
Abstract
Better biomarkers are needed to identify, characterize, and/or monitor drug-induced vascular injury (DIVI) in nonclinical species and patients. The Predictive Safety Testing Consortium (PSTC), a precompetitive collaboration of pharmaceutical companies and the U.S. Food and Drug Administration (FDA), formed the Vascular Injury Working Group (VIWG) to develop and qualify translatable biomarkers of DIVI. The VIWG focused its research on acute DIVI because early detection for clinical and nonclinical safety monitoring is desirable. The VIWG developed a strategy based on the premise that biomarkers of DIVI in rat would be translatable to humans due to the morphologic similarity of vascular injury between species regardless of mechanism. The histomorphologic lexicon for DIVI in rat defines degenerative and adaptive findings of the vascular endothelium and smooth muscles, and characterizes inflammatory components. We describe the mechanisms of these changes and their associations with candidate biomarkers for which advanced analytical method validation was completed. Further development is recommended for circulating microRNAs, endothelial microparticles, and imaging techniques. Recommendations for sample collection and processing, analytical methods, and confirmation of target localization using immunohistochemistry and in situ hybridization are described. The methods described are anticipated to aid in the identification and qualification of translational biomarkers for DIVI.
Introduction
The occurrence of drug-induced vascular injury (DIVI) is a significant cause of termination of candidate drugs in nonclinical assessment of safety, especially for vasoactive compounds, antineoplastic agents, and biotherapeutics. The clinical relevance of DIVI in nonclinical studies is not clear, as evidence of vascular injury (VI) is lacking in clinical populations treated with drugs demonstrating DIVI in nonclinical species. For example, minoxidil, caffeine, and theophylline, which cause DIVI in several nonclinical species, are not associated with clinical drug-induced vasculitis (CDIV), at least at therapeutic doses (Johansson 1981; Collins et al. 1988; Sobota 1989; National Toxicology Program 1998). In the absence of noninvasive methods to predict or monitor the onset of CDIV, the occurrence of DIVI in nonclinical studies can become an obstacle to the development of compounds that otherwise are predicted to be safe and efficacious in humans. The Vascular Injury Working Group (VIWG) primarily focused its research on acute DIVI because it is anticipated that the most robust alteration of the candidate biomarkers will occur in the acute stages after VI, and early detection for safety monitoring is desirable. This obstacle is encountered in the early phases of drug development, at which time it may be identified during histologic evaluation and/or the short-term in vivo cardiovascular studies for those compounds that cause systemic cardiovascular effects (Laverty et al. 2011). It also may occur at the level of the Phase I clinical trials, the Investigative New Drug application phase under U.S. Food and Drug Administration (FDA) jurisdiction, when the reviewing authorities evaluate the risk/benefit relationship in the context of a toxicity that may not be monitorable with biomarkers. As a result, pharmaceutical companies seldom advance into clinical trials compounds with a nonclinical DIVI liability, especially at or near clinically relevant exposure multiples (Sobota 1989; Kerns et al. 2005; Zhang, Hanig, and De Felice 2012).
CDIV is best known for its immune-mediated mechanisms and cutaneous manifestations (Jennette et al. 1994; Lee et al. 2010; Turk et al. 2013), which often occur for drugs without a DIVI signal in nonclinical studies (Kerns et al. 2005; Wong et al. 2008). CDIV also may affect the visceral vasculature independently of the skin, especially in the kidney and lung, with potentially life-threatening consequences (Cribier et al. 1999; Radic, Martinovic Kaliterna, and Radic 2012). In the absence of circulating biomarkers of VI, the diagnosis of noncutaneous CDIV in patients relies on nonspecific circulating biomarkers such as endothelial cell (EC) adhesion molecules or acute phase reactants (reviewed in Weaver et al. 2008; Zhang, Hanig, and De Felice 2012), or on nonspecific clinical signs such as hypertension, hemorrhage, and/or thromboembolism (Keefe et al. 2011).
The Critical Path Institute (C-Path) was established as a neutral third party, between the FDA and pharmaceutical companies, to improve health and save lives by accelerating the development of safe and effective medicines (Mattes and Walker 2009). The founding principle of C-Path is that the qualification of novel safety biomarkers is best tackled through precompetitive collaboration within the pharmaceutical industry. C-Path builds on the strengths of its members, including the FDA, to assemble teams of scientists that span the spectrum of skills and experiences available in the member pharmaceutical companies. These teams work toward drafting formal qualification submission packages for novel drug development tools, including safety biomarkers (Pharmaceuticals and Medical Devices Agency, Japan [PMDA] 2010; European Medicines Agency [EMA] 2012). The process includes a consultation phase with the FDA’s Drug Development Tools Qualification Program team, which is anticipated to lead to earlier and more widespread regulatory and scientific acceptance of novel safety biomarkers of drug-induced organ injury in the specified context of use (CoU). To this end, C-Path formed the Predictive Safety Testing Consortium (PSTC) in 2006 as a collaboration between C-Path, FDA, and 5 (now 18) member companies, which enables data sharing and reduced risk, cost, and potential duration of nonclinical and Phase I clinical trials (Food and Drug Administration [FDA] 2004). PSTC research is focused in the nonclinical arena with some clinical research pursued directly and some through collaborations, such as with the Safer and Faster Evidence-based Translation (SAFE-T) Consortium that is sponsored by Europe’s Innovative Medicines Initiative (Bendjama et al. in press). The VIWG of PSTC specifically aims at developing and qualifying translatable biomarkers for DIVI.
The objectives of this collaborative article are to outline the strategy of VIWG, to encourage further collaboration with basic research scientists and clinicians, and to highlight through copublication with SAFE-T, the complementary nature of our strategies to facilitate forward and reverse translation of novel DIVI safety biomarkers. In doing so, this document describes: Regulatory and clinical aspects of DIVI; Mechanisms of DIVI; A histomorphologic lexicon developed by the VIWG which enables a biostatistical integration with candidate circulating biomarkers of DIVI; Biological and analytical evaluation of assays for approximately 30 candidate circulating proteins with advanced validation completed for 9 candidate assays for qualification; Circulating endothelial microparticles (EMPs) and microRNAs (miRNAs), which are additional circulating candidate biomarkers of DIVI; Potential messenger RNA (mRNA) biomarkers;
In situ hybridization (ISH) methods used to confirm the association between DIVI and miRNA expression; Animal models of DIVI and spontaneous VI (also reviewed by Bishop 1989).
Candidate tissue and circulating biomarkers were selected based on literature review and results from exploratory qualification studies and ex vivo and in vivo imaging techniques. The results of these studies have been used to identify a proposed panel of circulating biomarkers. The strategy of the VIWG is to perform confirmatory biomarker qualification studies in rats using test articles that cause DIVI through diverse mechanisms and over a time course to enable biostatistical interrogation of the performance of the candidate circulating biomarkers against the morphologic end points of DIVI. These studies, in addition to the assay validation work, will provide the basis of the formal biomarker qualification submission package to regulatory agencies. The goal is to identify biomarkers of DIVI that are independent of mechanism and accordingly those that also will be useful as biomarkers of spontaneous VI. In vitro and ex vivo models of DIVI were not explored by the VIWG of PSTC, because they were generally not considered relevant to the two major mechanisms of DIVI in nonclinical species (i.e., DIVI caused by vasoactive compounds and/or immune complex [IC] deposition), and they were not comprehensive enough to capture the complexity of the vascular microanatomy and function. However, as tissue engineering methods continue to advance, it is possible that physiologically relevant in vitro systems will become available in the future for screening or other applications.
Prior to the formation of the VIWG, most DIVI biomarker investigative information was proprietary and/or fragmented. The information presented in this publication, along with the investigative and analytical methods developed to support the VIWG strategy are achievements that have ensured continuous support by the member pharmaceutical companies. The work of the VIWG is an ongoing effort with the conduct of confirmatory qualification studies, evaluation of additional safety biomarkers/methods, refinement of the lexicon, and the design of the statistical analysis plan still in progress.
Safety and Translational Principles
The hypothesis underlying the research and qualification efforts of the VIWG is that nonclinical biomarkers of DIVI will overlap those of CDIV because of overlapping molecular biology and histomorphologic features of VI among species (Guionaud et al. 2011). These histomorphologic similarities suggest that the cellular, biochemical, and molecular responses of DIVI might be sufficiently independent of the etiology, mechanisms, and sites of lesions and that some of the nonclinical biomarkers will overlap with the biomarkers of CDIV. These histomorphologic similarities were also central to the VIWG lexicon, biostatistical approach (in progress), and selection of cell type–specific biomarkers (i.e., for ECs and vascular smooth muscle cells [VSMCs]). Accordingly, the qualification strategies by PSTC and SAFE-T are complementary, as the circulating biomarkers are qualified against the vascular compartment/cell type of injury and inflammation rather than against the mechanism, etiology, or site of injury.
In humans, there are a large number of spontaneous and genetically determined vascular diseases that are not drug induced (reviewed by Bendjama et al. in press). The occurrence of these diseases has enabled the investigation of a large panel of candidate biomarkers in studies where disease status and biomarker levels can be correlated. The SAFE-T Consortium intends to qualify candidate biomarkers for clinical use in a series of exploratory and confirmatory clinical studies (Bendjama et al., this issue). Although some biomarkers proposed in the clinical studies cannot be preclinically back-translated because of species differences in biology or the lack of analytical reagents, the collaboration between PSTC and SAFE-T strengthens nonclinical and CDIV research through the use of shared methods (histomorphologic, analytical, and biostatistical), complementary list of candidate biomarkers, and compared study outcomes.
Mechanisms of DIVI
DIVI in nonclinical species has been recognized in mice, rats, dogs, pigs, and nonhuman primates. Multiple mechanisms have been proposed to explain DIVI, including hemodynamic alterations, altered endothelial nitric oxide synthase (eNOS) signaling, IC deposition, modulation of the immune system, and direct toxicity to ECs (Boor et al. 1995; Louden and Morgan 2001; Kerns et al. 2005; Guionaud et al. 2011; Tobin et al. in press). We summarize here the mechanisms commonly implicated in nonclinical DIVI and the recent advances in the field.
Hemodynamically Active Compounds
Hemodynamically active compounds cause the archetypical lesion of DIVI, namely vascular smooth muscle cell (VSMC) necrosis, and have been studied extensively (Supplemental Table 1). Hemodynamically active compounds cause systemic or local vasoconstriction or vasodilation, which may or may not be associated with changes in systemic and/or local blood pressure, turbulent blood flow, and/or eNOS signaling. The vascular beds most commonly affected by hemodynamically active compounds are the mesenteric arteries in rodents and the coronary arteries in nonrodent species (Joseph 2000; Clemo et al. 2003). These sites are subjected to a turbulent blood flow (Joseph et al. 1996a) and are the preferential site of occurrence of spontaneous vasculitis in both species. In addition, turbulent flow is a susceptibility factor for injury in these vessels (Kerns et al. 2005).
