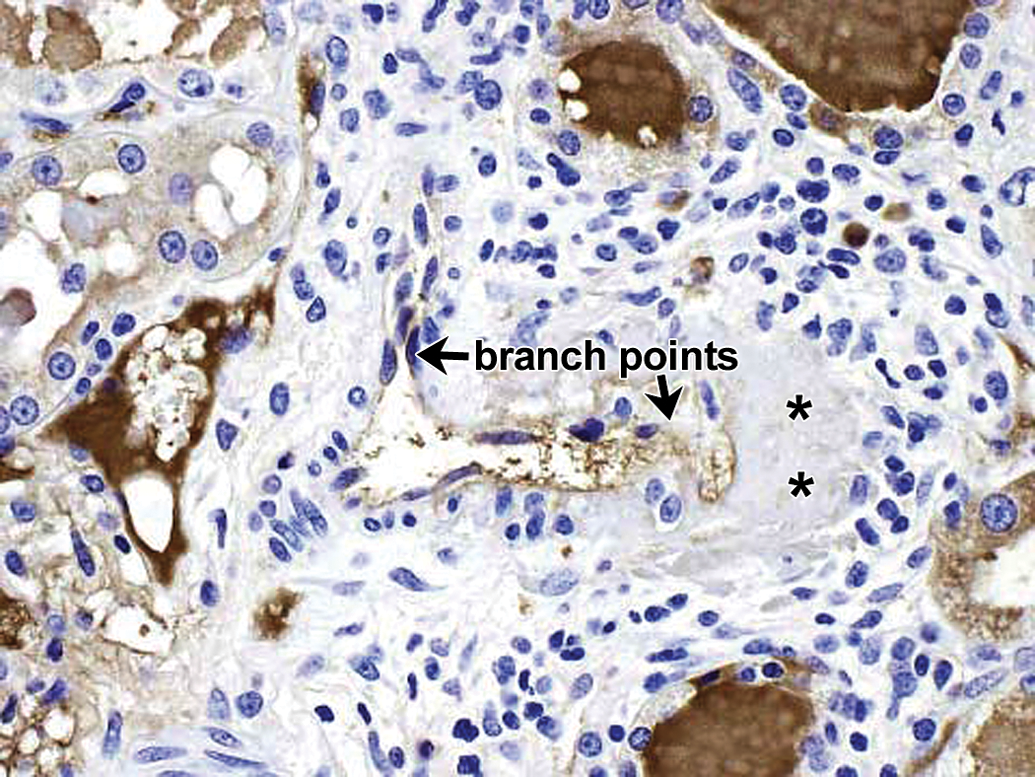
Drug-ADA-associated vascular/perivascular inflammation in kidney in a cynomolgus monkey. IHC staining for monkey IgG reveals little contribution of IgG to the IC-related granular deposits in vascular intima and media in this location in this animal (compare to Figures 8–9). Vascular branch points are evident in upper left and lower right of vessel. Asterisks indicate areas of smudginess in the vessel wall, which correspond to areas of fibrinoid/hyaline change demonstrated by hematoxylin and eosin (H&E). Increased monkey IgG staining in proximal tubular lumens with uptake (vesicular staining) by proximal tubular epithelium secondary to increased glomerular IgG loss in this monkey. 40×. ADA = antidrug antibody; IHC = immunohistochemistry.
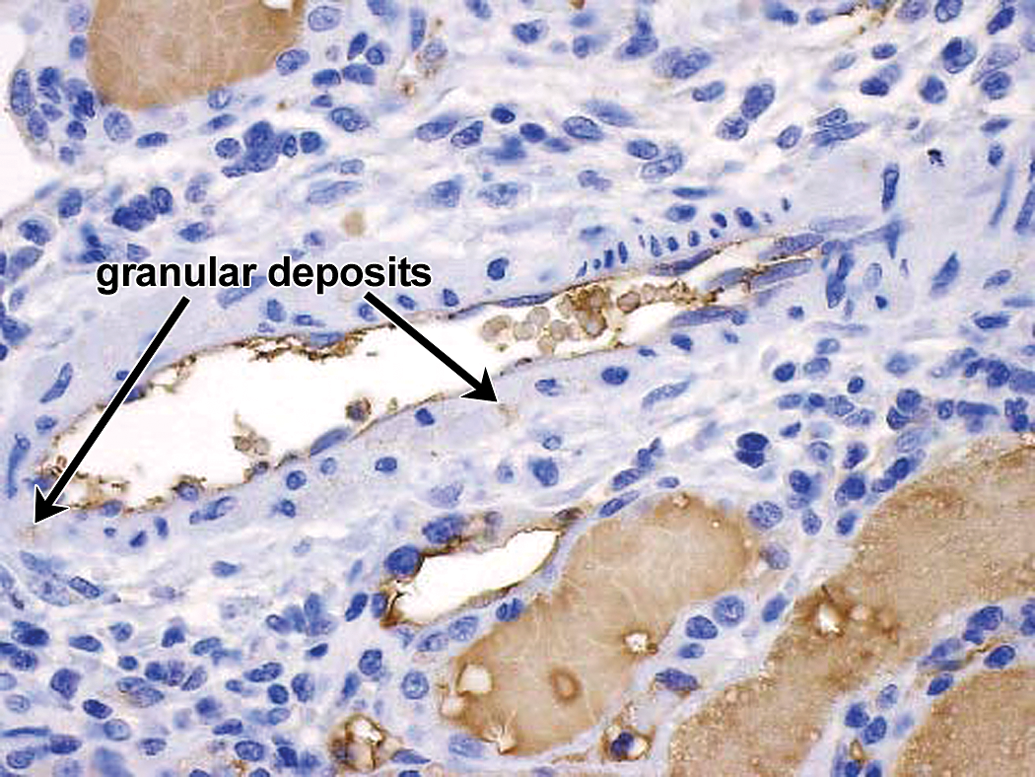
Drug-ADA-associated vascular/perivascular inflammation in kidney in a cynomolgus monkey. IHC staining for monkey C3 reveals fine-bore granular deposits at similar locations (arrows) as the drug and IgM-containing granular deposits, albeit very few in this photograph. Increased C3 staining in proximal tubular lumens with uptake (vesicular staining) by proximal tubular epithelium secondary to increased glomerular C3 loss in this monkey. 40×. ADA = antidrug antibody; IHC = immunohistochemistry.
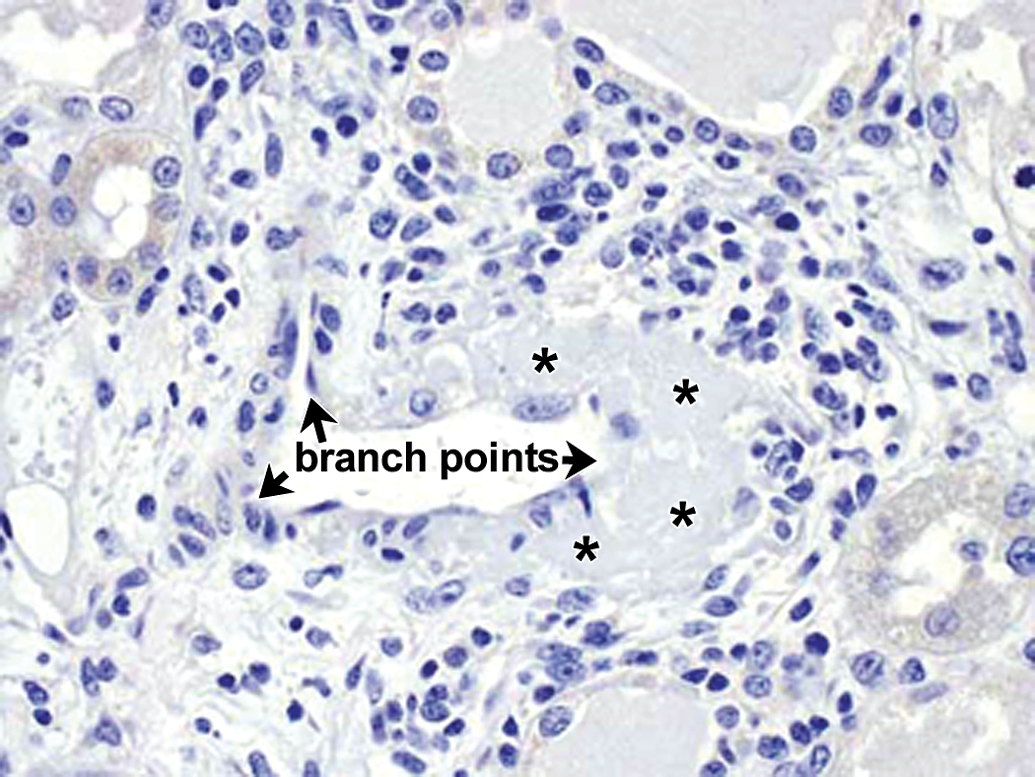
No staining of affected renal vessel with negative control antibody. Arrows identify vascular branch points evident at upper left, lower left, and lower right; asterisks identify smudginess in vascular wall. 40×.
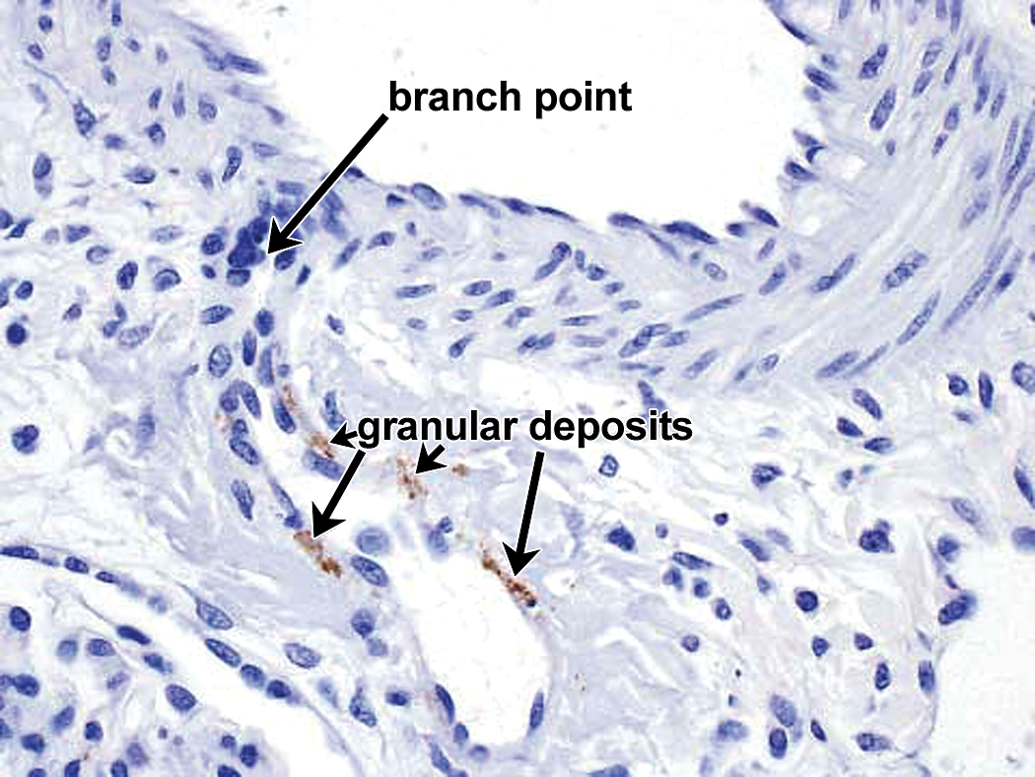
Drug-ADA-associated vascular/perivascular inflammation in nearby vessel in same kidney in same cynomolgus monkey as depicted in Figures 7 to 12. IHC staining for drug reveals fine-bore granular deposits containing drug and located in internal elastic lamina at vascular branch point (arrows). 40×. ADA = antidrug antibody; IHC = immunohistochemistry.
