
Abstract
Comparative nonclinical studies were conducted with the proposed biosimilar PF-05280586 and rituximab-EU (MabThera®). In side-by-side analyses, peptide maps and complement-dependent cytotoxicity assay results were similar. Sexually-mature cynomolgus monkeys were administered PF-05280586 or rituximab-EU as a single dose of 0, 2, 10, or 20 mg/kg on day 1 and observed for 92 days (single-dose study) or as 5 weekly injections of 0 or 20 mg/kg and necropsied on day 30, the day after the 5th dose, or on day 121 (repeat-dose study). The pharmacokinetic and pharmacodynamic profiles for both molecules were similar. Marked depletion of peripheral blood B cells 4 days after dosing was followed by near or complete repletion (single-dose study) or partial repletion (repeat-dose study). In the single-dose study, anti-drug antibodies (ADA) were detected by day 29 in all animals administered PF-05280586 or rituximab-EU and persisted through day 85, the last day tested. In the repeat-dose study, ADA were detected on day 121 in 50% of animals administered PF-05280586 or rituximab-EU. Both molecules were well tolerated at all doses. In all endpoints evaluated, PF-05280586 exhibited similarity to rituximab-EU.
Keywords
Introduction
Biosimilars are biologic drugs purposefully designed to have identical amino acid sequence; highly similar physiochemical and functional properties; and comparable pharmacologic activity, safety, and efficacy profiles to the approved marketed biologic. However, unlike generic small molecule drugs, creating an exact copy of the therapeutic biologic protein is impossible due to the complexity of the proprietary manufacturing process that is integral to the final drug product (Chirino and Mire-Sluis 2004; Schellekens 2004). Thus, the biosimilar can never be completely identical to the marketed biologic product. Consequently, regulatory agencies evaluate potential biosimilars based on their level of overall similarity to the marketed biologic product and specific regional biosimilar regulatory guidance (Kozlowski et al. 2011). For example, the European Medicines Agency (2005) requires in-depth comparative physicochemical and functional studies to “generate evidence substantiating the similar nature, in terms of quality, safety and efficacy, of the new similar biological medicinal product and the chosen reference medicinal product authorised in the Community” (Committee for Medicinal Products for Human Use [CHMP] 2005, 2012). The U.S. Food and Drug Administration (FDA 2012) defines biosimilarity as “the biological product is highly similar to the reference product, notwithstanding minor differences in clinically inactive components” with “no clinically meaningful differences between the biological product and the reference product in terms of the safety, purity, and potency of the product.” The FDA also requires the biosimilar to have the same amino acid sequence as the licensed product, notwithstanding minor modifications or truncations at the NH2- and COOH-terminus (commonly found in biologic manufacturing). The World Health Organization (WHO Expert Committee on Biological Standardization 2010, 2012), Canada (Canada Ministry of Health 2008), and several Asian (Korea Food and Drug Administration 2012; Government of India 2012) and South American countries such as Brazil (Castanheira, Barbano, and Rech 2011) have published guidelines for the regulatory approval of biosimilar products.
Rituximab (marketed as Rituxan® in the United States and MabThera® in the European Union) is a genetically engineered chimeric mouse/human IgG1 anti-CD20 monoclonal antibody (mAb) approved for the treatment of non-Hodgkin’s lymphoma, chronic lymphocytic leukemia, and moderate-to-severe rheumatoid arthritis (Genentech Inc 2010; Roche Pharma 2010; Reff et al. 1994). Rituximab binds to CD20 on the surface of B cells and elicits a signaling cascade resulting in cell death. Treatment with rituximab causes depletion of CD20+ B cells in peripheral blood and tissues, thereby reducing antigen presentation and activation of T cells, autoantibody production, and cytokine production (Edwards and Cambridge 2005, 2006; Edwards et al. 2004). PF-05280586 is a genetically engineered IgG1 anti-CD20 mAb with the same amino acid sequence as rituximab and is in clinical development as a potential biosimilar to rituximab-EU (MabThera®) and rituximab-US (Rituxan®). It is currently in a phase I/II clinical trial in patients with rheumatoid arthritis (REFLECTIONS B328-01; ClinicalTrials.gov, NCT01526057). In the studies reported here, we present comparative nonclinical data on the peptide maps and complement-dependent cytotoxicity (CDC) activity of PF-05280586 and rituximab-EU (MabThera®). We also present comparative pharmacokinetic (PK), pharmacodynamic (PD), and safety profiles of PF-05280586 and rituximab-EU following single- or repeat-dose administration to cynomolgus monkeys).