Vasodilators
The pathogenesis of DIVI caused by vasodilators is considered to be systemic (Dogterom and Zbinden 1992) and/or regional (Joseph et al. 1996b) alterations in blood flow. This hypothesis is supported by blood flow measurements in dogs administered minoxidil (Larson et al. 1996; Mesfin et al. 1989) and in rats administered the phosphodiesterase 3 inhibitor (PDE3i) SK F95654 (Joseph 2000), or the phosphodiesterase 4 inhibitor (PDE4i) CI-1044 (Korkmaz et al. 2009). Arterial vasodilation causes increased blood flow and wall stress. The increase in tensile stress imposed on the vessel during vasodilation has been hypothesized to cause the primary injury of medial hemorrhage and necrosis and secondary inflammation (Yuhas et al. 1985; Mesfin et al. 1989). In a less accepted hypothesis, the associated turbulent blood flow that causes EC shear stress is considered the pivotal early event leading to interendothelial gaps and subsequent medial injury and inflammation (Joseph et al. 1996b). Although medial injury may be compounded by the formation of gaps between ECs as a result of the vasoactive changes, ECs exposed to turbulent or oscillatory flow switch to an activated state and express proteins involved in inflammation, coagulation, platelet aggregation, and proliferation. Under these conditions, ECs may secrete factors involved in vascular remodeling, including matrix metalloproteinase 9 and bone morphogenetic protein 4 (BMP4). BMP4 in particular can modulate VSMC phenotype as well as induce the inflammatory cascade in a manner dependent on nuclear factor kappa light-chain enhancer of activated B cells (NFκB). Release of these factors by ECs may explain the observation that coronary arterial VSMCs exhibit injury prior to ECs in dogs administered the adenosine receptor agonist CI-947 (Metz et al. 1991).
The predominance of specific vasoactive receptors in certain vascular beds may render these vessels more vulnerable to DIVI. For example, rats administered fenoldopam, a selective dopaminergic D1 receptor (DA1) agonist, develop vasodilation and DIVI mainly in the medium size mesenteric arteries, which highly express DA1 receptors (Kerns et al. 1989a; Kerns, Arena, and Morgan 1989b). In comparison, rats administered dopamine, a DA1 agonist and an α- and β-adrenergic receptor agonist, develop DIVI in the larger mesenteric arteries and also in small arteries of multiple tissues that express α- and β-adrenergic receptors (Kerns et al. 1989a; Kerns, Arena, and Morgan 1989b). Similarly, higher expression of the endothelin receptor B (ETRB) in VSMCs of the right compared to the left coronary artery of dogs was proposed to predispose the right coronary to DIVI after the administration of an endothelin receptor A (ETRA)/ETRB dual antagonist (Louden et al. 2000). However, a similar right-sided predilection was seen with two highly selective ETRA antagonists, which indicates that additional factors are involved (Jones et al. 2003).
Vasoconstrictors
Compounds and endogenous substances with vasoconstrictor activity and DIVI potential include noradrenaline (Sandusky et al. 1992; Sandusky, Means, and Todd 1990), endothelin-1 (EDN1; Louden and Morgan 2001), angiotensin II (ANG2; Wilson and Heptinstall 1983; Jacobsen et al. 2002), digoxin (Teske et al. 1976; Bourdois et al. 1982), methoxamine, and midodrine (Dalmas et al. 2011). These agents act on multiple vascular compartments through α-adrenergic, ANG2, or ETRs, increases in intracellular free calcium, activation of ρ kinase, or inhibition of the Na+/K+ ATPase pump. In an acute ANG2 infusion model in rats, vascular damage was preceded by segmental hypercontraction causing vascular wall instability (Wilson and Heptinstall 1983; Jacobsen et al. 2002). At a molecular level, acute vasoconstriction is mediated through increases in intracellular free calcium and activation of ρ kinase, which increase VSMC contraction by phosphorylation of myosin light chain (Nguyen Dinh Cat and Touyz 2011). Compounds acting through the ANG2 receptor may cause EC apoptosis and promote oxidative stress and inflammation via uncoupling of eNOS and activation of mitogen-activated protein kinase kinase kinase 5 (Yamamoto et al. 2007), and nicotinamide adenine dinucleotide phosphate oxidase (Mehta and Griendling 2007). Finally, ANG2 activates members of the mitogen-associated protein kinase family, which contributes to intimal and VSMC growth and vascular remodeling (Mehta and Griendling 2007) as chronic effects of vasoconstriction. The function of the Na+/K+ ATPase can be modulated by both 8-Br-cAMP and L-NAME (Chen, Chen, and Wu 2005), suggesting a direct connection with primary EC damage.
Compounds Directly Toxic to ECs
Direct toxicity to ECs is thought to be the initiating mechanism of DIVI for some cancer drugs and certain immunomodulators (Bregman et al. 1987; Pisoni, Ruggenenti, and Remuzzi 2001; Wang et al. 2002; Wu et al. 2002; Rezzani 2004; Trapp and Weis 2005; Fujino, Kim, and Ito 2007; Mikaelian et al. 2010). Damage to ECs by cancer drugs is thought to be related to their pharmacologic activity, although hemodynamic factors may also be involved (Kumar, Hysmith, and Boor 1990; Miyauchi et al. 1993; Nishida et al. 2004; Guionaud et al. 2011).
Vascular Injury Secondary to Inflammation and ICs
Nonclinical DIVI may occur as the result of inflammation, especially with angiocentric inflammation, and in the case of IC deposition secondary to an immune response to therapeutic proteins, antibodies, and small molecules (Sams 1985; Vugmeyster et al. 2012). As an example of angiocentric inflammation, a generalized vascular leakage syndrome has been described after administration of interleukin 2 (Anderson and Hayes 1989). In this brief review, IC-mediated glomerulonephritis was considered vasculitis, because, in essence, it is a microangiopathy specific to the kidney.
Three mechanisms have been implicated in IC-mediated vasculitis in nonclinical species (reviewed by Bussiere and Johnson 2011). The first mechanism is the result of recognition by a therapeutic antibody of an epitope expressed by ECs. In this mechanism, antidrug antibodies (ADA) bind to the therapeutic antibody immobilized on ECs, resulting in complement activation and ensuing vascular damage. The second mechanism of IC-mediated vasculitis is the result of therapeutic antibody binding to soluble target followed by the formation of ICs by ADA, and subsequent complement activation. The third mechanism of IC-mediated vasculitis is the result of the binding of aggregates of therapeutic antibody by ADA, with ensuing activation of complement and deposition of the ICs.
In humans, the hypothesis of vasculitis originating from the induction of antineutrophil cytoplasm antibody (ANCA) antibodies has received much attention. The mechanism of ANCA-mediated vasculitis implies direct activation of cytokine-primed neutrophils and monocytes by ANCAs, which results in degranulation and subsequent EC damage by both cell types. (Jennette et al. 1995; Gao and Zhao 2009; Radic, Martinovic Kaliterna, and Radic 2012; Zhang, Hanig, and De Felice 2012). The availability of nonclinical models of ANCA-mediated VI may facilitate immediate forward and reverse translation of novel DIVI biomarkers (Weening, Hoedemaeker, and Bakker 1981; Mathieson et al. 1993; Kettritz et al. 1995; Specks 2000; Savige et al. 2002; Kallenberg, Heeringa, and Stegeman 2006; Jennette, Xiao, and Falk 2006; van der Geld et al. 2007; Kallenberg 2010; Salama and Little 2012).
Role of eNOS Signaling in DIVI
At a molecular level, there is evidence to suggest that altered eNOS signaling is a common pathogenic factor for a number of vasodilator compounds (Slim et al. 2003; Zhang et al. 2006; Weaver et al. 2008; Weaver et al. 2010). The ability to modulate the severity of DIVI lesions by modulating eNOS supports a central role of eNOS in DIVI caused by vasoactive compounds (Sheth et al. 2011; Brott, Richardson, and Louden 2012). There is also limited evidence suggesting altered signaling around eNOS with some vasoconstrictors (Tobin et al. this issue). It is not clear whether physical injury and eNOS signaling are either a single or two distinct processes. A definitive understanding of these processes requires advances in our basic knowledge of the molecular details of vascular physiology.
Assessment of DIVI by Histopathology
To assess DIVI and the performance of potential candidate biomarkers in biomarker qualification studies, the VIWG herein presents a histopathology approach, including tissue collection, a lexicon that captures lesion characterization relative to vascular compartment and duration, and biomarker target validation in tissues by immunohistochemistry (IHC) and ISH.
This section provides basic information on the rationale informing the tissue collection in DIVI biomarker qualification studies as defined in the CoU statement (Supplemental Appendix 1) and on the histomorphologic lexicon developed by the VIWG of PSTC for identifying, characterizing, and scoring DIVI. The tissue collection list was primarily designed for the rat, because the rat is the standard rodent species recognized by both United States and international regulatory agencies for use in toxicity studies, and because this species is affected by DIVI. For completeness, additional information related to the anatomy of the vascular system and most of the references used to draft this section are presented in Supplemental Appendix 2 and Appendix 3, respectively. This section also reviews lesion characterization over the time relative to the test article to support time point selection for circulating biomarker evaluation and the use of IHC and ISH to support biomarker target validation.
Extent of Tissue Collection
To identify and characterize DIVI appropriately, the tissue selection for routine examination should consider the specifics of the compounds, such as tissue expression of the target, target organs of VI, and non-VI and mechanisms of DIVI, as well as the differences among species in the vascular beds affected by spontaneous VI and DIVI and tissue size (Table 1). Of note, 1 of the 2 liver samples collected and evaluated must encompass the hilus, which is a common site of DIVI in rodents (unpublished observation). The mesentery is evaluated more comprehensively than in standard toxicity studies (Bregman et al. 2003), because the rat mesenteric arteries are frequent sites of DIVI. The evaluation of the rat mesentery is facilitated by the practicality of processing it in a limited number of blocks/slides using the roll method (Supplemental Appendix 4; Dalmas et al. 2008). In the dog, DIVI must be differentiated from spontaneous Beagle Pain Syndrome; therefore, the cervical spinal cord meninges may be included for evaluation. The heart will include multiple sections to increase the likelihood of identifying coronary lesions for the compounds that predominantly affect this vascular bed. The sampling strategy also accounts for the predilection of some compounds for specific vascular beds, such as the peri-islet capillaries for an undisclosed compound class (Brenneman et al. 2014), the heart valves and renal arcuate arteries for some oligonucleotides (Frazier 2013), the heart capillaries for tubulin binders (Mikaelian et al. 2010), and the vessels with slow blood flow (e.g., renal glomerulus, choroid plexus, epididymis, and/or the pampiniform plexus) for compounds causing IC-mediated DIVI.