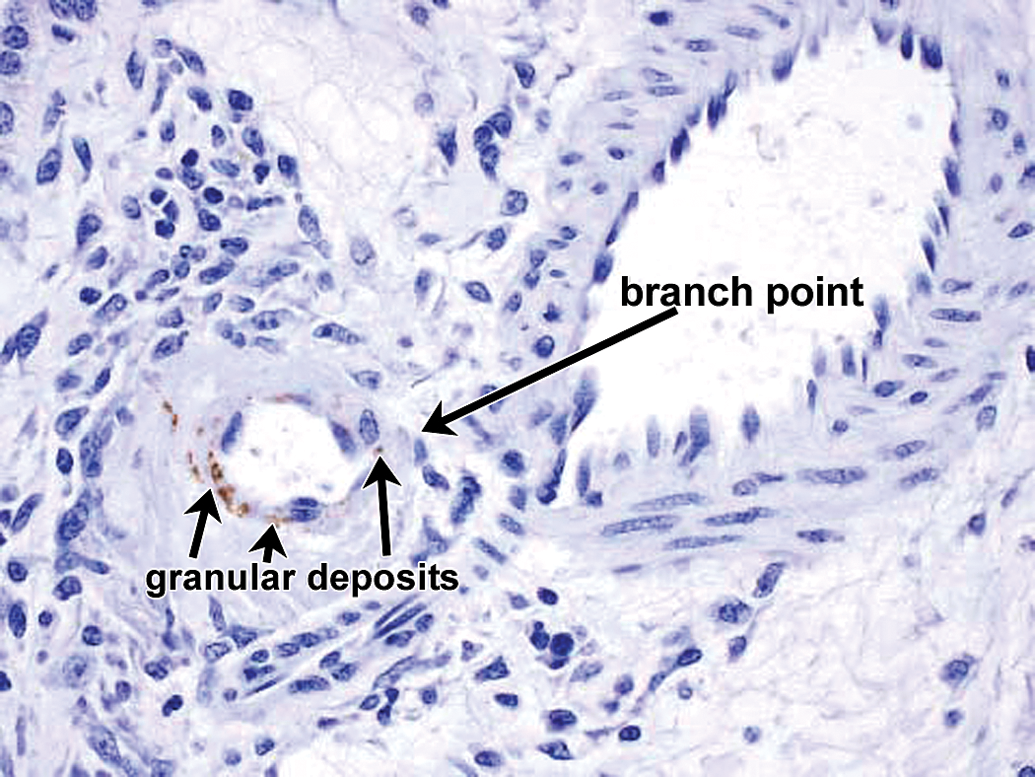
Drug-ADA-associated vascular/perivascular inflammation in nearby vessel in same kidney in same cynomolgus monkey as depicted in Figures 7 to 12; branch point different from that in Figure 13. IHC staining for drug reveals fine-bore granular deposits containing drug and located in internal elastic lamina at adjacent vascular branch point (branch point not contiguous in section). 40×. ADA = antidrug antibody; IHC = immunohistochemistry.
The histologic features and IHC staining patterns in monkey vessels with vascular/perivascular inflammation are consistent with IC
In case 5, two high
Finally, one or more small foci of vascular/perivascular inflammation can be evident in rare undosed monkeys on toxicity studies and have been attributed to low
Systemic or Localized Drug-ADA IC Formation and Localized Deposition in Kidney
Case 5 monkey 5G and case 6 monkey 6D provide examples of relatively localized kidney (Table 3) IC deposition (drug in interstitium but not glomeruli; Tables 3–4; Figures 15
–36). Other examples of kidney IC deposition in association with systemic IC formation and/or deposition are found in cases 2, 4, 5, 6, and 7 (Table 4). In these monkeys, the glomerular change is usually diagnosed as membranous and/or membranoproliferative glomerulopathy or GN although other diagnoses may be more descriptive and refer to thickening of the basement membrane and/or mesangial matrix. TEM may demonstrate subepithelial, subendothelial, and/or mesangial electron-dense deposits (e.g, monkeys 5F, 5G from case 5 listed as “TEM-positive for IC” in Tables 3–4). However, in many monkeys with early or limited (nonkidney) IC disease, the glomerular change is not uniform and the TEM samples may be negative (e.g., monkeys 5E, 5H, 5J, 5K, and 5L from case 5 listed as “TEM-negative for IC” in Table 3), particularly if the electron microscope (EM) sample does not contain any affected glomeruli. In monkeys with IC
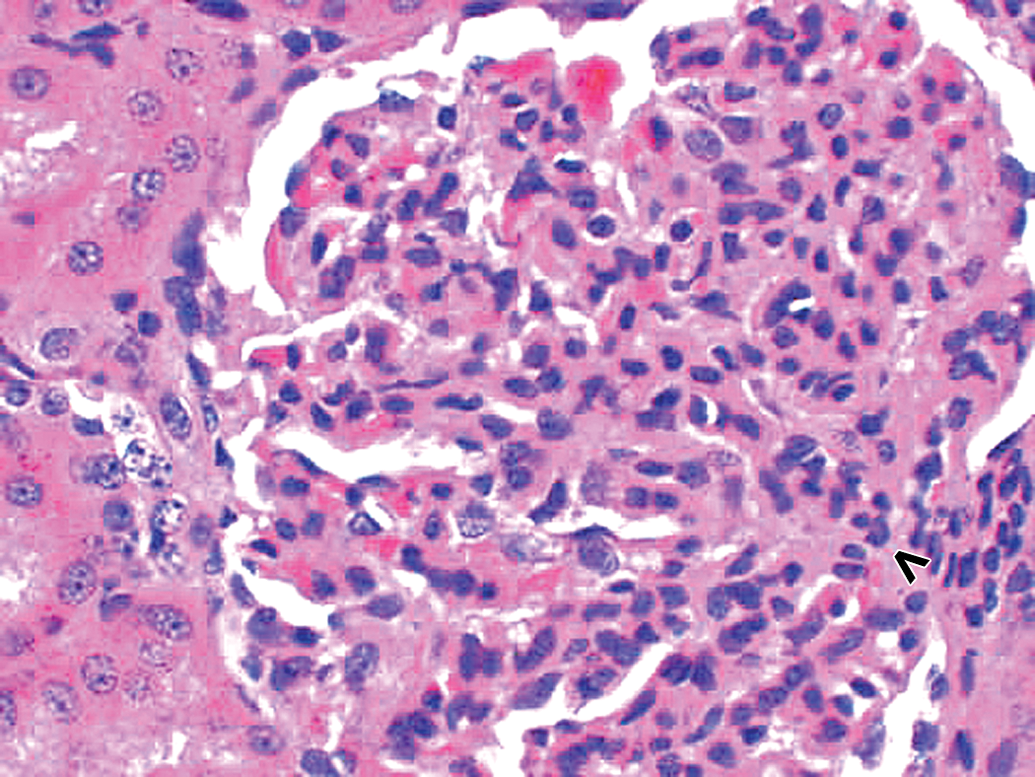
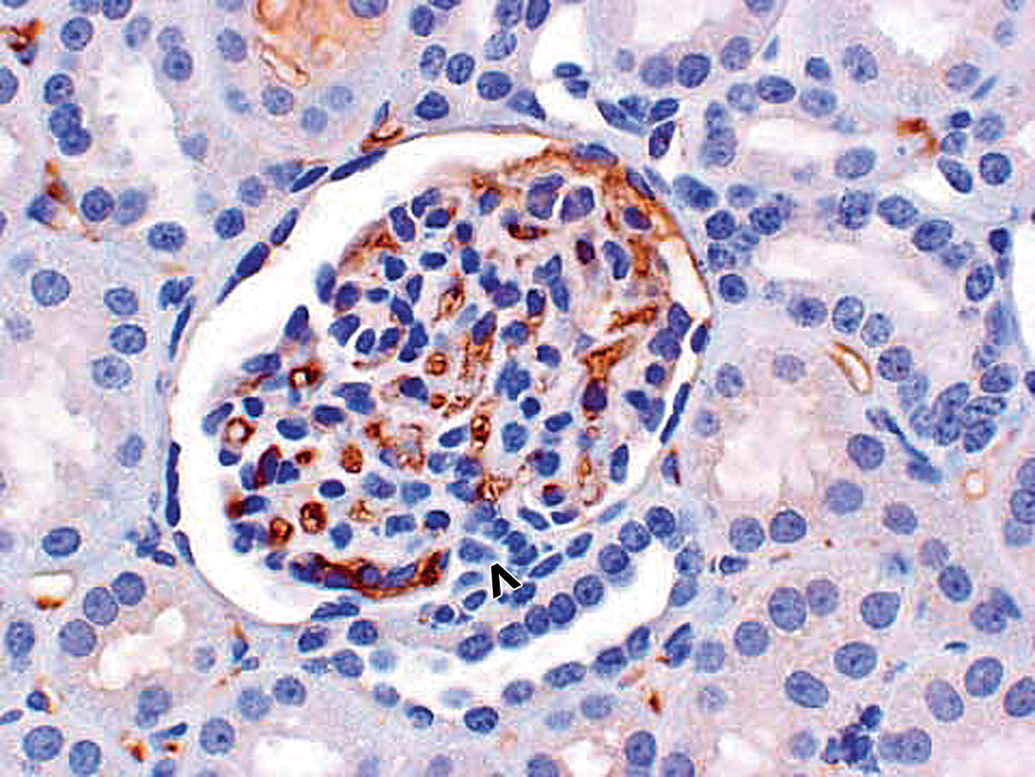
Spontaneous glomerulopathy in a control cynomolgus monkey. Hematoxylin and eosin (H&E) stain reveals glomerular hypercellularity as well as increased mesangial matrix. Vascular pole of glomerulus is identified by an open arrowhead. 40×.