Methods
Comparative Biochemical and Functional Characterization
To determine the level of physiochemical similarity, tryptic peptide maps were generated for both PF-05280586 and rituximab-EU and resolved by reverse-phase high-performance liquid chromatography (LC) on a BioSuite C18 (2.1 × 150 mm) column (Waters Corporation, Milford, MA) and monitored at 214-nm ultraviolet absorbance. The identity of each peptide was determined by LC/mass spectrometry (MS) using an ultra–high-resolution quadrupole time-of-flight MS. To determine the level of functional similarity, both PF-05280586 and rituximab-EU were analyzed simultaneously using an optimized complement-dependent cytotoxicity (CDC) assay with Ramos cells, a human B lymphoblast cell line that expresses the CD20 antigen. The level of cell lysis was determined using the ToxiLight™ bioluminescence assay kit (Lonza Nottingham Ltd., Nottinghamshire, UK), which measures the amount of adenylate kinase released from lysed cells.
Comparative Nonclinical in Vivo Study Designs
Two comparative nonclinical in vivo studies were conducted: a single-dose PK study and a repeat-dose study. These studies were designed to identify any biologically meaningful differences between PF-05280586 and rituximab-EU. Because these studies utilized nonhuman primates, they were not powered for statistical analysis.
In both studies, PF-05280586 and rituximab-EU were administered by bolus intravenous injection into the saphenous vein at a dose volume of 2 ml/kg. In the single-dose study (Figure 1A), 3 animals/sex/group were administered 0, 2, 10, or 20 mg/kg of PF-05280586 or rituximab-EU and observed for 92 days. At the end of the study, animals were returned to the colony. In the repeat-dose study (Figure 1B), 7 animals/sex/group were administered 0 or 20 mg/kg of PF-05280586 or rituximab-EU once weekly for 4 weeks (5 doses). Of these, 4 animals/sex/group were necropsied on day 30, 24 hr after the last dose. The remaining 3 animals/sex/dose were necropsied following a 92-day observation phase (study day 121).