Tissues collected for histomorphologic evaluation in the context of the studies for the VIWG of PSTC.

Note: PSTC = Predictive Safety Testing Consortium; VIWG = Vascular Injury Working Group.
aCross and longitudinal. bTo include the hilus in rat. cUsing the mesenteric roll methods in rat (Supplemental Appendix 4). dGastrocnemius muscle. eBased on the clinical findings, which may include changes for the mucocutaneous junctions. fDuodenum, jejunum, and ileum.
Histomorphologic Lexicon
Development of a histomorphologic lexicon was one of the first endeavors of the VIWG, because histology is considered the basis for the identification, characterization, and semiquantification of DIVI and is the gold standard against which circulating biomarker performance will be evaluated for qualification. The primary goal of the lexicon is to standardize the recording of histomorphologic end points of acute DIVI among institutions and pathologists in a format that enables biostatistical interrogation against the circulating candidate biomarkers (CoU Statement—Supplemental Appendix 1). This lexicon includes recommended terminology for the identification and characterization of degenerative and hyperplastic intimal, medial, and adventitial findings and inflammatory changes (Table 2). The vascular bed and the types of vessels affected by DIVI also are recorded (Table 3), the recording of this information will enable further interrogation of the data, as the data set of studies expands. Because the lexicon was designed to support the biomarker strategy of the VIWG, which is to identify biomarkers of acute DIVI, changes against which biomarker performance is not evaluated may be recorded but not used in the biostatistical analysis. Such changes include those that may be too subtle to be toxicologically relevant, are of chronic duration, or are indirect/secondary sequelae to DIVI (Table 4). The spontaneous background findings/conditions such as polyarteritis nodosa, Beagle Pain Syndrome, fibrinoid necrosis, and plexiform vasculopathy, will be graded using the lexicon and identified as spontaneous changes in comments (Zeek, Smith, and Weeter 1948; Kelly 1989; Westwood, Iswaran, and Greaves 1990; Clemo et al. 2003).
The lexicon developed by the VIWG of PSTC aims at enabling a statistical association between histomorphologic findings and clinical pathology parameters.
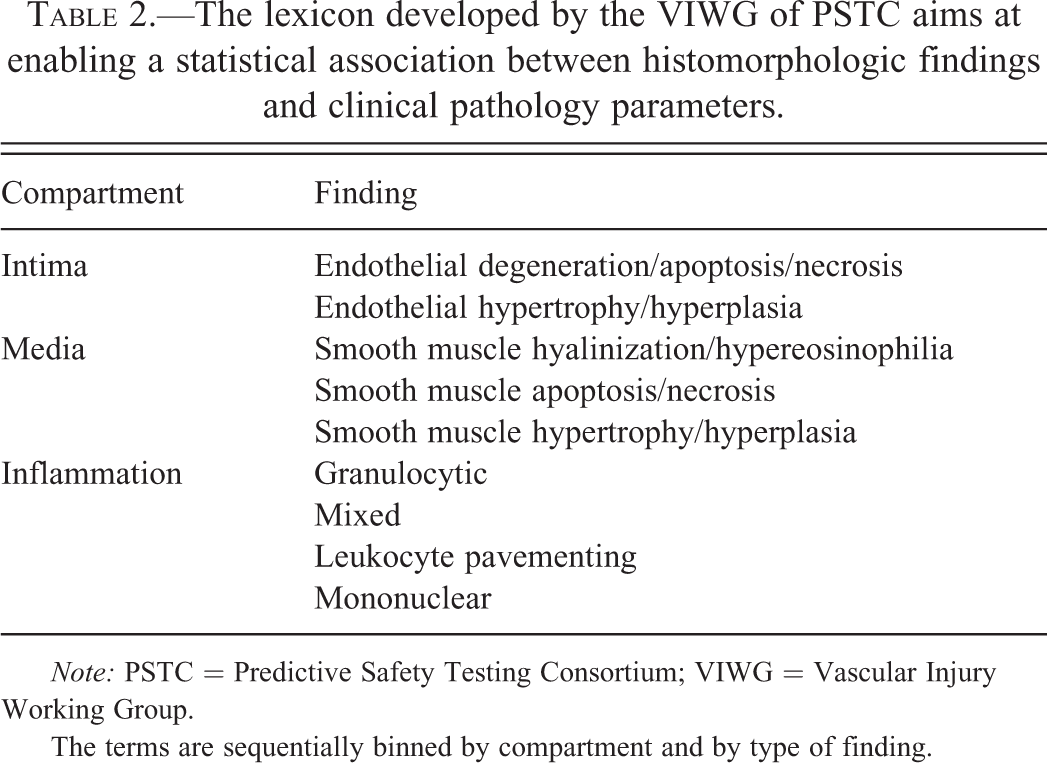
Note: PSTC = Predictive Safety Testing Consortium; VIWG = Vascular Injury Working Group.
The terms are sequentially binned by compartment and by type of finding.
Vascular beds and vessel type categories tracked by the Lexicon of PSTC.
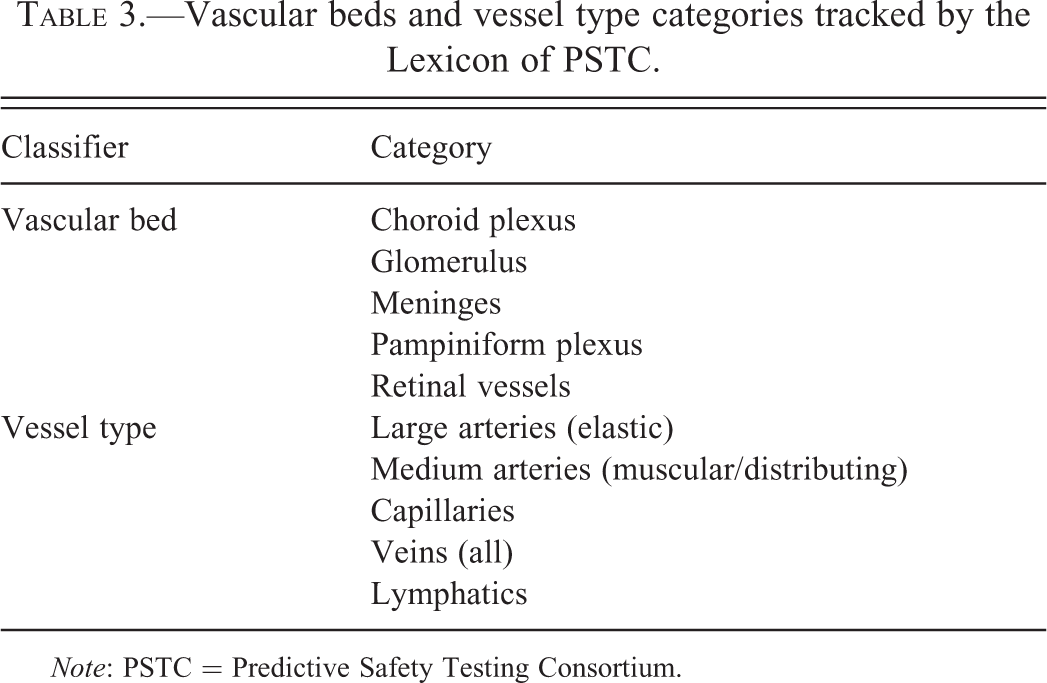
Note: PSTC = Predictive Safety Testing Consortium.
Histomorphologic end points against which biomarkers are not currently sought.

Note: These findings are recorded and graded independently of the end points against which a biomarker is sought.
The lexicon enables analyses outside the initial CoU Statement, because it captures end points other than those against which biomarker performance is evaluated. The lexicon adheres to the regulatory guidelines for approval of biomarker submissions by PSTC. It also follows the best practices recommended by the Society of Toxicologic Pathology and regulatory guidelines for histomorphologic evaluation in biomarker qualification studies (Burkhardt et al. 2011). The findings are graded by the pathologist from 0 (absence) to 4 (marked/severe), and a peer review ensures consistency of criteria and grades across studies. In addition to the grading of the histomorphologic findings in individual organs/tissues, the VIWG will assess the impact of multiorgan involvement on biomarker performance during the qualification phase.
Endothelium, Degeneration/Apoptosis/Necrosis
Endothelial cell degeneration/apoptosis/necrosis (DAN) is a grouped diagnosis to encompass all changes describing EC injury, because these types of injury cannot be rigorously differentiated in routine toxicology studies. In addition, it is not the strategy of the VIWG to identify biomarkers that differentiate along this continuum of histomorphologic changes. EC DAN is a transient process and appears as apoptotic debris sloughing off into the vascular lumen (Figure 1A); therefore, it cannot be identified unless the tissues are collected proximate to the onset of damage. Also, definitive identification of EC damage may require electron microscopy (Ownby, Bjarnason, and Tu 1978; Sharma and Kiyatkin 2009), immunolabeling of apoptotic cells (Lim, DeLano, and Schmid-Schonbein 2001; Mikaelian et al. 2010), quantification of ECs through immunolabeling (Copple et al. 2002), or assessment of ethidium bromide uptake (Lim, DeLano, and Schmid-Schonbein 2001) or vascular leakage (Wiener and Giacomelli 1973; Joris et al. 1982; Pino and Thouron 1983; Turek and Schoenlein 1993; Ohno et al. 1995; Gyomber et al. 1996; Joseph et al. 1996a; Albassam et al. 1999; Albassam et al. 2001; Schuettauf et al. 2006). Thrombi/thrombosis may be the sole manifestation of EC damage. However, thrombi/thrombosis was not included in the primary lexicon, because direct biomarkers of this end point were not sought.