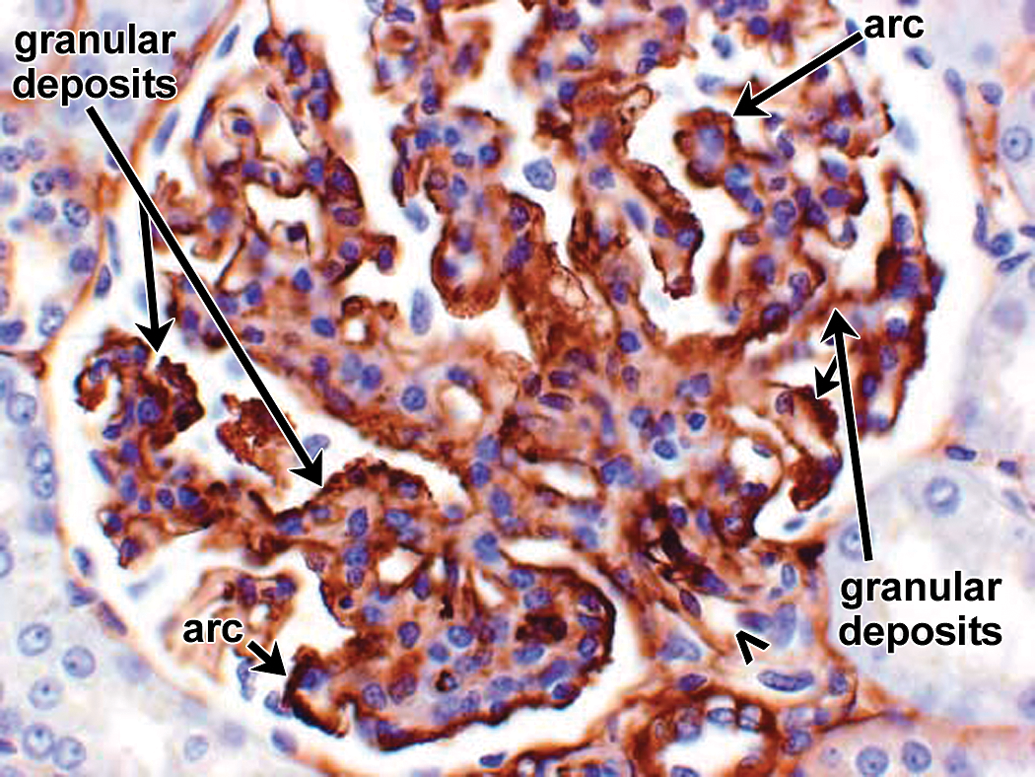
Spontaneous glomerulopathy in a control cynomolgus monkey. IHC staining for monkey IgM reveals granular deposits in mesangial, subendothelial, and subepithelial locations. Vascular pole of glomerulus is identified by an open arrowhead. Other arrows identify multiple granular deposits as well as arc shapes formed by confluent granular deposits. 40×. IHC = immunohistochemistry.
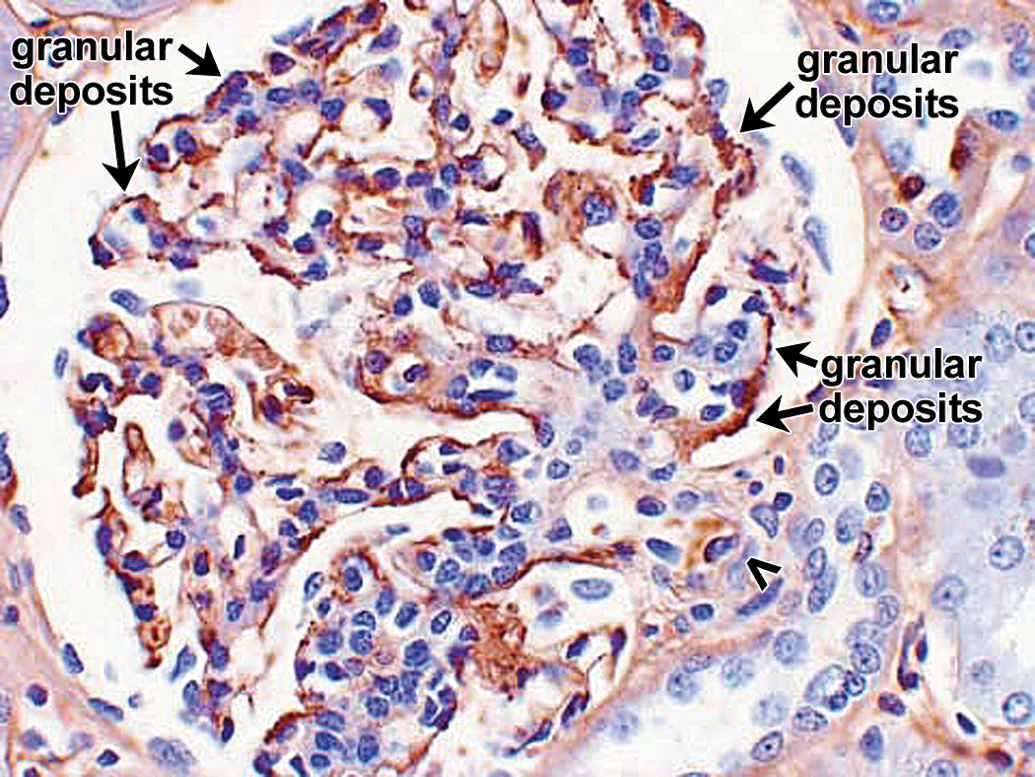
Spontaneous glomerulopathy in a control cynomolgus monkey. IHC staining for monkey IgG reveals granular deposits primarily in subepithelial locations. There are fewer monkey IgG granular deposits in this glomerulus (arrows) than there are monkey IgM granular deposits (compare to Figure 16). Vascular pole of glomerulus is identified by an open arrowhead. 40×. IHC = immunohistochemistry.
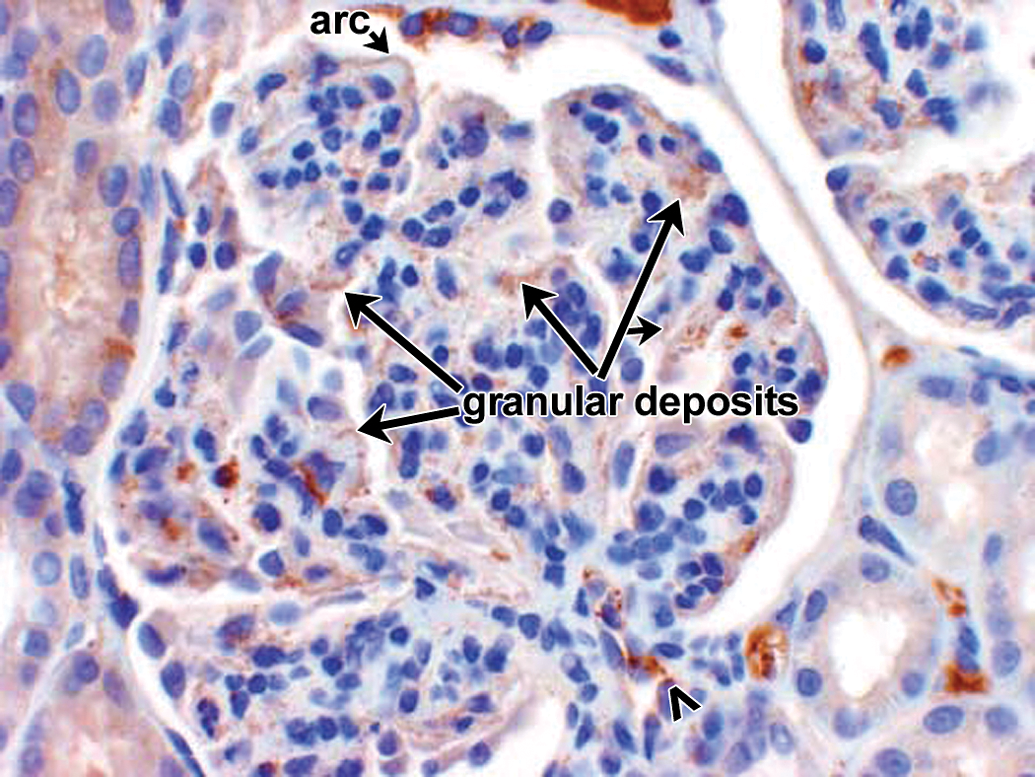
Spontaneous glomerulopathy in a control cynomolgus monkey. IHC staining for monkey C3 reveals granular deposits in a few subepithelial locations, including as a short beaded arc (arrow). Compared with Figures 16 and 17, there are fewer monkey C3 granular deposits in this glomerulus than there are monkey IgM and monkey IgG granular deposits. Vascular pole of glomerulus is identified by an open arrowhead. 40×. IHC = immunohistochemistry.