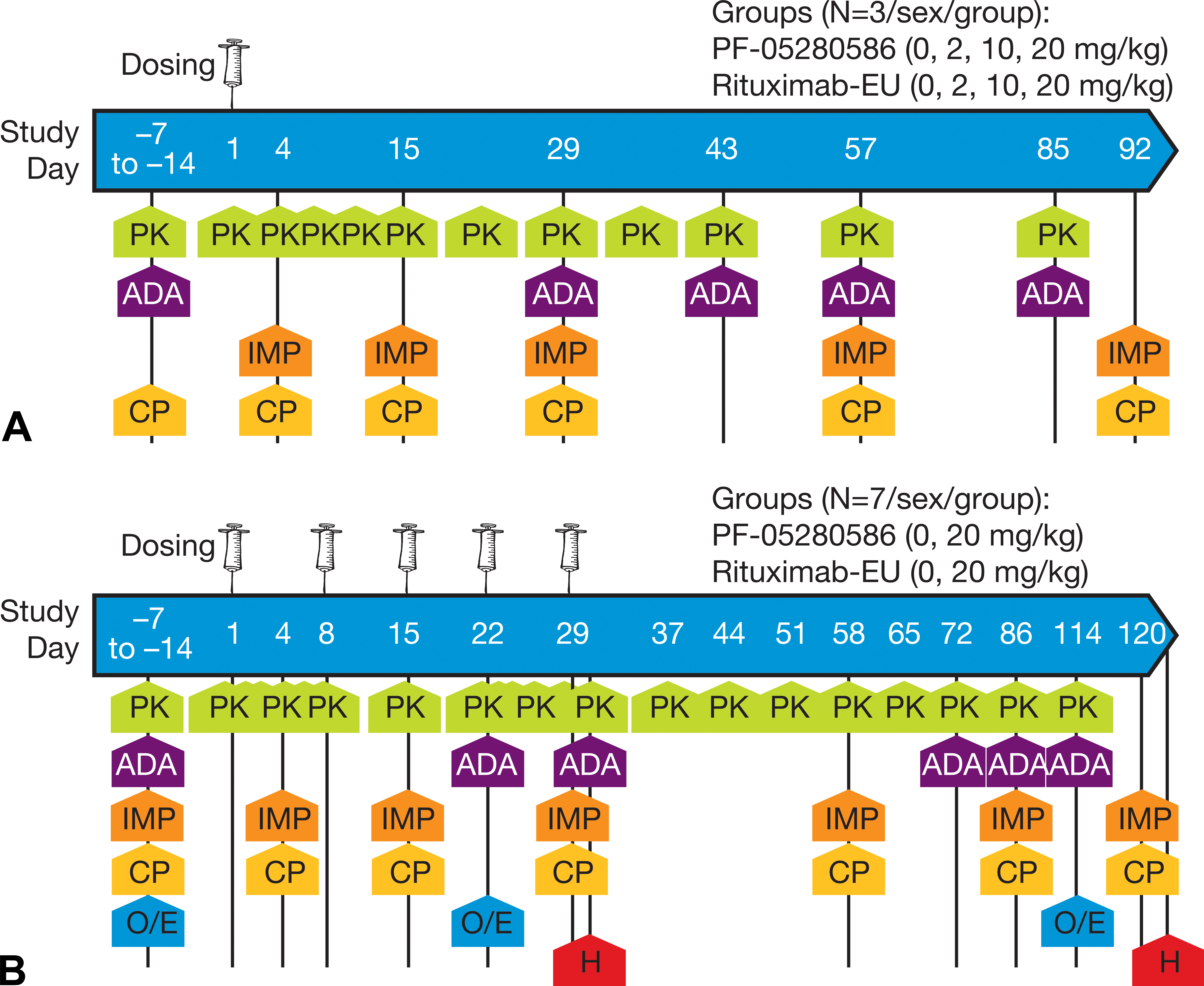
Study design schematic for single-dose (A) and repeat-dose (B) studies. Clinical observations, food consumption, and body weights were recorded in both studies. Other endpoints in both studies included pharmacokinetics (PK), clinical pathology (CP), immunophenotyping (IMP), and anti-drug antibody (ADA). The repeat-dose study also included ophthalmology and electrocardiography (O/E) and histopathology, organ weigh measurements, and immunohistochemistry (H).
Study Animals
Sexually-mature (4–5 years old) male and female cynomolgus monkeys (Macaca fascicularis; SNBL, Alice, TX) were acclimated for at least 3 weeks under approved environmental controls prior to study initiation. At study initiation, body weights ranged from 4.6 to 9.0 kg for males and 3.0 to 6.4 kg for females. Individual animal body weights, clinical pathology, and B cell parameters were collected prestudy (week -2 and week -1) as a baseline. Animals were fed Certified Primate Diet #2055C (Harlan Laboratories, Inc, Indianapolis, IN) 1 or 2 times daily unless fasted for study procedures; water was provided ad libitum. These animal studies were conducted in accordance with Good Laboratory Practice guidelines, and all animal procedures were approved by the Covance-Madison Laboratories Animal Care and Use Committee.
Dose Formulation, Preparation, and Analysis
For both studies, PF-05280586 was diluted in a formulation-based buffer in water, and rituximab-EU was diluted in a citrate-based buffer in water to the appropriate concentration for each dose level. These diluents were chosen to match the approved formulation (rituximab-EU) or a standard platform formulation for mAb programs (PF-05280586). In both studies, each vehicle control was specific to rituximab-EU or PF-05280586.