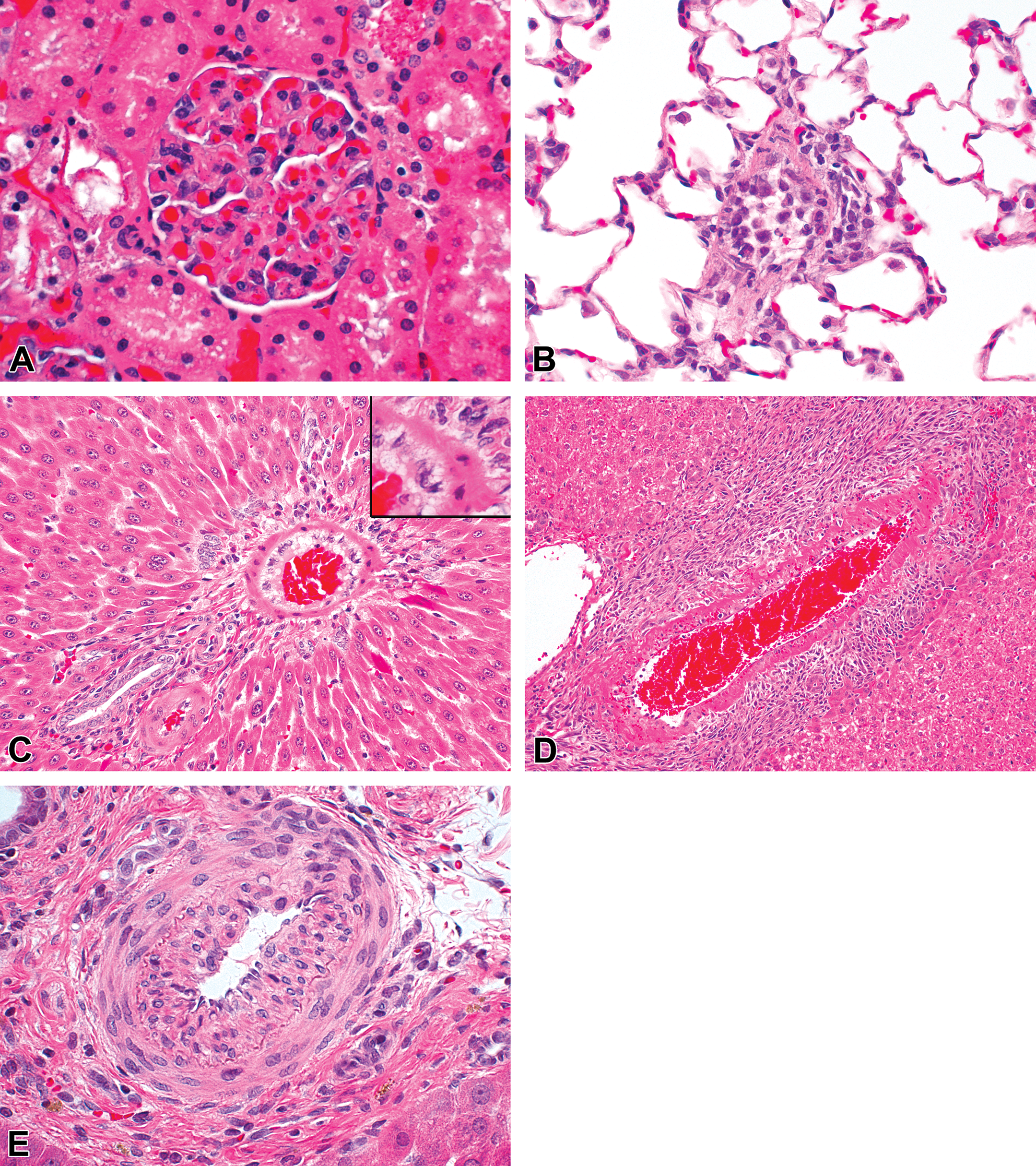
The main histomorphologic end points tracked by the lexicon of the VIWG of PSTC are degenerative and hyperplastic changes of ECs and VSMCs. Inflammation may be superimposed over these changes. The changes illustrated here consist of apoptosis of ECs in a glomerulus associated with microthrombosis of the glomerular loops secondary to necrosis of the wall of the afferent artery (A); endothelial hyperplasia/hypertrophy in the pulmonary artery and capillaries associated with monocytosis and adventitial histiocytic infiltrations (B); hyalinization of the VSMCs associated with endothelial hyperplasia/hypertrophy and vacuolation (C); VSMC necrosis in the portal space of the liver associated with endothelial hyperplasia/hypertrophy, and adventitial fibroplasia and histocytic, neutrophilic, and eosinophilic inflammation (D); spontaneous medial and intima VSMC hypertrophy in one of the main portal spaces of the liver associated with disruption of the internal elastic lamina, and pigment deposition and minimal mononuclear inflammation in the adventitia (E). All illustrations are courtesy of Hoffmann-La Roche, Inc., from 8- to 11-week-old Wistar Han rats administered undisclosed compounds (A, B, D), recombinant interleukin 2 (C), or the vehicle (E). EC = endothelial cell; PSTC = Predictive Safety Testing Consortium; VIWG = Vascular Injury Working Group; VSMC = vascular smooth muscle cell.
Endothelium, Hypertrophy/Hyperplasia
EC hypertrophy/hyperplasia (HH) is a grouped diagnosis that refers to an increase in size and/or number of ECs, respectively. It is characterized by rounded, larger, and more numerous ECs, often with a visible amount of cytoplasm that may be vacuolated (Figure 1B and C). As hypertrophy and hyperplasia often develop concurrently and are difficult to grade separately by routine histology, they are graded as a single finding. In addition, it is not the current strategy of the VIWG to differentiate along this continuum of morphologic changes. Special methods, such as immunolabeling to assess bromodeoxyuridine incorporation, may be needed to identify EC hyperplasia (Breider et al. 1999). Of note, EC HH generally is not graded in toxicology studies, because it is often secondary to primary findings affecting the parenchymal cells. However, it needs to be graded in DIVI biomarker qualification studies, as endothelial change is a primary end point for assessing biomarker performance. It is the canonical morphologic adaptive or physiological response of ECs, and it may be induced by a variety of stimuli, including administration of large amounts of saline (Morton et al. 1997), hypertension (Peach and Loeb 1987), exposure to cytokines (Raines and Ferri 2005), or primary injury to the media and/or adventitia. It may recover or proceed along the continuum of DAN (Zhang, Hanig, and De Felice 2012). Due to the fact that HH may progress to DAN, any putative biomarker for either of these two changes is more likely to inform about the onset of DIVI rather than inform about which of these two processes is occurring in vivo.
Vascular Smooth Muscle Cell Hyalinization
Vascular smooth muscle cell hyalinization is included in the lexicon to identify the early degenerative changes of VSMCs that may precede VSMC apoptosis/necrosis. At the ultrastructural level, it is characterized by vacuolation of VSMCs (Kobori et al. 1979; Joris and Majno 1981b). On routine histology, it is characterized by hypereosinophilia of the cytoplasm of VSMCs (Figure 1C). Hyalinization of the cytoplasm of VSMCs occurs prior to the onset of karyorrhexis/karyolysis, although the nucleus may appear condensed with ruffled borders. Vascular smooth muscle hyalinization is anticipated to be associated with leakage of constitutive proteins of these cells. It may be accompanied by dilation of the vasa vasorum and, unlike VSMC necrosis, the layered structure of the vessel is preserved.
Vascular Smooth Muscle Apoptosis/Necrosis
Vascular smooth muscle cell apoptosis/necrosis is the archetypical hallmark of DIVI caused by vasoactive compounds and hypertension. In the literature, this finding often is called medial arterial necrosis and arteritis/periarteritis. It is characterized in its acute stages by hypereosinophilia of the cytoplasm and karyorrhexis/karyolysis (Figure 1D). It may be accompanied by vasodilation of the vasa vasorum and medial hemorrhage. The change may be concomitant or followed by the deposition in the media and at times in the adventitia of a brightly acidophilic fibrillar to amorphous material representing fibrin admixed with various proportions of other serum proteins, and by inflammation which is graded separately. This appearance is commonly referred to as “fibrinoid necrosis.”
Vascular Smooth Muscle Hypertrophy/Hyperplasia
Hypertrophy/hyperplasia (HH) of VSMC may occur in the media and intima (Figure 1E) and is a common end-stage consequence of DIVI and of chronic hypertension (Limas, Westrum, and Limas 1980; Giacomelli, Juechter, and Wiener 1972). Similar to EC HH, VSMC HH often develop concurrently and cannot be graded separately by routine histology; therefore, they are graded as a single finding. Hypertrophied VSMCs often have a large vesicular nucleus with a ruffled nuclear membrane.
Inflammation, Granulocytic/Mixed/Mononuclear
Inflammation associated with DIVI is characterized by the severity and the specific leukocytes present in the inflammatory infiltrate. The types of inflammation in DIVI are granulocytic (neutrophils and/or eosinophils), mixed, and mononuclear (histiocytic and/or lymphocytic). Neutrophils, eosinophils, and/or macrophages tend to be the predominant leukocytes in the acute and subacute stages of DIVI. The diagnosis “leukocyte pavementing” was added to this list to describe leukocytes distributing to the periphery of the vessels and lining along the EC surface regardless of the type of leukocyte. This diagnosis was added, because rolling, arrest, and transmigration associated with leukocyte recruitment may be associated with endothelial activation, for which specific biomarkers are sought.
Other Changes
Other changes may include hemorrhage and extracellular matrix (ECM) deposition that is reported in some articles as fibroplasia/fibrosis, neovascularization, mineralization, rupture of the internal elastic lamina, and pigment deposition. These findings are recorded but are not used in the biostatistical analysis at this stage, because there is no attempt to find biomarkers specific for these findings. Of note, fibroplasia/fibrosis and neovascularization primarily affect the adventitia rather than the media.
Time Course of Progression of DIVI Caused by Vasoactive Compounds
The histopathologic time course of DIVI caused by vasoactive compounds is best documented in rats. It differs across drug classes, may be affected by route of administration, may be better identified by gene expression analysis than by histopathology in its earliest stages (Dagues et al. 2007), and appears to progress similarly in rats and in larger nonclinical species (Albassam et al. 1999; Albassam et al. 2001; Joseph et al. 1996b; Losco et al. 2004). Of note, the severity, but not the progression over time of the histomorphologic changes, is observed to be dose-dependent (Zhang et al. 2008).
In rodents, the onset of changes is acute for the PDE3i, SKF and 95654, and occurs as early as 1 hr postdose progressing over 24 hr (Zhang et al. 2002). The onset of changes is similar but progresses more slowly with the PDE4i, SCH351591, as medial necrosis of the mesenteric arteries occurs 72 hr after dosing (Zhang et al. 2008). Long-term dosing with the PDE3i ICI 153.110 results in severe HH and a plexiform appearance of the arterial vasculature (Westwood, Iswaran, and Greaves 1990). Long-term dosing with a PDE4i SCH 351591 in primates results in both acute type lesions and in medial hypertrophy and fibrosis (Losco et al. 2004). Fibrosis and VSMC HH are the anticipated sequelae (Albassam et al. 1999; Albassam et al. 2001; Bendeck et al. 2002), although complete to near complete resolution has been reported after short-term dosing with other vasomodulators causing similar initial injury (Albassam et al. 2001; Ikegami et al. 2001). It is thus inferred that some lesions of DIVI may fully resolve with time; however, the threshold of severity above which vascular sequelae occur is not known.
The local application in vivo on muscular arteries of the vasoconstrictor
Characterization of DIVI and Biomarker Target Validation by IHC and ISH
Knowledge of the distribution of the biomarkers in the healthy and damaged tissues is essential to biomarker qualification (Dunstan et al. 2011). The major advantage of IHC and ISH is their ability to localize the biomarker to specific cell types compared to Western blot, enzyme-linked immunosorbent assay, and quantitative reverse transcription polymerase chain reaction (qRT-PCR). Immunohistochemistry and ISH, however, may be less sensitive and specific and are not as amenable to precise quantification as other molecular methods. IHC and ISH are particularly indicated to monitor the expression of biomarkers that are highly expressed in the tissue/cell of interest and that may be released as the result of cellular damage, such as smooth muscle actin (ACTA2), transgelin (TGLN), or miR-145 (Wiener et al. 1996; Albassam et al. 1999; Brott et al. 2005; Supplemental Figure 1). These methods also may be used for biomarkers that are upregulated, that denote inflammatory cells, or markers that are deposited in the damaged vessels, such as complement, fibrin, or ICs (Supplemental Table 2).