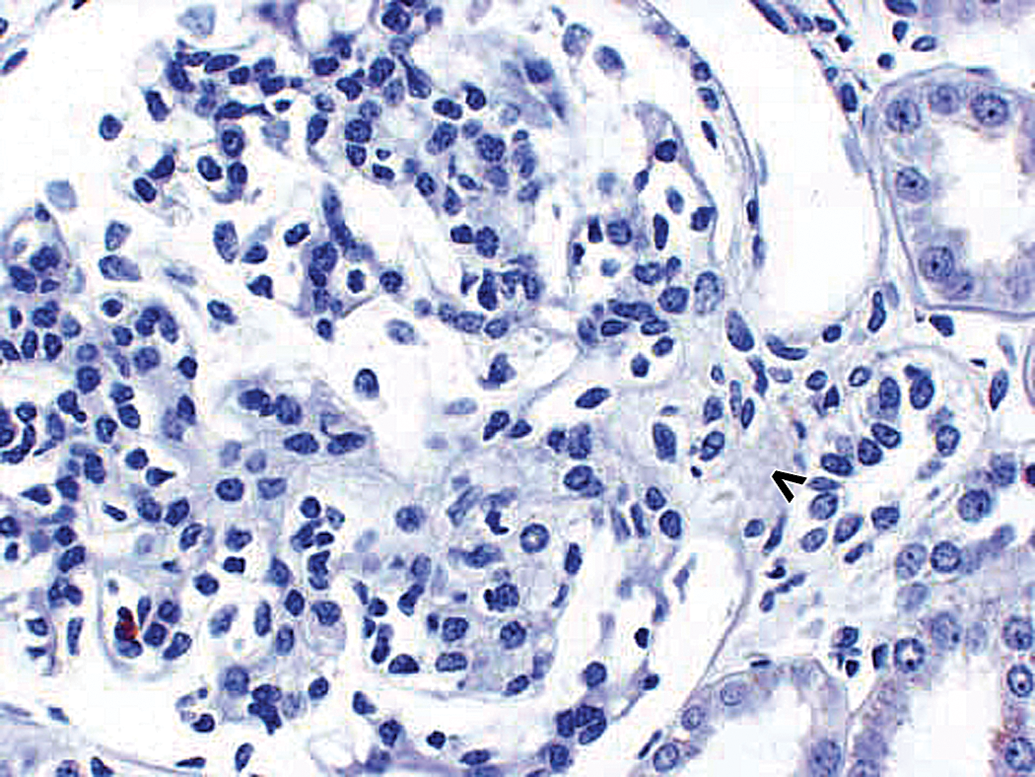
Spontaneous glomerulopathy in a control cynomolgus monkey. There is no staining with the negative control antibody. Vascular pole of glomerulus is identified by an open arrowhead. 40×.
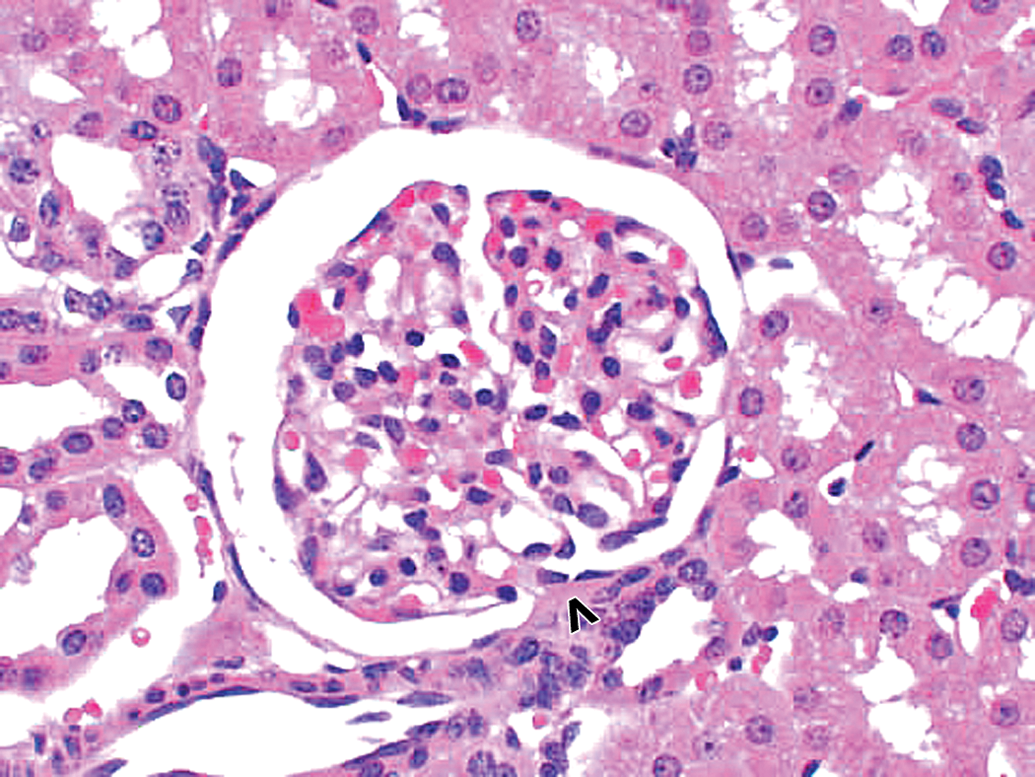
Morphologically normal kidney glomerulus in a control cynomolgus monkey. Control monkey kidney. Hematoxylin and eosin (H&E) stain reveals morphologically normal glomerulus. Vascular pole of glomerulus is identified by an open arrowhead. 40×.
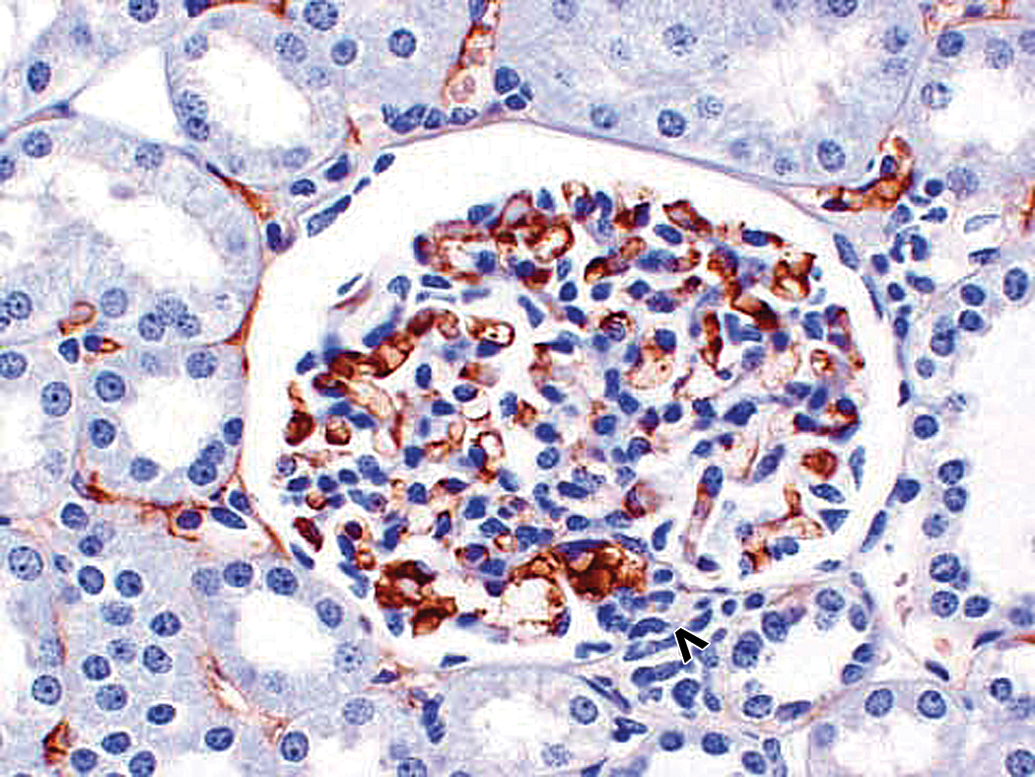
Morphologically normal kidney glomerulus in a control cynomolgus monkey. Control monkey kidney. IHC staining reveals physiologic monkey IgM staining with faint stippling in mesangium; majority of monkey IgM staining is observed intravascularly in glomerular and peritubular capillaries. Vascular pole of glomerulus is identified by an open arrowhead. 40×. IHC = immunohistochemistry.
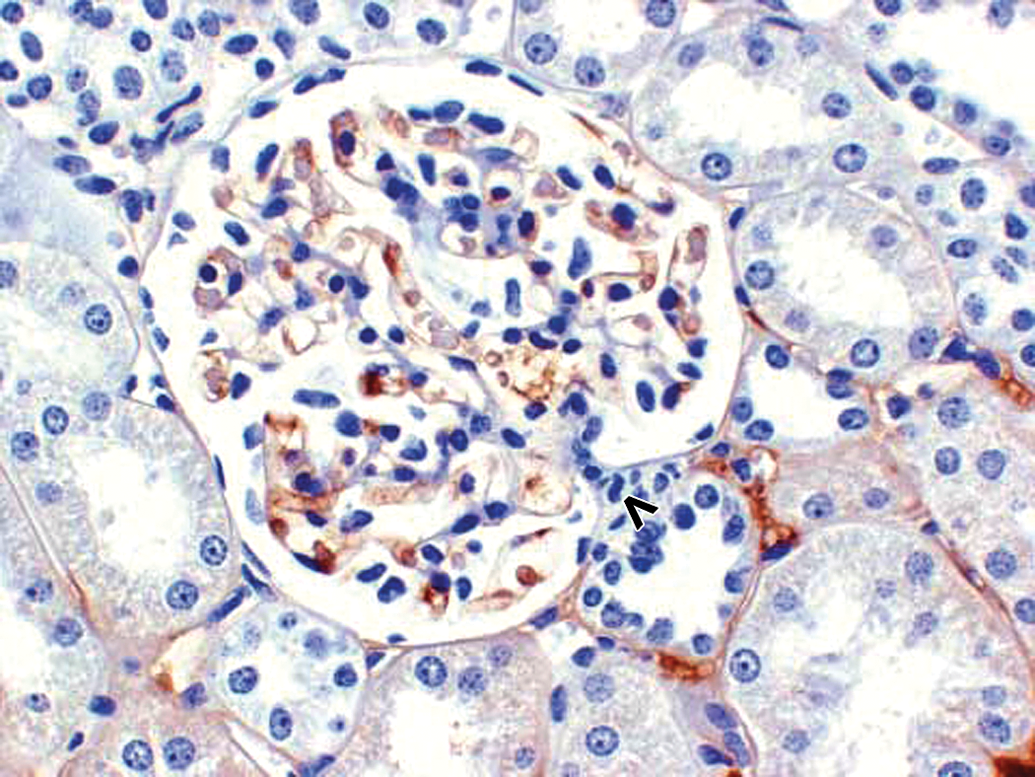
Morphologically normal kidney glomerulus in a control cynomolgus monkey. Control monkey kidney. IHC staining reveals physiologic monkey IgG staining with majority of monkey IgG staining observed intravascularly in glomerular and peritubular capillaries. Faint monkey IgG staining of proximal tubular epithelium in a finely vesicular pattern, consistent with physiologic resorption. Vascular pole of glomerulus is identified by an open arrowhead. 40×. IHC = immunohistochemistry.