Dose preparations were performed weekly and stored at 2° to 8°C. Stability of the formulations under these conditions was confirmed; concentrations were verified by spectrophotometry for each dose preparation and were documented to be within 10% of the targeted concentrations.
A high dose of 20 mg/kg was selected for both studies because it approximated the approved dose of the drug for human use (Cohen et al. 2006). The low- and mid-dose levels in the single-dose PK study (2 and 10 mg/kg) were chosen to evaluate a range of dose levels to compare the PK and PD properties of PF-05280586 and rituximab-EU.
Clinical and Laboratory Assessments
In both studies, clinical and laboratory assessments included observations of mortality, clinical signs, and qualitative food consumption; measurement of body weight; clinical pathology parameters (hematology, clinical chemistry, coagulation, and urinalysis); immunophenotypic analysis of peripheral blood lymphocytes, including B cell subsets as a PD marker; determination of PK parameters; and the induction of anti-drug antibodies (ADA; Figure 1). The repeat-dose study included the additional assessments of electrocardiograms, ophthalmology examinations, necropsy, histopathology, and CD20 immunohistochemistry on lymphoid tissue sections (spleen and mesenteric and axillary lymph nodes).
Comparative Lymphocyte Immunophenotyping and Pharmacodynamic Effects on B Cells
After overnight fasting, samples for peripheral blood immunophenotyping were collected in potassium-ethylenediaminetetraacetic acid (EDTA) via the femoral vein. Total lymphocyte populations, total T cells (CD3+), helper T cells (CD3+CD4+), cytotoxic T cells (CD3+CD8+), natural killer cells (CD3−CD16+), and B cells (CD3−CD20+) were determined using flow cytometry (FACSCanto II, Becton Dickenson and Co., Franklin Lakes, NJ). Because B cell depletion is a recognized PD marker for rituximab activity (Genberg et al. 2006, 2007) and rituximab is an anti-CD20 mAb with the potential to compete for binding with the anti-CD20 reagent used for immunophenotyping, additional B cell subsets were also characterized by flow cytometry: CD−CD20+CD40+ (mature B cells), CD3−CD19+ (immature and mature B cells), and CD3−CD40+ (immature and mature B cells). The relative percentage of each phenotype was multiplied by the absolute total lymphocyte count from the hematology analysis to calculate absolute (cells/μL) values for each lymphocyte subset.
Comparative PKs and Immunogenicity Evaluation
Serum samples were analyzed using a validated sandwich enzyme-linked immunosorbent assay specific for each molecule. A monoclonal rat anti-rituximab antibody was used to capture PF-05280586 or rituximab-EU that was then detected using horseradish peroxidase–conjugated goat anti-human IgG and 3,3′,5,5′-tetramethylbenzidine as a substrate, generating a colorimetric signal. Sample concentrations were determined by interpolation from the calibration curve generated using a 4-parameter logistic equation. PK parameters were determined from individual animal data using noncompartmental analysis (WinNonlin® version 5.2, Mountain View, CA). Samples assaying below the limit of quantitation (<15.0 ng/ml) were assigned a value of 0 ng/ml for the analysis. PK evaluations included area under the curve from drug administration until time of evaluation (AUC0-t) and the maximum serum concentration of the drug (C max). Samples for ADA were analyzed using electrochemiluminescence assays (Meso Scale Discovery, Rockville, MD) validated for PF-05280586 or rituximab-EU.
Comparative Histopathology and Immunohistochemistry (Repeat-Dose Study Only)
Animals were fasted overnight prior to necropsy on day 30 or day 121. At necropsy, organs were weighed and tissues were collected in 10% neutral buffered formalin, embedded in paraffin, sectioned at 5 μm, and stained with hematoxylin and eosin for microscopic examination. Additional tissue sections from spleen, mesenteric lymph node, and axillary lymph node were evaluated using an antibody against an intracellular epitope of CD20 (L26 clone; Ventana Medical Systems, Tucson, AZ) followed by a secondary antibody conjugated with horseradish peroxidase.