The assessment of ECs is rendered difficult by the small size and the relatively small amount of cytoplasm of ECs. In addition, ECs are highly heterogeneous, as supported by the following observations: (1) most proposed markers of ECs, including miRNAs, are expressed differentially among vascular beds (Mandriota et al. 2001; Pusztaszeri, Seelentag, and Bosman 2006; Valadon et al. 2006) and ECs from different vascular beds respond differently to stimuli (Bindewald et al. 2004); (2) the common EC markers are dysregulated during vasculitis (Figure 2); and (3) there are disease-specific EC markers (Mandriota et al. 2001; Mutuberria et al. 2004; Liang et al. 2006). Thus, an important future step in the identification of novel safety biomarkers of DIVI will be the understanding of their expression as it relates to the physiologic status of individual ECs (Valadon et al. 2006). Conversely, the assessment of VSMCs by IHC and ISH is facilitated by the large size of these cells and/or the availability of multiple well-characterized markers, which are expressed homogenously in all VSMCs such as ACTA2, TGLN, and high-molecular weight caldesmon 1 (h-CALD1). The ease of assessing VSMCs damage by histomorphology is in contrast to the difficulty of developing corresponding circulating biomarkers. The assessment of the ECM and of the inflammatory cells relies on commercially available antibodies and special stains that are solidly established in the literature, and includes collagen types, vimentin, desmin, fibronectin, picrosirius red, Masson’s trichrome, and reticulin silver stain.
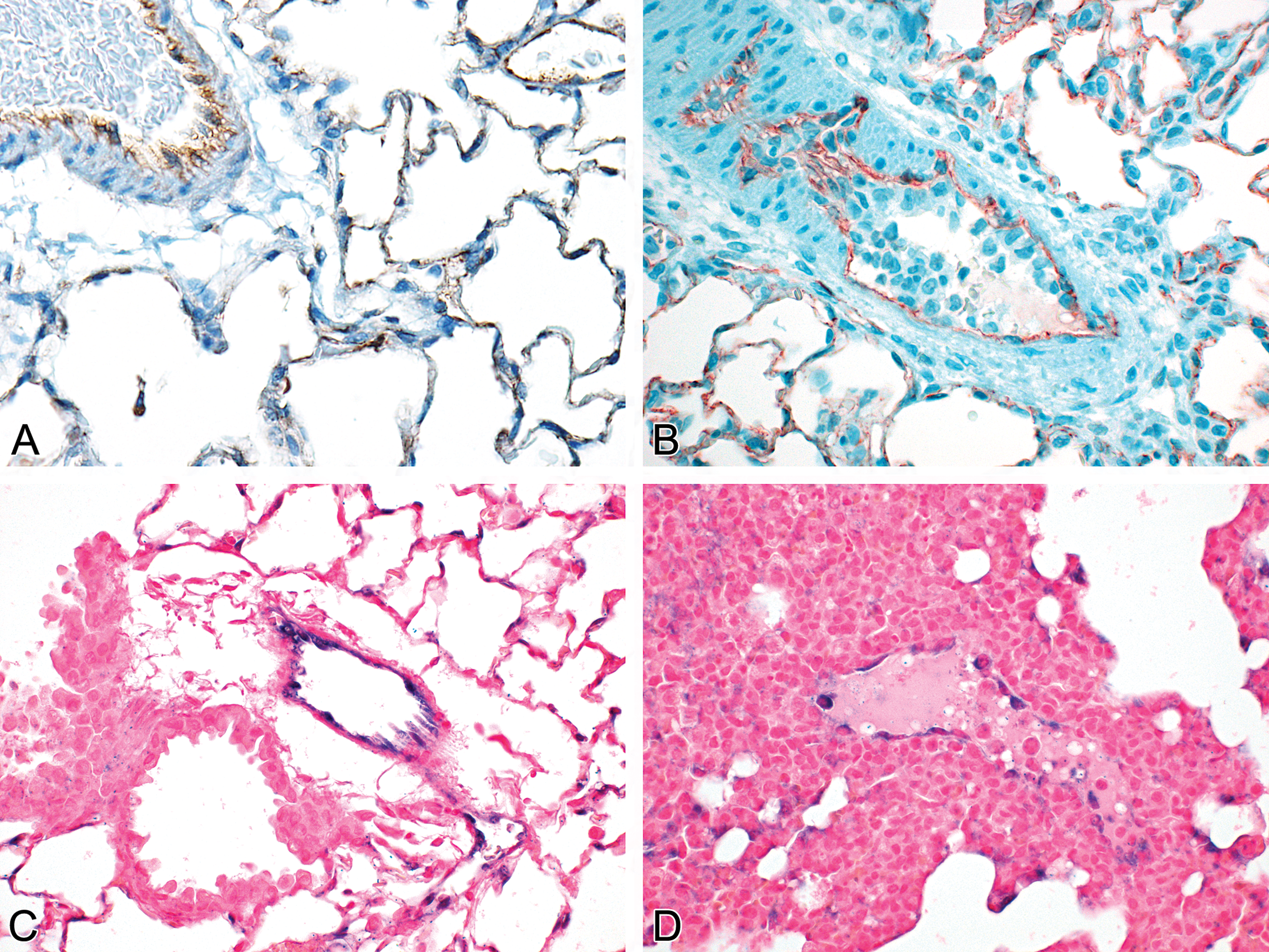
The expression of the hallmark proteins and miRNAs of ECs generally is dysregulated in DIVI. For example, CD31 in rat is upregulated in the alveolar capillaries during systemic endothelial activation (B) compared to a vehicle-treated animal (A). miR-126 expression in the lung of normal rats is nuclear and cytoplasmic and is observed in ECs of the pulmonary veins as well as in the alveolar capillaries (C); in contrast, miR-126 expression in activated ECs is predominantly cytoplasmic in a pulmonary vein and it is lost in most alveolar capillaries (D). All illustrations are courtesy of Hoffmann-La Roche, Inc., from animals administered undisclosed compounds. DIVI = Drug-Induced Vascular Injury; EC = endothelial cell; miRNA = microRNA.
Candidate Biomarkers
A list of potential biomarkers was collated from the literature, internal expertise, and past experience (Butcher 1991; Bevilacqua and Nelson 1993; Kerns et al. 2005; Dagues et al. 2007; Krupinski et al. 2007; Lin et al. 2008; Weaver et al. 2008; Martinez-Lemus, Hill, and Meininger 2009; Tummers et al. 2010; Weaver et al. 2010; Monach et al. 2011; Monach et al. 2012; Zhang, Hanig, and De Felice 2012; Supplemental Table 3). The biomarkers were categorized according to their association with compartments/inflammation of the vasculature and their potential expression or release during DIVI. The cellular response to degenerative processes in DIVI overall follows similar progression for all compounds although the mechanism of toxicity and the timing of the sequence of events may vary by compound (Mecklenburg et al. 2006; Tesfamariam and DeFelice 2007; Zhang et al. 2008). Thus, regardless of sequence of events and their pathophysiology, activation and damage to ECs, damage to VSMCs, and inflammation, as characterized by vascular leakage, inflammatory cell infiltrates, and modification of the ECM, are consistent features of all major types of DIVI. In light of these acknowledged features of DIVI, the list was narrowed to candidate biomarker constituents of ECs and VSMCs or biomarkers associated with inflammation and ECM homeostasis/remodeling (Sprague and Khalil 2009; Table 5). In addition to proteins/peptides, cytokines, chemokines, and biochemical biomarkers, consideration was given to circulating EMPs and miRNAs, which are addressed in separate sections of this article. Circulating mRNAs also were considered but not currently pursued because of limited supportive literature. Similarly, coagulation parameters such as platelet count, D-Dimer, Factor VII, and prothrombin time, which were historically used as biomarkers of VI, were not pursued, because they indicate consumption of coagulation factors rather than damage to a specific compartment/compartments of the vascular wall.
Final list of candidate rat biomarkers for selection by the VIWG of PSTC for DIVI qualification.

Note: DIVI = Drug-Induced Vascular Injury; PSTC = Predictive Safety Testing Consortium; VIWG = Vascular Injury Working Group.
aEC = endothelial cell. bVSMC = vascular smooth muscle cell.
The criteria for prioritizing biomarkers for qualification were as follows: (1) weight and breadth of evidence that a biomarker is specific and sensitive in at least one nonclinical species or, if not specific, can be used in combination with specific biomarker/biomarkers to add signal robustness, or sensitivity to the diagnosis; (2) evidence suggesting the biomarker, or its ortholog, is reflective of VI in other nonclinical species and humans; (3) availability of analytical tools to measure the biomarker in the serum or the plasma, and (4) the temporal and physiological robustness of the biomarker in vivo and ex vivo.
Exploratory (i.e., proof of concept) studies were conducted using reference vascular toxicants, which resulted in the elimination of some candidate biomarkers (Supplemental Table 3). Examples of excluded biomarkers include C-reactive protein, because it is a poor acute phase biomarker in rat (Watterson et al. 2009; Giffen et al. 2003); tumor necrosis factor α, because it was not temporally robust in rats administered the PDE4i CI-1044; monocyte chemotactic protein 1 (MCP1), because the magnitude of the change was minimal (data not shown); and proteins associated with VSMCs or with the remodeling of the ECM (with the exception of H1 calponin (CNN1), and tissue inhibitor of metalloproteinase 1(TIMP1)), because reliable rat assays could not be identified or developed.
In the sections on EMPs, EC, VSMC, and inflammation biomarkers, we present the results of an exploratory study conducted in rats administered 1 or 2 doses of the PDE4i, CI-1044, at 60 mg/kg. Histomorphologic and clinical pathology end points were evaluated at 4, 8, 16, and 24 hr after administration of the first dose and at 24, 48, and 192 hr after administration of the second dose (Supplemental Appendix 5).
Biomarkers of EC Activation/Damage
The strategy to identify biomarkers of EC activation/damage was guided by the significant body of literature that describes EC activation/damage ultrastructurally and mechanistically in CDIV as well as in animal models (Zhang et al. 2002; Zhang et al. 2008; Mecklenburg et al. 2006; Dietsch et al. 2006; Koshi et al. 1993; Bannerman and Goldblum 2003; Rao et al. 2007; Chowdhary et al. 2007; Reidy and Schwartz 1983; Dosso, Leuenberger, and Rungger-Brandle 1999; Ashford and Frieiman 1967; Zhang et al. 2011). Biomarkers of EC damage are anticipated to lack specificity for DIVI, because EC activation also occurs in other diseases or toxicities, most notably inflammation and cancer.