Morphologically normal kidney glomerulus in a control cynomolgus monkey. Control monkey kidney. IHC staining reveals physiologic monkey C3 staining with majority of monkey C3 staining observed intravascularly. Faint monkey IgG staining of proximal tubular epithelium in a finely vesicular pattern, consistent with physiologic resorption; slightly greater C3 (physiologic) staining in lumen of tubule near top center of photograph. Vascular pole of glomerulus is identified by an open arrowhead. 40×. IHC = immunohistochemistry.
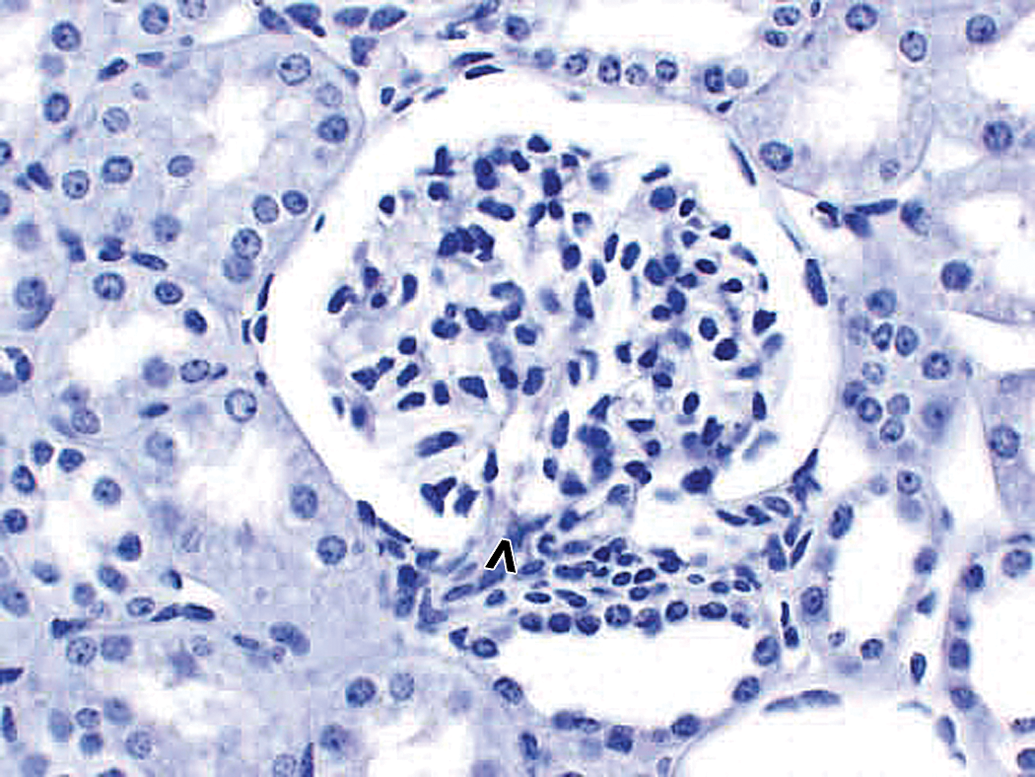
Morphologically normal kidney glomerulus in a control cynomolgus monkey. Control monkey kidney. No staining of glomerulus with negative control antibody. Similar lack of staining was observed when this kidney was stained for drug. Vascular pole of glomerulus is identified by an open arrowhead. 40×.
Drug/ADA-containing ICs in acute glomerulopathy and associated glomerular arteriole inflammation in a cynomolgus monkey. Drug-dosed monkey with glomerulopathy and inflammation of glomerular arteriole at vascular pole (open arrowhead). Hematoxylin and eosin (H&E) stain reveals a fibrinoid change (asterisks) in glomerulus, associated glomerular arteriole at vascular pole (open arrowhead), and extension into glomerular capillary loops. 40×. ADA = antidrug antibody; IC = immune complex.
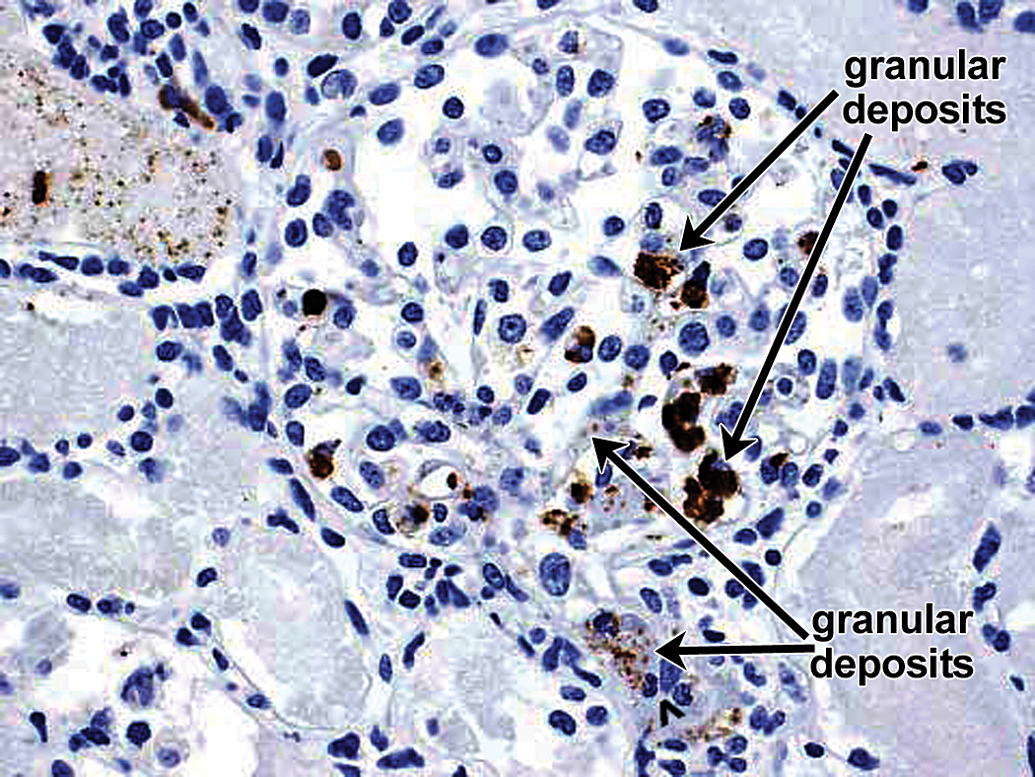
Drug/ADA-containing ICs in acute glomerulopathy and associated glomerular arteriole inflammation in a cynomolgus monkey. Drug-dosed monkey with glomerulopathy and inflammation of glomerular arteriole at vascular pole (open arrowhead). IHC staining for drug reveals drug-containing IC-related granular deposits (arrows) in the wall of glomerular arteriole with extension into subendothelial locations in adjacent capillary loops (arrows). Other arrows identify drug-containing IC-related granular deposits associated with intravascular and/or intraglomerular macrophages and/or neutrophils. 40×. ADA = antidrug antibody; IC = immune complex; IHC = immunohistochemistry.
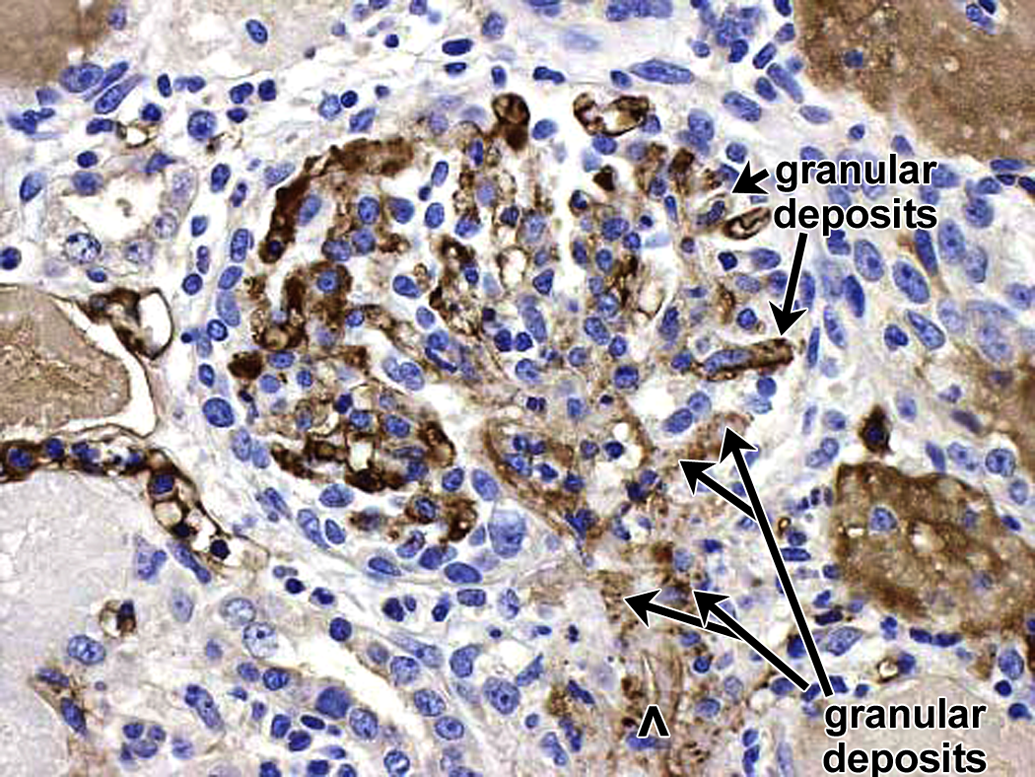
Drug/ADA-containing ICs in acute glomerulopathy and associated glomerular arteriole inflammation in a cynomolgus monkey. Drug-dosed monkey with glomerulopathy and inflammation of glomerular arteriole at vascular pole (open arrowhead). IHC staining for IgM reveals IgM-containing IC-related granular deposits (arrows) in the wall of glomerular arteriole with extension into subendothelial locations in adjacent capillary loops (arrows) which generally colocalize with drug-containing granular deposits (compare to Figure 26). Other arrows identify rare granular deposits in intraglomerular macrophages and/or neutrophils. 40×. ADA = antidrug antibody; IC = immune complex; IHC = immunohistochemistry.