Results
Comparative Biochemical and Functional Assessment
Comparison of the tryptic peptide map of PF-05280586 with that of rituximab-EU demonstrated that the 2 chromatographic profiles were essentially superimposable (Figure 2). Peptide identifications by MS were consistent between PF-05280586 and rituximab-EU, with 98.6% of the overall amino acid sequence of rituximab detected in this method. The remaining portion of the sequence that was not detected corresponds to small peptides that were not retained on the chromatographic column. The complete amino acid sequence, including those peptides not observed in the peptide map, was confirmed in a separate LC/MS analysis at the subunit level (data not shown). These results confirmed that the primary sequence of PF-05280586 and rituximab-EU were identical.
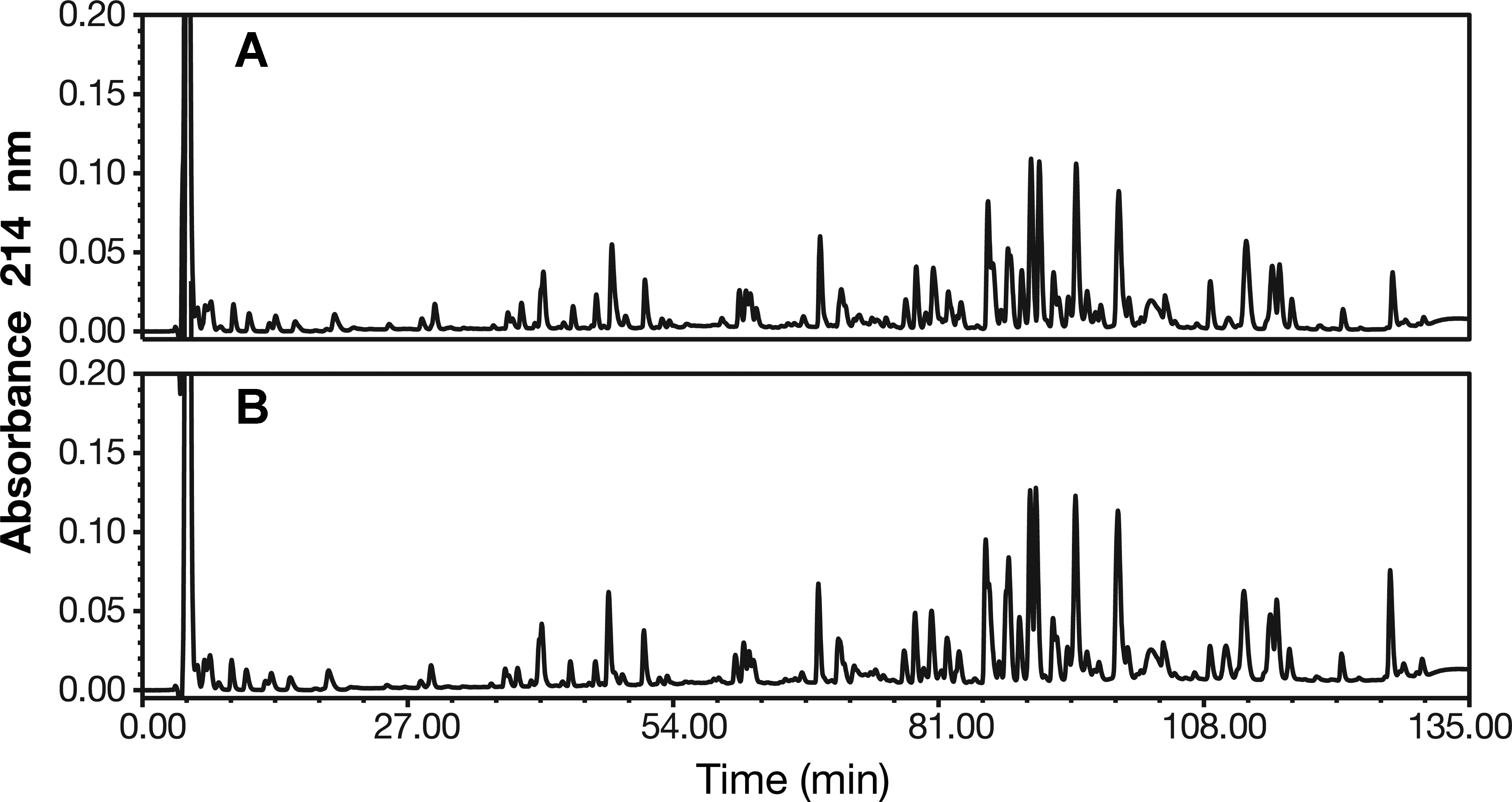
Tryptic peptide maps of PF-05280586 (A) and rituximab-EU (B) resolved by reverse-phase high performance LC and monitored at 214 nm ultraviolet absorbance. The 2 chromatographic profiles were essentially superimposable.