The activation of ECs is associated with leukocyte rolling and adhesion mediated primarily by the expression on ECs of vascular cell adhesion molecule 1 (VCAM1), intercellular adhesion molecule 1 (ICAM1), and E-selectin (SELE; Zhang et al. 2011; Bratt and Palmblad 1997; Salmi and Jalkanen 2001; Butcher 1991; Bevilacqua and Nelson 1993). The initial phase of the DIVI response is followed by a phase of angiogenesis involving pro- and antiangiogenic signals. In response to the production of endothelial-derived nitric oxide (NO; see Inflammation Biomarkers section), ECs produce EDN1, which causes vasoconstriction and proliferation of VSMCs (Yanagisawa et al. 1988; Suzuki et al. 2006). This proliferative response is balanced by the release of antiangiogenic factors, including prostacyclin that causes vasodilation (Dusting, Moncada, and Vane 1977), angiopoietin 2 (ANGPT2) that is released in the presence of vascular endothelial growth factor A (VEGFA) from the Weibel–Palade bodies of ECs, and thrombospondin 1 (THBS1).
In the exploratory study with CI-1044, the activation phase of ECs was not identified through evaluation of serum concentrations of VCAM1 and ICAM1, because their levels were unchanged in rats with DIVI. The angiogenesis phase was identified by marked increases in serum ANGPT2 and slight increases in VEGFA and EDN1 24 hr after administration of the first dose, and 24 and 48 hr after administration of the second dose (Figure 3). Other changes consistent with endothelial damage consisted of marked decreases of SELE in serum samples collected 24 and 48 hr after the second dose, a marked increase of serum total nitrite (NO2 −) 48 hr after the second dose, and a slight decrease in THBS1 48 hr after the second dose.
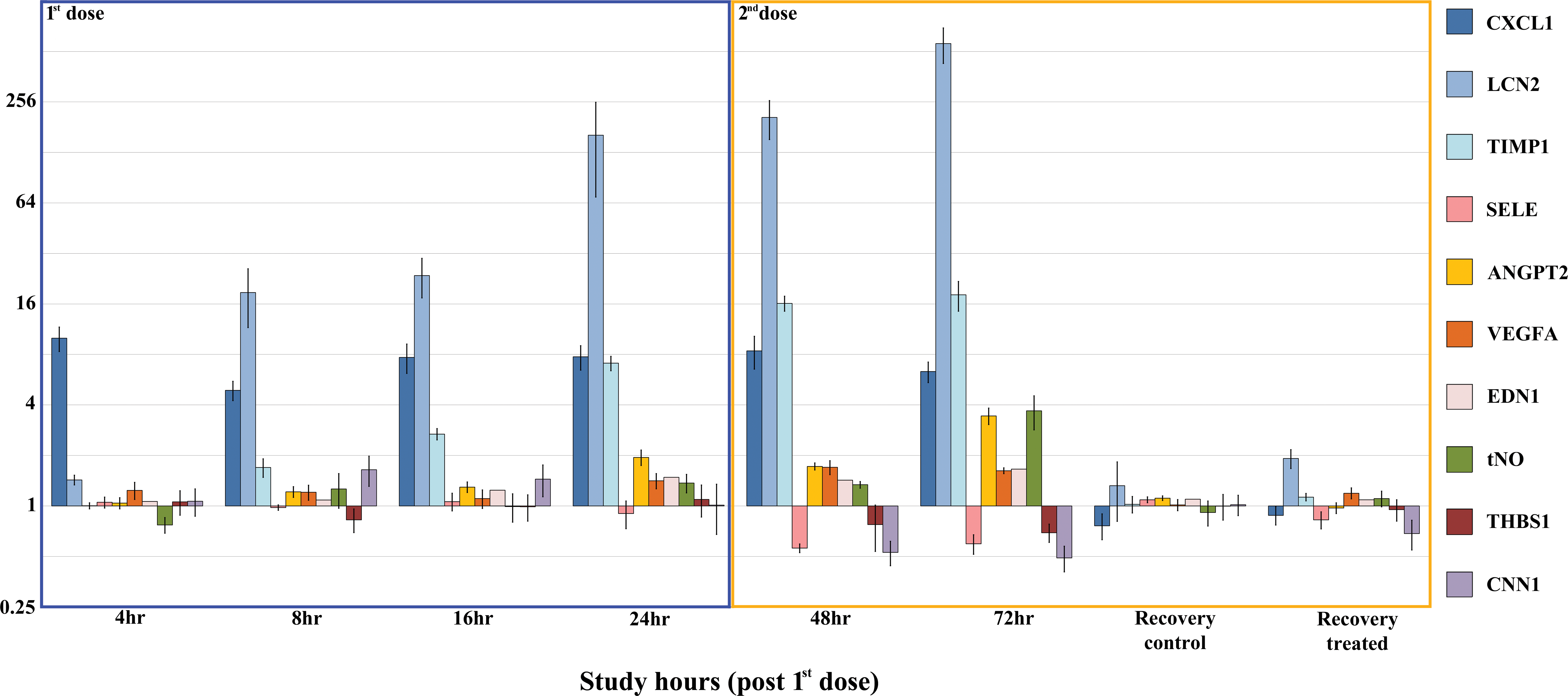
Biomarker fold change from mean of the vehicle-treated rats (Log2) ± standard error in male Sprague-Dawley (SD) rats administered 60 mg/kg of the PDE4 inhibitor CI-1044. The rats were dosed via oral gavage with either vehicle (5% methylcellulose) or 60 mg/kg CI-1044 (Pfizer, Inc). Terminal plasma samples were collected from 4 or 5 treated and 5 control animals at 4, 8, 16, and 24 hr after a single dose and 24, 48, and 192 hr after a second dose of CI-1044. Microscopic evaluation of the mesenteric arteries revealed mild perivascular inflammation in 5 of the 5 animals by 24-hr postdose (although minimal inflammation was observed microscopically as early as 8 hr) and fibrinoid necrosis in 1 of the 5 animals. All animals were maintained according to the NIH standards established in the Guide for the Care and Use of Laboratory Animals. The Pfizer Animal Care and Use Committee (IACUC) approved all experimental protocols. NIH = National Institutes of Health; PDE4 = phosphodiesterase 4.
Biomarkers of VSMC Damage
Most candidate VSMC biomarkers were selected based on their high expression in VSMCs. Their expression is reported to decrease in VSMC in the prototypical VI, a process that is anticipated to be concurrent with their leakage into the circulation (Albassam et al. 1999; Nagel et al. 2011; Kitagaki et al. 2012; Supplemental Figure 1). These candidate biomarkers are ACTA2, smoothelin, TGLN, CNN1, and h-CALD1. The expression of some of these proteins is influenced by the maturity and degree of hypertrophy of VSMCs (Walsh 2000; Matthew et al. 2000; van Eys, Niessen, and Rensen 2007; Frid, Moiseeva, and Stenmark 1994; Frid et al. 1997; Hu et al. 2008). In response to the altered redox state of the tissues, caveolin 1 (CAV1) is produced and released from ECs, pericytes, and VSMCs leading to eNOS downregulation, which leads to vasoconstriction. Except for CNN1, which currently is undergoing analytical validation, the development of assays for these candidate biomarkers in rats was unsuccessful thus far. In rats administered CI-1044, there was modulation of serum CNN1 with an early mild increase 8 hr after administration of the first dose, followed by a robust decrease 24 and 48 hr after administration of the second dose (Figure 3).
Inflammation Biomarkers
Candidate biomarkers for the inflammatory component of DIVI are released primarily by inflammatory cells in response to tissue stimulation/damage and remodeling. However, the source of some inflammation biomarkers is not clear, as they may be released by cells other than the inflammatory cells. For example, interleukin 6 (IL6) may be released by ECs. Many inflammation-related markers were considered with the understanding that inflammation biomarkers are not specific for DIVI. In the exploratory study with CI-1044, an inflammation biomarker signature was identified and correlated well with the pathologic process of DIVI (Figure 3). At 4 hr after administration of the first dose, serum chemokine (C-X-C motif) ligand 1 (CXCL1; KC/Gro/Cinc) concentrations were increased robustly, initiating the inflammatory process by recruiting and activating neutrophils. Serum lipocalin-2 (LCN2) concentrations were increased markedly 8 hr after administration of the first dose, suggesting the continuation of the inflammatory process, as LCN2 is produced predominantly by activated neutrophils. TIMP1 is a modulator of LCN2 and matrix metalloprotease, and it increases in serum of rats prior to and at the onset of DIVI caused by administration of a single high dose of CI-1044 (Dagues et al. 2007). In parallel with the increase in LCN2 serum TIMP1 concentrations were slightly increased 8 hr after administration of the first dose and continued to increase over time and after the second dose. As already mentioned, serum NO2 − was increased after administration of the second dose.
Other inflammation-related markers were assessed in this study, including the proinflammatory acute phase cytokines interleukin-1β and IL6, and MCP1 and macrophage inflammatory protein 3a (MIP3A). Macrophage inflammatory protein 3a and IL6 were noticeably increased 24 hr after the second dose (data not shown). In addition, others have reported increases in CXCL1, TIMP1, α-1 acid glycoprotein 1 (AGP1, rat oromuscoid 1, i.e., Orm1), and serum NO2 − in rats with histologically confirmed DIVI after administration of PDE4 inhibitors (Weaver et al. 2008; Dagues et al. 2007). The temporal and robust response of CXCL1, LCN2, TIMP1, AGP1, and serum NO2 − met the early decision criteria for robust inflammation biomarkers that may be used together with more specific markers of VI, or in the context of a compound causing DIVI. Serum total NO2 − was grouped with the inflammatory biomarkers as per the literature although activated ECs are a major source of NO2 −.
Endothelial Microparticles
Microparticles (MPs) are small membrane vesicles (0.1–1 µm in diameter) that are shed from cells in response to activation, injury, inflammation, and/or apoptosis and can be detected in plasma using flow cytometry in combination with specific cell surface antibodies (Jimenez et al. 2001; Rautou et al. 2011; Combes et al. 1999). The formation of MPs by necrotic and apoptotic cells, especially for EMPs, is considered a process that prevents the formation of microemboli (Xu et al. 2005). Because MPs are derived from the cell membrane they contain antigens specific to the cell of origin and also may display antigens that indicate the overall physiological status of the cell of origin (Jimenez et al. 2003). MPs also contain cellular constituents from the cell of origin and may participate as a vector in the exchange of biological information between cells, including through the transfer of miRNAs (Mause and Weber 2010; Diehl et al. 2012; Andriantsitohaina et al. 2012; Orozco et al. 2008). The presence of MPs has been described in a number of biofluids (Smalley et al. 2008; Berckmans et al. 2011; Berckmans et al. 2005; Mrvar-Brecko et al. 2010). The sources of MPs in the blood include leukocytes, platelets, red blood cells, and ECs.