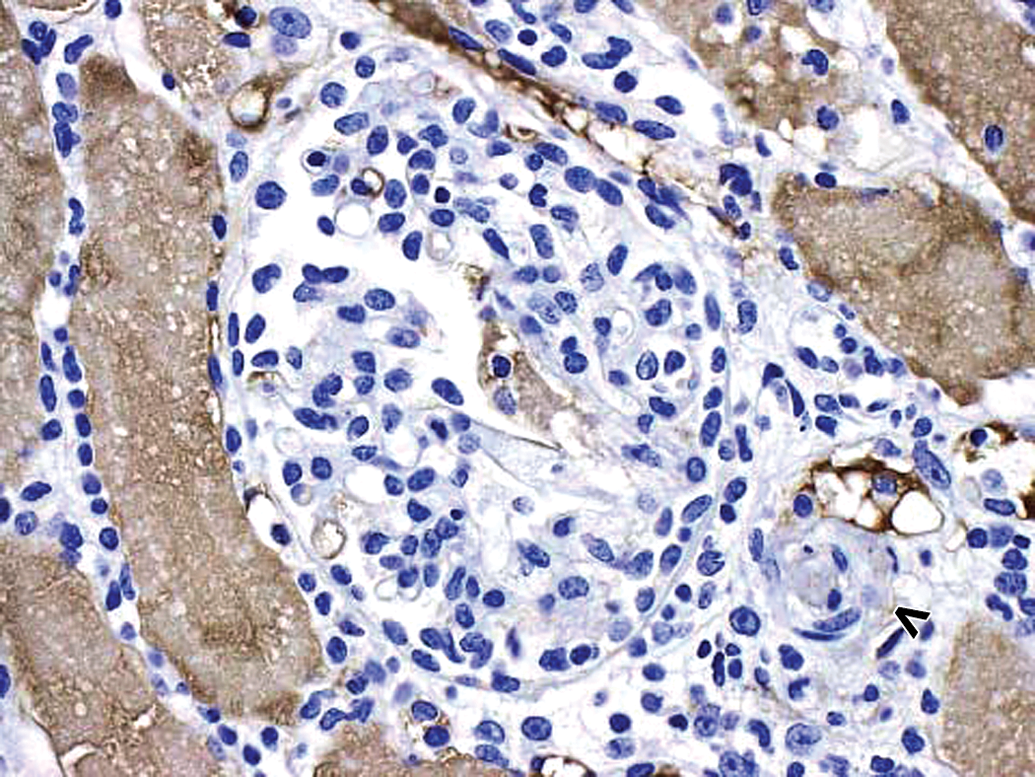
Drug/ADA-containing ICs in acute glomerulopathy and associated glomerular arteriole inflammation in a cynomolgus monkey. Drug-dosed monkey with glomerulopathy and inflammation of glomerular arteriole at vascular pole (open arrowhead). IHC staining for monkey IgG does not reveal prominent monkey IgG-containing IC-related granular deposits in this photograph. Prominent monkey IgG staining in adjacent proximal tubular lumens (indicative of increased glomerular loss). 40×. ADA = antidrug antibody; IC = immune complex; IHC = immunohistochemistry.
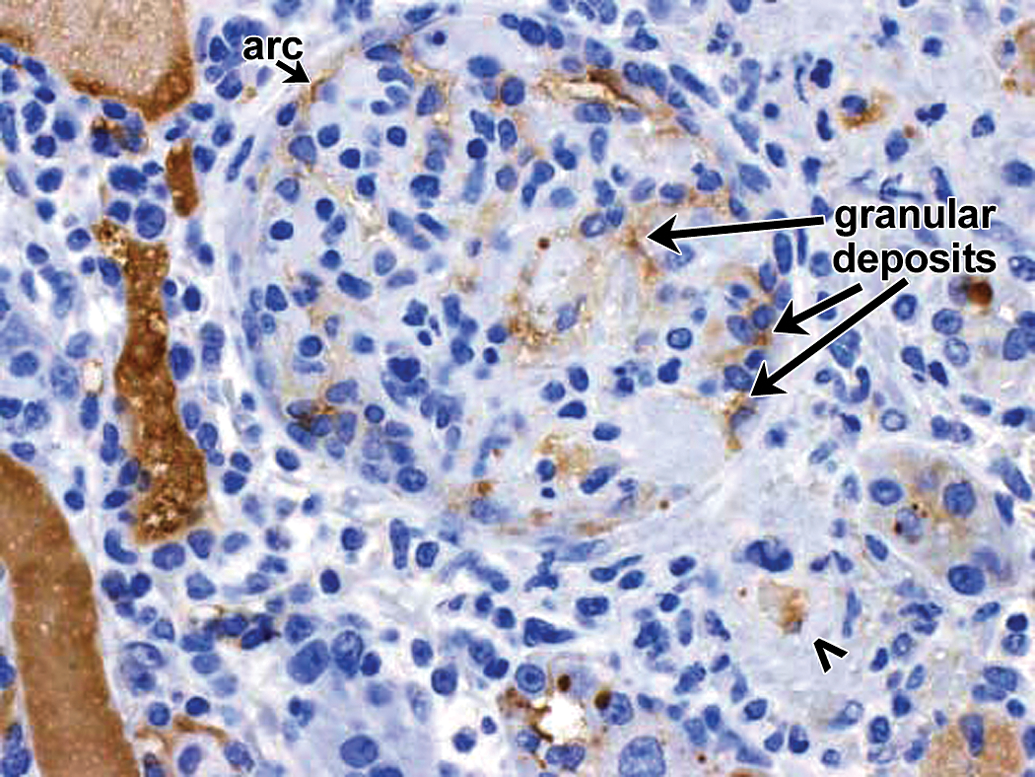
Drug/ADA-containing ICs in acute glomerulopathy and associated glomerular arteriole inflammation in a cynomolgus monkey. Drug-dosed monkey with glomerulopathy and inflammation of glomerular arteriole at vascular pole (open arrowhead). IHC staining for monkey C3 reveals a few scattered C3-containing IC-related granular deposits. Prominent C3 staining in adjacent proximal tubular lumens (indicative of increased glomerular loss). 40×. ADA = antidrug antibody; IC = immune complex; IHC = immunohistochemistry.
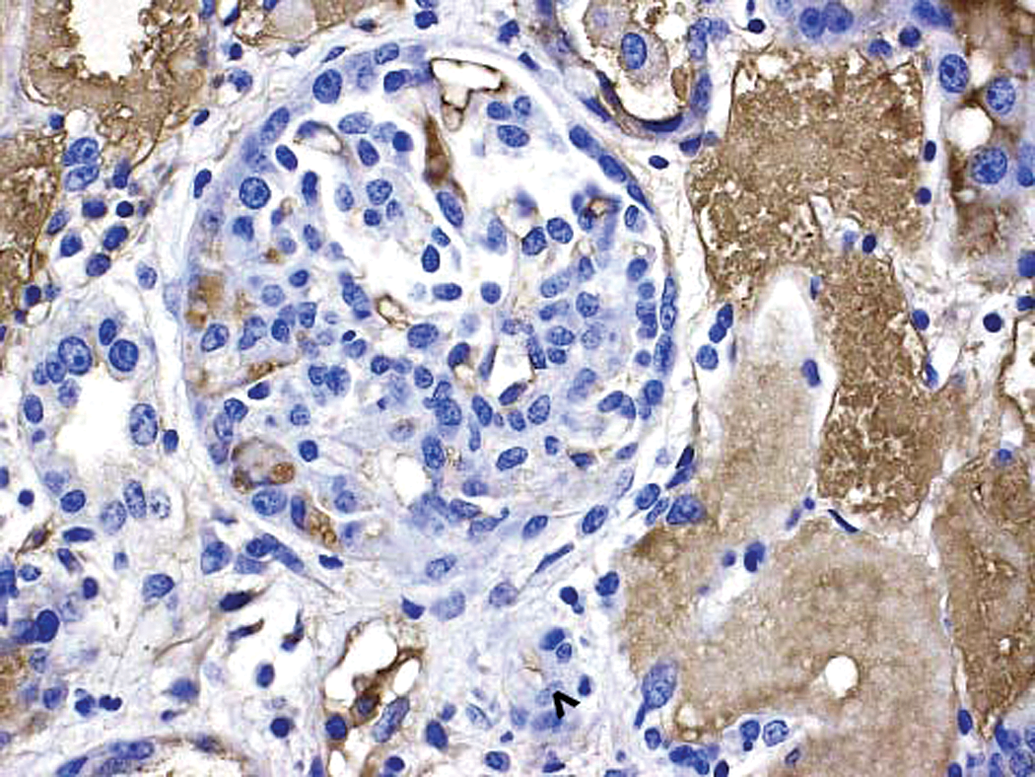
Drug/ADA-containing ICs in acute glomerulopathy and associated glomerular arteriole inflammation in a cynomolgus monkey. Drug-dosed monkey with glomerulopathy and inflammation of glomerular arteriole at vascular pole (open arrowhead). IHC staining for monkey albumin was included as a control for leakage and did not reveal albumin-containing IC-related granular deposits. Prominent albumin staining in adjacent proximal tubular lumens (indicative of increased glomerular loss). 40×. ADA = antidrug antibody; IC = immune complex; IHC = immunohistochemistry.
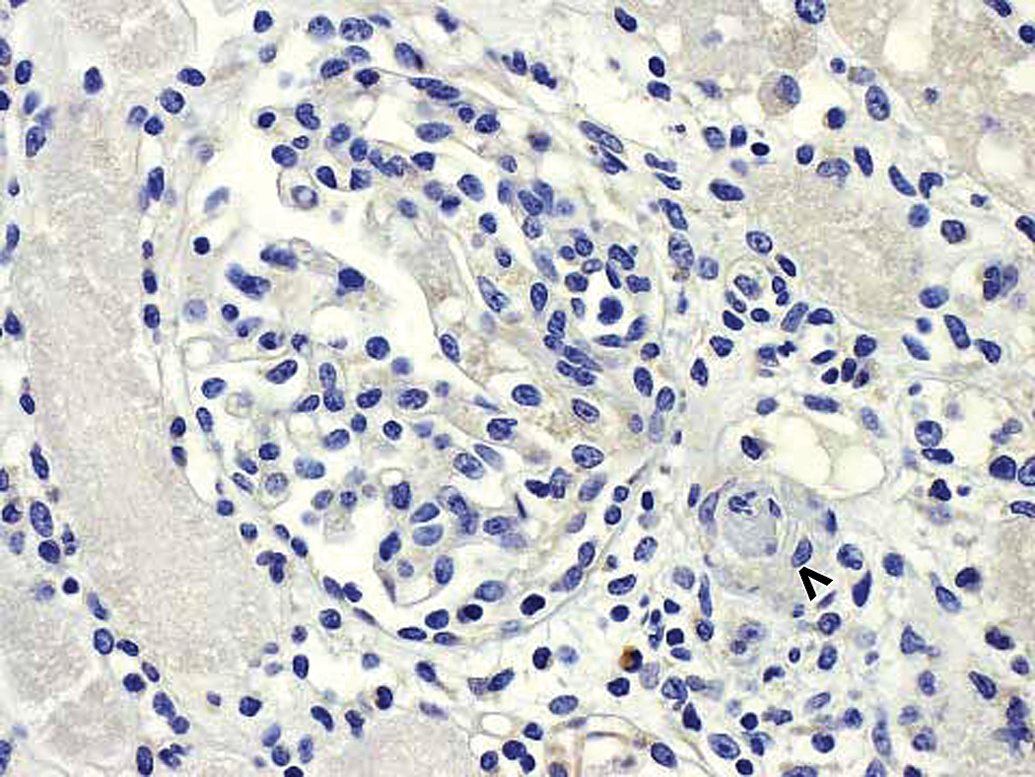
Drug/ADA-containing ICs in acute glomerulopathy and associated glomerular arteriole inflammation in a cynomolgus monkey. Drug-dosed monkey with glomerulopathy and inflammation of glomerular arteriole at vascular pole (open arrowhead). There is no staining with the negative control antibody. 40×. ADA = antidrug antibody; IC = immune complex.