Molecular in vitro function, as determined by the biological activity of PF-05280586 or rituximab-EU in an optimized CDC assay, demonstrated that the 2 molecules were similar and the functional curves were essentially superimposable across the concentration range examined (Figure 3). The reported activity for each molecule was within the variability of the assay.
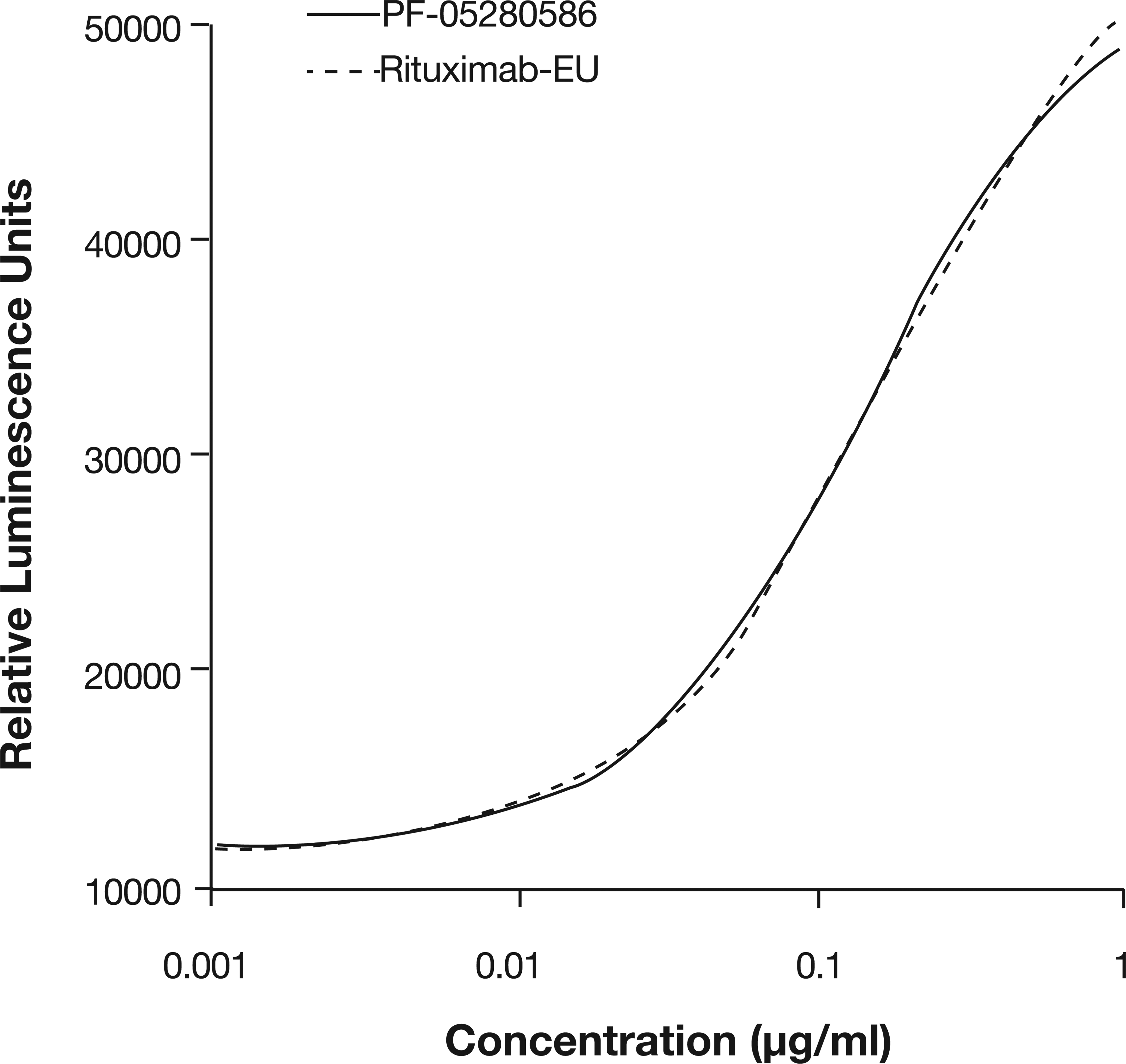
Complement-dependent cytotoxity (CDC) activity of PF-05280586 and rituximab-EU using CD20-expressing Ramos human B-lymphoblast cell line and human serum complement. Cell lysis, quantitated by measuring the amount of adenylate kinase released from lysed cells, demonstrated that the in vitro CDC function of the 2 molecules was similar across the range of concentrations examined.
Clinical Assessments
In both studies, all animals survived until the end of their designated study phases and both molecules were well tolerated at doses up to 20 mg/kg. In both studies, there were no effects on food consumption, body weight, body weight gain, coagulation, urinalysis, or clinical chemistry parameters. There were no cardiac or ophthalmologic effects noted in the repeat-dose study (these were not evaluated in the single-dose study).