The evaluation of EMPs as biomarkers of nonclinical DIVI is supported by an abundance literature that reports elevated or altered EMP levels in patients with vasculitides (Mallat et al. 2000; Bernal-Mizrachi et al. 2003; Williams et al. 2007; Arteaga et al. 2006; Sabatier et al. 2002; Preston et al. 2003; Jimenez et al. 2001; Amabile et al. 2008; Bakouboula et al. 2008; Koga et al. 2005; Erdbruegger et al. 2008; Brogan et al. 2004; Combes et al. 1999). However, there are challenges associated with the use of EMPs as biomarkers of DIVI, including the assay itself, because there are no standard laboratory methods for EMP analysis (Mrvar-Brecko et al. 2010; Orozco and Lewis 2010; Ahn et al. 2007), and studies assessing the specificity of these EMPS for DIVI are lacking (Sharma and Kiyatkin 2009; Jimenez et al. 2003).
In the exploratory study with CI-1044, significant increases in VCAM1+ (CD106+) EMP absolute counts occurred at 8, 24, and 48 hr after the administration of CI-1044 and returned to baseline during the 30-day recovery phase (Enerson et al. 2010). The changes in absolute counts of VCAM1+ EMPs also correlated with microscopic observations of VI.
MicroRNAs
MicroRNAs are small noncoding RNAs that regulate gene expression and play critical roles in many biological and pathological processes (Mikaelian et al. 2012; Chen et al. 2008; Baehrecke 2003; He et al. 2009; Krichevsky et al. 2003). MicroRNAs inhibit translation of target mRNA and/or promote degradation of mRNA, thereby modulating targeted pathways. A single miRNA/miRNA cluster may act to control expression of several genes in concert, acting as a master switch. For example, the miR-17-92 cluster promotes angiogenesis in tumors (Urbich, Kuehbacher, and Dimmeler 2008) by targeting several antiangiogenic proteins (Dews et al. 2006). In addition to their pleiotropic effects on mRNAs, individual miRNAs may influence the expression of other miRNAs (Tuccoli, Poliseno, and Rainaldi 2006). The biogenesis, processing, and functional characteristics of miRNA have been reviewed extensively (Liu et al. 2010b; Boyd 2008; Choudhuri 2010).
MicroRNAs can be detected using qRT-PCR or hybridization-based technologies, which are simple to set up and have a high level of sensitivity and specificity. MicroRNAs are prospective noninvasive biomarkers for diseases/conditions beyond DIVI, because of their relative stability and presence in all tested body fluids, including blood (Saunders and Lim 2009). The stability of miRNAs in RNAse-rich environments is conferred by their encapsulation in membrane-bound vesicles such as exosomes and/or their association with proteins such as Argonaute 2, which also affords them their function (Arroyo et al. 2011; Valadi et al. 2007; Holford et al. 2008; Wang et al. 2010; Jung et al. 2010; Patnaik, Mallick, and Yendamuri 2010; Mraz et al. 2009; Chen et al. 2008). Another advantage of miRNAs as biomarkers compared with proteins is that miRNAs offer reduced complexity in body fluids and the absence of posttranslational modifications. Nonclinical and clinical work, however, to validate miRNAs as biomarkers is in its early stages, and the members of the VIWG are actively pursuing miRNAs as circulating and tissue biomarkers of DIVI. Also, the methods for collection, measurement, and interpretation of these data are still in development.
Release of miRNAs into the circulation is likely to be cell-, time-, and/or miRNA-specific. Therefore, the relationship between tissue and circulating miRNA levels is presently poorly understood. MicroRNAs are released passively during cell death and actively during cell–cell signaling. Factors affecting stability, complex formation, and release of miRNAs may also differentially affect the extractability of a particular miRNA in circulation. Increased tissue expression of certain miRNAs may correlate with an increase in circulating levels (Caporali et al. 2011; Di Stefano et al. 2011), while the phenomenon of inverse correlation between tissue and peripheral miRNA changes also has been reported (Bostjancic et al. 2010; D’Alessandra, Pompilio, and Capogrossi 2011; Wang et al. 2010; Thomas et al. 2012).
MicroRNAs have been implicated in the responses of ECs, VSMCs, and the adventitia to injury (Albinsson and Sessa 2011). They have been described as regulators of key signaling pathways involved in vascular and cardiovascular biology (Small and Olson 2011; Di Stefano et al. 2011; Fish and Cybulsky 2012; Hartmann and Thum 2011; Santoro 2011; Urbich, Kuehbacher, and Dimmeler 2008) and pathologic response, to include regulation of inducible adhesion molecules (Sen et al. 2009); EC activation via the NFκB signal transduction pathway (Fish and Cybulsky 2012); antiproliferative/antiangiogenic signaling via attenuation of eNOS expression (Chan et al. 2012; Chan et al. 2009; Katare et al. 2011; Liu et al. 2010a; Rippe et al. 2012); and control of arterial vascular tone through a paracrine role for periadventitial adipose tissue (Auger, D’Orleans-Juste, and Germain 2007; Gollasch 2012; Gollasch and Dubrovska 2004; Verlohren et al. 2004; Estep et al. 2010; Ugras et al. 2011). Dysregulation of several of the miRNAs that are key players in cardiovascular biology and pathology occurs at the site of DIVI and in circulation in several models of acute DIVI in the rat (Thomas et al. 2012).
In situ hybridization is a key follow-up to microarray and/or qRT-PCR experiments to identify expression of miRNAs at a cellular level (Zimmerman et al. 2003). The locked nucleic acids technology (LNA™, Exiqon, Woburn, MA) applied to ISH has enabled the localization of miRNAs in tissues. miR-134 and miR-126 dysregulation was identified using this technology on tissues with DIVI from multiple animal species and models (Thomas et al. 2012 this report; Supplemental Figure 1). Of note, miR-134 was upregulated in the endothelium, the media, and adventitia in the areas of DIVI, but not in the adjacent seemingly unaffected tissues and in unaffected areas of the same vessel (Thomas et al. 2012). Finally, an exploratory study demonstrated that the expression of miR-145, a marker of VSMC, is markedly decreased in VSMC at the onset of DIVI in several animal models with a pattern similar to that of the loss of ACTA2 and TGLN (Supplemental Figure 1).
Candidate Genomic Tissue Markers of Nonclinical DIVI
Identification of a signature mRNA profile of DIVI in rat
Several alternative strategies have been employed to enhance the odds of successfully identifying new biomarkers to distinguish nonclinical DIVI from other toxicities and/or inflammatory conditions. In particular, laser capture microdissection recently was used to identify candidate genomic markers of DIVI in the endothelial and medial compartments of the mesenteric arteries of rats following administration of 28 different compounds, including some causing VSMC necrosis (Dalmas et al. 2008; Dalmas et al. 2011). In rats administered fenoldopam, a genomic profile was identified, and 92% of the genes in this profile were dysregulated prior to the onset of histomorphologic changes (Table 6; Dalmas et al. 2011). The specificity of this gene panel for DIVI is supported by the observation that these genes were not dysregulated in rats administered yohimbine, an α 2-adrenergic receptor antagonist that produces marked vasodilation but no VI as assessed by histology. Functionally, most genes differentially regulated in both ECs and VSMCs correspond to proteins involved in protein transport, cellular metabolism/energy, and cell movement; only 3% of the genes of ECs were associated with immune function and inflammation. Thus, the candidate genes were considered to represent signals derived from the vasculature itself and not from the secondary inflammatory and acute phase responses as in earlier studies (Dagues et al. 2007).
Candidate genomic markers of medial arterial necrosis (p < 0.01, fold change ≥ 1.7) in the intima- and/or media-enriched samples of mesenteric arteries following in vivo treatment of rats for 4 days with known vascular toxicants and nontoxicants (negative controls).
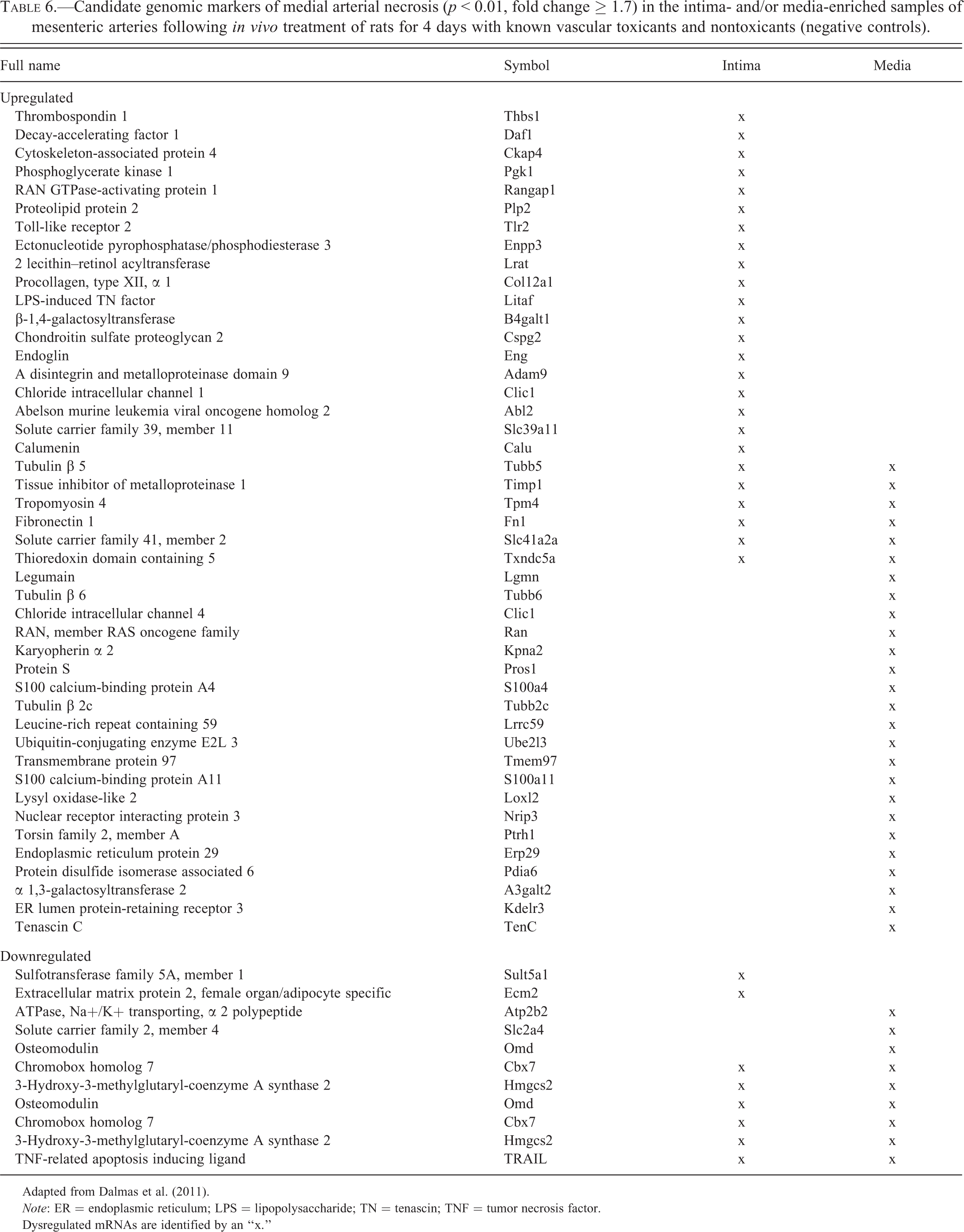
Adapted from Dalmas et al. (2011).