Glomerulopathy with monkey Ig-containing granular deposits but no evidence of drug in granular deposits in a cynomolgus monkey. Drug-dosed monkey with glomerulopathy. Hematoxylin and eosin (H&E) stain reveals glomerular hypercellularity. Vascular pole of glomerulus is identified by an open arrowhead. 40×.
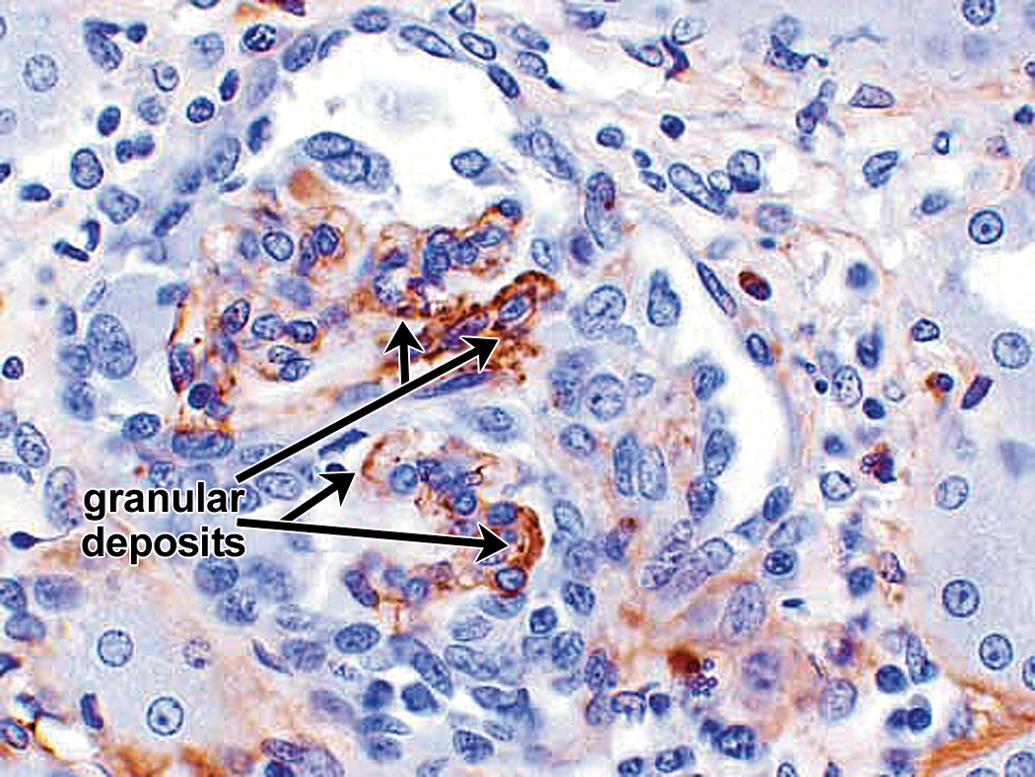
Glomerulopathy with monkey Ig-containing granular deposits but no evidence of drug in granular deposits in a cynomolgus monkey. Drug-dosed monkey with glomerulopathy. IHC staining for monkey IgM reveals IgM-containing IC-related granular deposits primarily in subepithelial locations (arrows). There were no drug-containing IC-related granular deposits in glomerulus, but there were prominent drug-containing IC-related granular deposits on phagocytes (not shown in this photograph). Vascular pole of glomerulus is identified by an open arrowhead. 40×. IC = immune complex; IHC = immunohistochemistry.
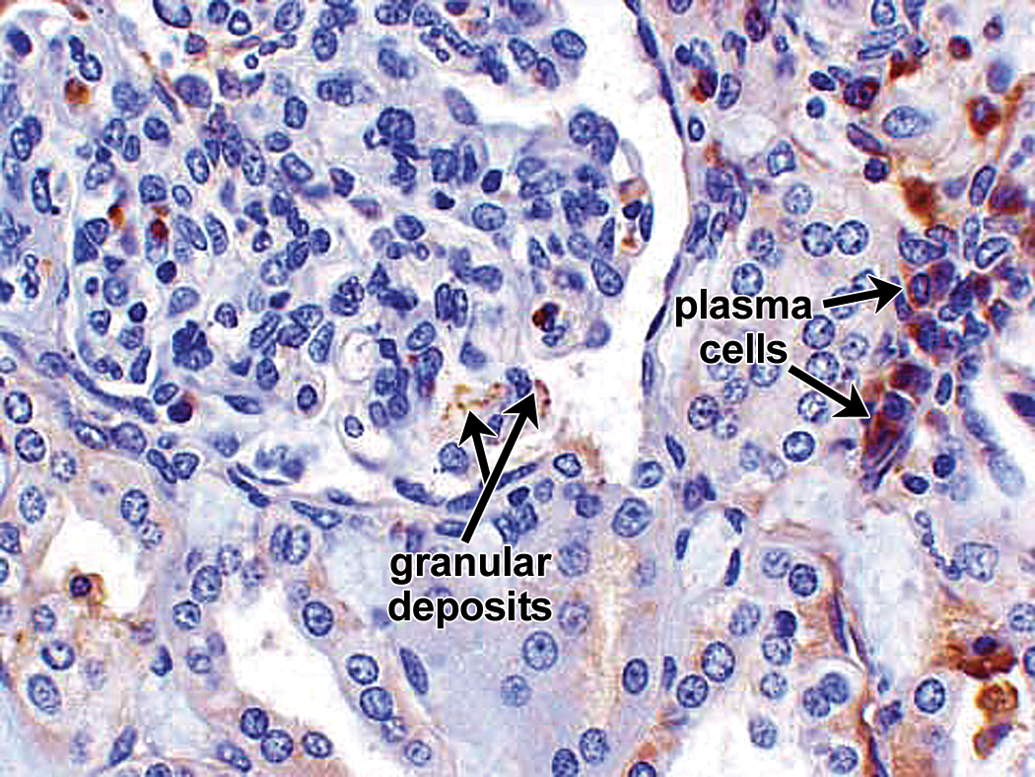
Glomerulopathy with monkey Ig-containing granular deposits but no evidence of drug in granular deposits in a cynomolgus monkey. Drug-dosed monkey with glomerulopathy. IHC staining for monkey IgG reveals focal subepithelial IgG-containing IC-related granular deposits (arrows). Other arrows identify monkey IgG-containing plasma cells to the right of the glomerulus. 40×. IC = immune complex; IHC = immunohistochemistry.
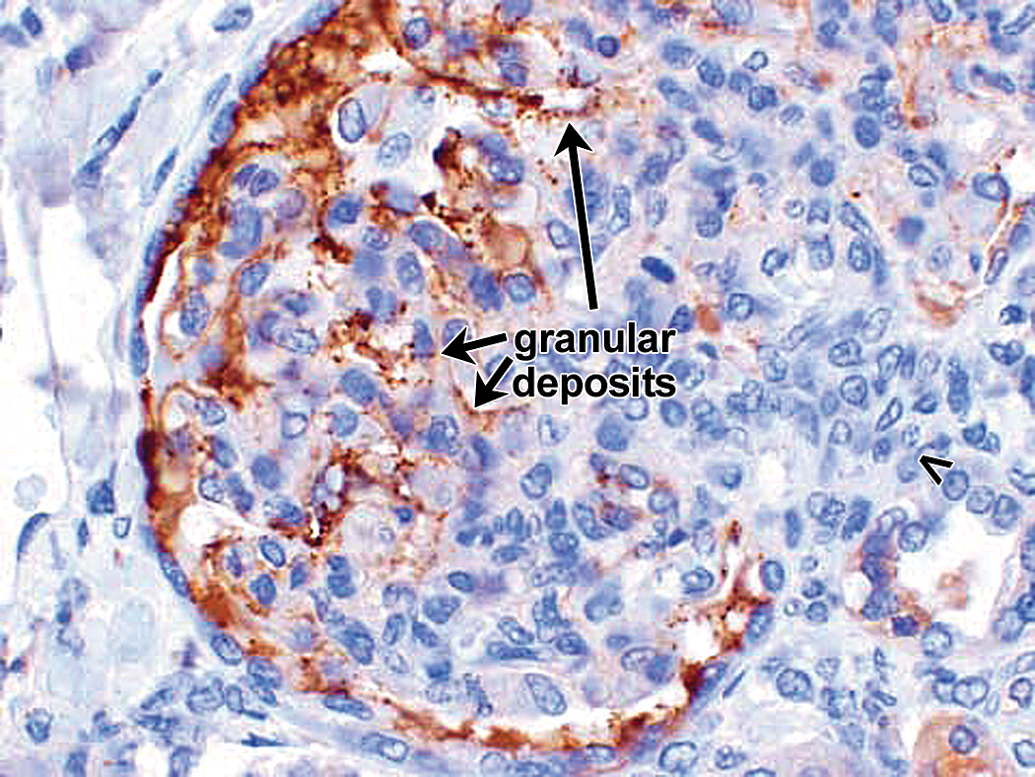
Glomerulopathy with monkey Ig-containing granular deposits but no evidence of drug in granular deposits in a cynomolgus monkey. Drug-dosed monkey with glomerulopathy. IHC staining for monkey C3 reveals subepithelial C3-containing IC-related granular deposits (arrows) which colocalized primarily with the IgM-containing granular deposits. C3 leakage into urinary space also evident, particularly along the left half of the glomerulus in the photograph. 40×. IC = immune complex; IHC = immunohistochemistry.
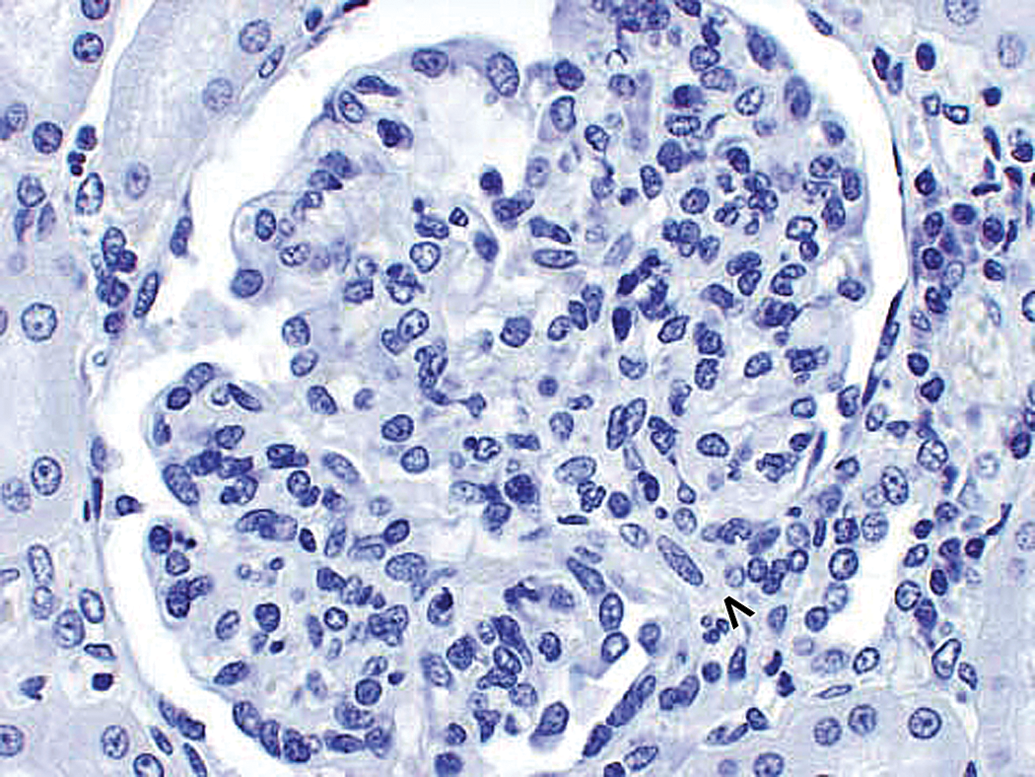
Glomerulopathy with monkey Ig-containing granular deposits but no evidence of drug in granular deposits in a cynomolgus monkey. Drug-dosed monkey with glomerulopathy. There is no staining with negative control antibody. 40×.
Mesangial IgM granular deposits are the most frequent finding in drug
Summary of relationship between glomerular disease and IC deposition in nonhuman primates.

Note. BUN = blood urea nitrogen; C1q, C4, and C3 = complement proteins or fragments thereof; IC = immune complex; TEM = transmission electron microscopy.
IgM transport across the glomerulus
IgM is transported across the glomerulus but is too large to cross via the podocyte slit pores which exclude molecules of 70 kDa (e.g., albumin) or larger in health. In health, themesangium/macula densa/distal convoluted tubule pathway is important in clearing IgM from the blood. At the centripetal portion of the glomerular capillary loops (i.e., at the glomerular angles), the fenestrated glomerular endothelium may be interrupted and Fcα/µ receptor
IHC findings in monkey glomerular disease
The relationship between naturally occurring IC deposition and GN/glomerulopathy has been studied in nonhuman primates including cynomolgus monkeys, pigtailed macaques, squirrel monkeys, common marmosets, and baboons (Table 5). The most frequent IHC finding is IgM granular deposits in mesangium. Deposition in extraglomerular mesangium usually precedes deposition in intraglomerular mesangium. In monkeys, mesangial IC less often contain IgG, IgA, C3, or sC5b
When sequential studies of spontaneous IC deposition and GN/glomerulopathy are performed, it appears that mesangial granular IgM deposits precede histologic evidence of pathology (Brack 1995; Brack and Weber 1995). Giddens and colleagues (1981) reported that 14% of adult nemestrine macaques dying from spontaneous disease had GN and concurrent IgM glomerular deposits. However, very limited IgM deposits were found in clinically normal nemestrine macaques without renal disease consistent with Brack’s marmoset studies, similar to the very limited IgM deposits observed in control monkeys from case 5 (Table 1). Antigen
Often, the antigen is unknown in the IC-related granular deposits in spontaneous glomerular disease in monkeys. In contrast, antigen may be demonstrated in glomerular granular deposits in experimental/toxicity studies involving administration of foreign proteins (e.g., BGG or BSA, or human mAbs or other rh proteins). When antigen is demonstrated, it may be seen in mesangial, subendothelial, and/or subepithelial granular deposits (Rojko, Price, and Raymond, unpublished; Cosio, Hebert, et al. 1987; Cosio, Birmingham, et al. 1987; Hebert, Allhiser, and Koethe 1978; Hebert 1991; Hebert, Birmingham, Mahan, et al. 1994; Hebert, Birmingham, Shen, et al. 1994).
As discussed in the Introduction section, BGG
Other IC-related IHC findings in kidney
Some monkeys with IC
Residual IC-mediated glomerular disease following drug withdrawal
Case 6 monkey 6D was a recovery mid-dose animal with glomerular disease, azotemia, hypoproteinemia, proteinuria, and IC
Transient IC-mediated glomerular disease has been described in a child with Pompe’s disease treated with rh acid α
Systemic or Generalized Drug-ADA IC Formation Associated with Type 3 HSRs; Often Involving Systemic Complement Activation
The case studies provide multiple examples of systemic or generalized drug
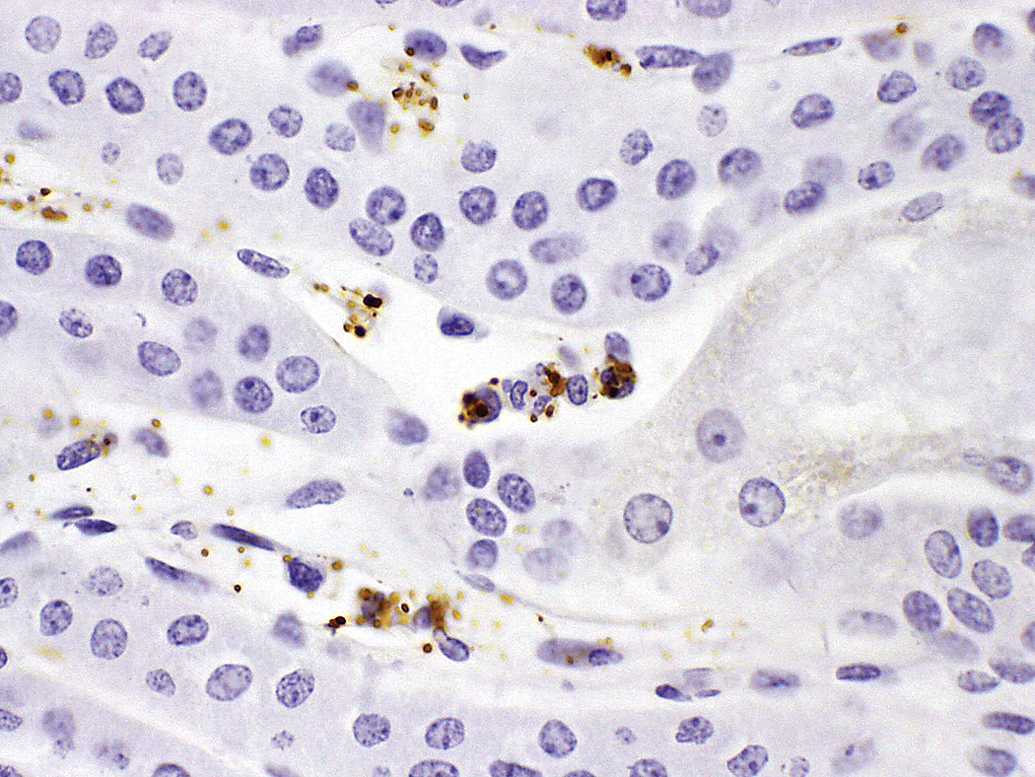
Drug-containing IC-related granular deposits associated with platelets, neutrophils, and monocytes/macrophages in peritubular capillaries in kidney. 40×. IC = immune complex.
Histopathology may be negative or may reveal minor findings (localized or systemic congestion or hemorrhage). Other times, histopathology reveals microthrombi and/or leukocyte sequestration, particularly in lung capillaries, and liver or spleen sinusoids. Leukocyte sequestration was particularly prominent in monkeys 9A-E in case 9 although microthrombi were not evident in those animals. These locations, particularly the lung capillaries, may reflect the sites of drug and ADA interaction, as the blood flow slows and the drug and ADA come into close proximity. Different scenarios can be proposed, based on antigen or antibody excess conditions. The most consistent IHC findings include granular deposits containing drug, IgM, monkey IgG, and/or C3 associated with neutrophils and/or monocytes/macrophages in multiple tissues (Figures 27 and 37), often liver or spleen sinusoids, alveolar capillaries in lung, or liver Kupffer cells. Within or on phagocytic cells, larger granular deposits may represent IC
The interrelationship and cross-activation of the complement, coagulation, and kinin systems
The interaction of the complement, coagulation, and kinin systems can often occur as a sequela to the activation of complement and can be a significant contributing factor to a moribund state or to mortality in monkeys (unpublished observations). The formation of C3b through complement activation plays a central role in linking to the coagulation system through the combination with factor B (factor IX) and with factor D activity that forms the alternative pathway C3 convertase (C3bBb; Warren and Ward 2001). C3 convertases generated by either pathway (classical or alternative) can cleave C3 to form C3a and C3b that can further lead to the conversion of C5 to C5a, C5b, and C5b
Relevance of IC Formation and HSRs in Animals on Toxicity Studies to Human Risk in Clinical Trials
The purpose of this article was to review the formation, clearance, deposition, and pathogenicity of biopharmaceutical-related ICs in monkeys and other toxicity study species; to illustrate many of the possible findings associated with generalized or localized IC formation with the proteins or mAbs in toxicity study animals; and to illustrate the challenges associated with demonstrating IC
Footnotes
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The author(s) received no financial support for the research, authorship, and/or publication of this article.