Note: ER = endoplasmic reticulum; LPS = lipopolysaccharide; TN = tenascin; TNF = tumor necrosis factor.
Dysregulated mRNAs are identified by an “x.”
Gene panel predictivity
The panels of genomic biomarkers ideally should be predictive of DIVI, that is, they should respond before the onset of histomorphologic changes. Thus, the usefulness of the nonclinical candidate gene panel, identified from both single dose (1- and 4-hr postdose) and repeat dose (4 daily doses) administration of fenoldopam to rats, was evaluated in samples of mesentery.
Thirty-one percent of the genes identified in a single-dose fenoldopam study were also differentially regulated prior to the onset of histomorphologic findings in rats administered multiple doses of fenoldopam (Dalmas et al. 2011). Based on the regulation of genes at time points prior to histologic vascular damage, these genes are likely to predict the occurrence of mesenteric DIVI in rats. Many of these genes are consistent with the circulating proteomic biomarkers proposed by Kerns et al. (Kerns et al. 2005) and by the VIWG of PSTC (Table 6) as candidate nonclinical biomarkers of DIVI. In EC-enriched samples, these genes include von Willebrand Factor, THBS1, and tissue plasminogen activator, which were upregulated in ECs and/or VSMCs at 1 and/or 4 hr after dosing and prior to histologic evidence of vascular damage (Dalmas et al. 2008). In VSMC-enriched samples, these genes included smooth muscle α actin and CAV1, which may also originate from the fibroblasts and myofibroblasts (Dalmas et al. 2008).
Although further validation still is required to qualify genomic candidate tissue markers, a gene panel now is available to aid in the prediction and identification of mesenteric DIVI in the rat (Dalmas et al. 2008; Dalmas et al. 2011). This gene list provides an opportunity to identify additional circulating proteomic biomarkers of DIVI, because several of these genes correspond to secreted proteins.
In Vivo and Ex Vivo Imaging of DIVI
While many platforms and methods are available for imaging blood vessels in basic cardiovascular research, ultrasound imaging is the primary noninvasive platform used both nonclinically and clinically. The ultrasound modalities for imaging blood vessels include Doppler blood flow measurement, shear (frictional) stress assessment, and molecular imaging. Ultrasound imaging is a relatively inexpensive technique with widespread clinical availability and does not require radio tracers. Computed tomography (CT) and magnetic resonance imaging (MRI), although used in the clinic, are not used routinely in nonclinical species; therefore, CT and MRI are not presented here. In addition to imaging, the measurement of systemic arterial and venous blood pressure is an important tool in the identification of cardiovascular toxicity, including DIVI.
The strategy of the VIWG is to use in vivo imaging ad hoc to document the onset of damage in tissues. In vivo imaging also may be used as a screening tool to differentiate among drug candidates in a series, and some modalities of imaging have accepted translational application. In vivo imaging, however, is not anticipated to be used as a routine biomarker in support of the qualification studies performed by the VIWG. Ex vivo imaging may be used on a case-by-case basis to confirm the distribution and quantify the expression of certain circulating candidate biomarkers (Gonzalez et al. 2013).
Changes in systemic and regional hemodynamics have long been associated with nonclinical DIVI, and familiar cases include DIVI in rat mesentery and canine coronary arteries after administration of vasodilators such as minoxidil, adenosine agonists, and PDE3is. In these instances, the hemodynamic changes can be linked to the vascular pathology and serve as translatable biomarkers. Other compounds, however, can cause DIVI in these same arteries without the associated change in systemic hemodynamics. In some of these cases, such as with endothelin receptor agonists and the PDE4i CI-1044, it has been demonstrated that significant regional changes in blood flow are present in arteries at doses that lack systemic hemodynamic changes (Korkmaz et al. 2009; Louden et al. 2000), indicating that systemic hemodynamic parameters lack sensitivity in specific instances. Because of this limitation, the VIWG is not using systemic hemodynamic parameters as primary biomarkers of DIVI or as qualification end points. In these instances, Doppler blood flow measurement by ultrasonography may be used to monitor regional blood flow alterations in large-caliber vessels prior to the occurrence of histomorphologic findings or in the absence of systemic hemodynamic changes. It is still not possible, however, to reliably image medium- and small-caliber vessels, which, therefore are monitored indirectly by measuring the blood flow of the arteries that feed into the affected vascular beds as a surrogate marker. Quantitative alterations in shear stress also may be used to monitor DIVI and are performed in nonclinical species using high-frequency ultrasonography (Niu et al. 2012).
Dynamic measurement of EC dysfunction, as performed by ultrasonography, refers to an imbalance between vasodilating and vasoconstricting substances produced by (or acting on) the endothelium (Faulx, Wright, and Hoit 2003; Munzel et al. 2008; Deanfield et al. 2005). High-resolution ultrasound has become a standard noninvasive technique for clinical assessment of endothelial dysfunction and cardiovascular disease risk through quantification of flow-mediated dilation of the brachial artery (Celermajer et al. 1992). Endothelial dysfunction, as measured by reduced flow-mediated dilation, has been associated with human vasculitides (Filer et al. 2003; Noto et al. 2009) and tracks with other markers associated with cardiovascular inflammation (Konduracka et al. 2008). Techniques for measuring flow-mediated dilation in nonclinical species have been described for dog and rat (Heiss et al. 2008; Puglia et al. 2006; Knight et al. 2013). Despite the potential for easy translation into the clinic, there are no published studies of flow-mediated dilation in nonclinical animal models of DIVI.
Vascular lesions are associated with expression of markers that are potential targets for contrast molecular ultrasound imaging. Ultrasonographic molecular imaging is enabled by conjugating ligands or antibodies directed against targets expressed in the vascular lumen to ultrasound contrast agents. The contrast agents usually comprise encapsulated gas-filled microbubbles detected by the acoustic signal produced when they oscillate in an acoustic field. With this technology, VCAM1-targeted microbubbles were able to detect early inflammatory processes in aortas of apolipoprotein E1−/− mice and discriminate the severity of the inflammatory burden (Kaufmann et al. 2007). Other relevant published imaging targets include the adhesion molecules P-selectin, ICAM1, and the integrin adhesion receptor αVβ3 (Weller et al. 2003; Ellegala et al. 2003; Kaufmann et al. 2010). Of note, the targets generally can be visualized only in the vascular lumen; therefore, DIVI primarily affecting the media and adventitia may not be detectable by contrast-enhanced U.S. microimaging. In addition, while nonclinical applications exist, there are no molecular contrast agents approved for human use (Chadderdon and Kaul 2010), and ultrasound-generated microbubbles may themselves induce vascular lesions (Miller et al. 2008).
The use of fluorescently labeled probes also has emerged as a promising ex vivo approach for the visualization and quantification of DIVI in the mesentery of rats (Gonzalez et al. 2013). The utility of these probes in the rat was demonstrated in hemodynamic-mediated injury caused by fenoldopam, and also by allylamine, which causes VSMC toxicity as a result of its degradation into acrolein intermediates (Conklin et al. 2001). The degree of matrix metalloproteinase activity and increases in vessel permeability, using MMPSense™ and AngioSense™, respectively, in mesenteric arteries correlated well with the severity of histopathologic findings (Gonzalez et al. 2013), namely perivascular inflammation and medial degeneration. These findings were further supported by IHC staining for matrix metalloproteinase protein 3, which exhibited a clear increase in abundance in most arterial wall smooth muscle cells following allylamine treatment and throughout the perivascular connective tissue for both fenoldopam and allylamine compared to the vehicle control.
Summary and Future Prospects
The VIWG was founded to identify and qualify circulating safety biomarkers of nonclinical DIVI. The first step of this effort, which is summarized previously, entailed the development of a strategy and a toolbox of investigative and analytical methods by the PSTC’s members. This toolbox includes a common understanding of the pathophysiology and time course of DIVI, a biostatistically enabled histomorphologic lexicon, and a succinct list of candidate biomarkers tested in exploratory studies using analytically validated assays. These candidate biomarkers are in the process of being evaluated through the qualification process, with the aim of accessing a complementary clinical profile through collaboration with SAFE-T. In addition, there has been and continues to be a significant amount of research focused on understanding the mechanisms of DIVI as well as on investigating and developing additional tools and technologies to identify the initiation and progression of DIVI. This research has produced significant advances in our understanding of the pathogenesis of DIVI and promises to bring additional biomarkers to a state of maturity sufficient to enter the formal qualification process.
The second phase of the activities of the VIWG is under way and consists of the formal biological qualification of these candidate biomarkers using multiple confirmatory studies performed across the member companies. The VIWG will submit the results and their interpretation to the 3 major regulatory agencies (FDA, the EMA, and the PMDA) to apply for biomarker qualification. The anticipated outcome of this feedback is that, following the example of the Nephrotoxicity Working Group of PSTC (Dieterle et al. 2010), the VIWG will have formally qualified biomarkers of DIVI as outlined by the FDA (FDA 2010), EMA (EMA 2012), and the PMDA (Ichimaru, Toyoshima, and Uyama 2010; PMDA 2010). The DIVI safety biomarker/biomarkers and/or biomarker panel/panels are not intended to replace or supersede histological evaluation and thus would not be used in isolation to define a no observed effect level and safety margin (i.e., not to serve as a predictive biomarker). However, in the future, the nonclinical and clinical validation of these makers may result in their use to guide drug safety decisions. PSTC will collaborate with the SAFE-T Consortium and other research groups to achieve translation from nonclinical species to humans. Overall, the VIWG has leveraged internal and external resources to develop a solid program of nonclinical DIVI biomarker research that is anticipated to aid in the identification and qualification of translatable biomarkers for DIVI.
Footnotes
Authors’ Note
This contribution does not necessarily reflect the official views, policies, or positions of the companies or institutions that employ any of the authors. The authors are listed in alphabetical order and contributed through a collaborative effort.
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Abbreviations
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.