In the repeat-dose study, emesis was reported in 6 and 7 animals administered PF-05280586 and rituximab-EU, respectively, compared with 1 and 2 animals, respectively, in the control groups. Emesis was primarily observed on day 22, a day with numerous study procedures, and was not associated with sex or ADA status. A similar increased incidence of emesis in monkeys has been reported for the marketed product (European Medicines Agency 2005).
Peripheral B Cell Depletion and Repletion
There was a slight decrease in mean total lymphocyte counts on day 4 following administration of PF-05280586 or rituximab-EU. However, no further decreases in total lymphocyte counts were noted at subsequent time points in either study (Figure 4A and B). In addition, no changes were observed in total T cells, helper T cells, cytotoxic T cells, or natural killer cell counts (data not shown).
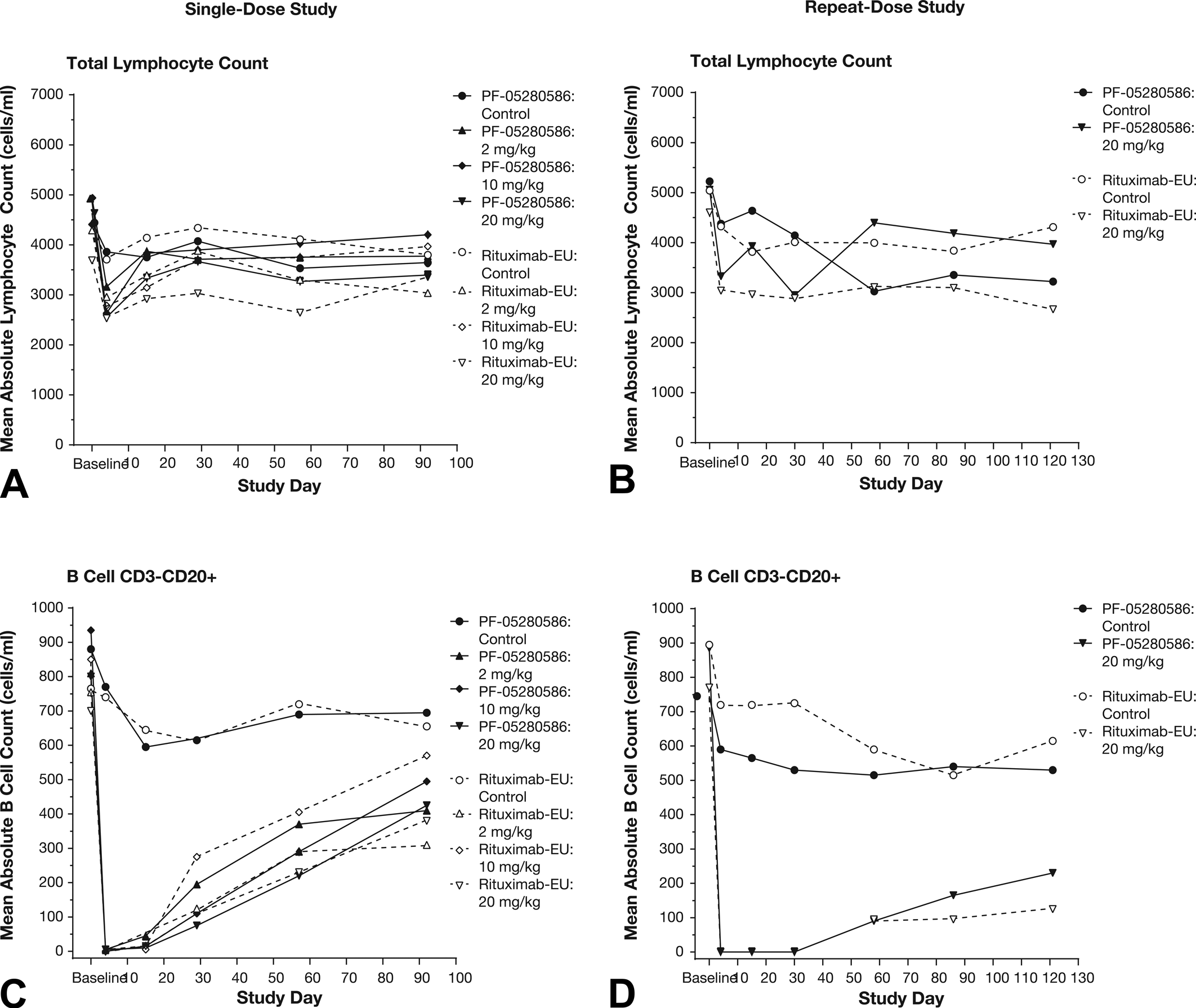
Effect of administration of PF-05280586 or rituximab-EU on mean total lymphocyte (A and B) and mean total CD3-CD20+ B cell (C and D) counts in the single- (A and C) and repeat-dose (B and D) studies. There was a slight decrease in total lymphocyte count on day 4 with no further decreases noted at subsequent timepoints. Marked to complete depletion in mean CD3−CD20+ B cells were observed on day 4 (2, 10, and 20 mg/kg groups), consistent with the expected pharmacology of anti-CD20 mAbs. CD3−CD20+ B cells were repleted by day 92 in some animals at all dose levels in the single-dose study, but, as expected, only partially repleted by day 121 in the repeat-dose study.
In the single-dose study (Table 1 and Figure 4C), marked to complete depletion in mean absolute CD3−CD20+ B cell values (relative to baseline) were observed on day 4 (2, 10 , and 20 mg/kg groups) following PF-05280586 or rituximab-EU administration, consistent with the expected pharmacology of anti-CD20 mAbs. B cell repletion, defined as B cells reaching 25% of baseline levels (Vaidyanathan et al. 2011), was present by day 92 in some animals in all dose groups (Table 1). In the repeat-dose study (Table 1 and Figure 4D), marked to complete depletion in peripheral blood mean absolute CD3−CD20+ B cells were observed on days 4 to 30. By study day 121, CD3-CD20+ B cells were partially repleted (Table 1 and Figure 4D), as might be expected following repeated administration of PF-05280586 or rituximab-EU.
Percentage decrease of absolute B cell subsets in the single- and repeat-dose studies.
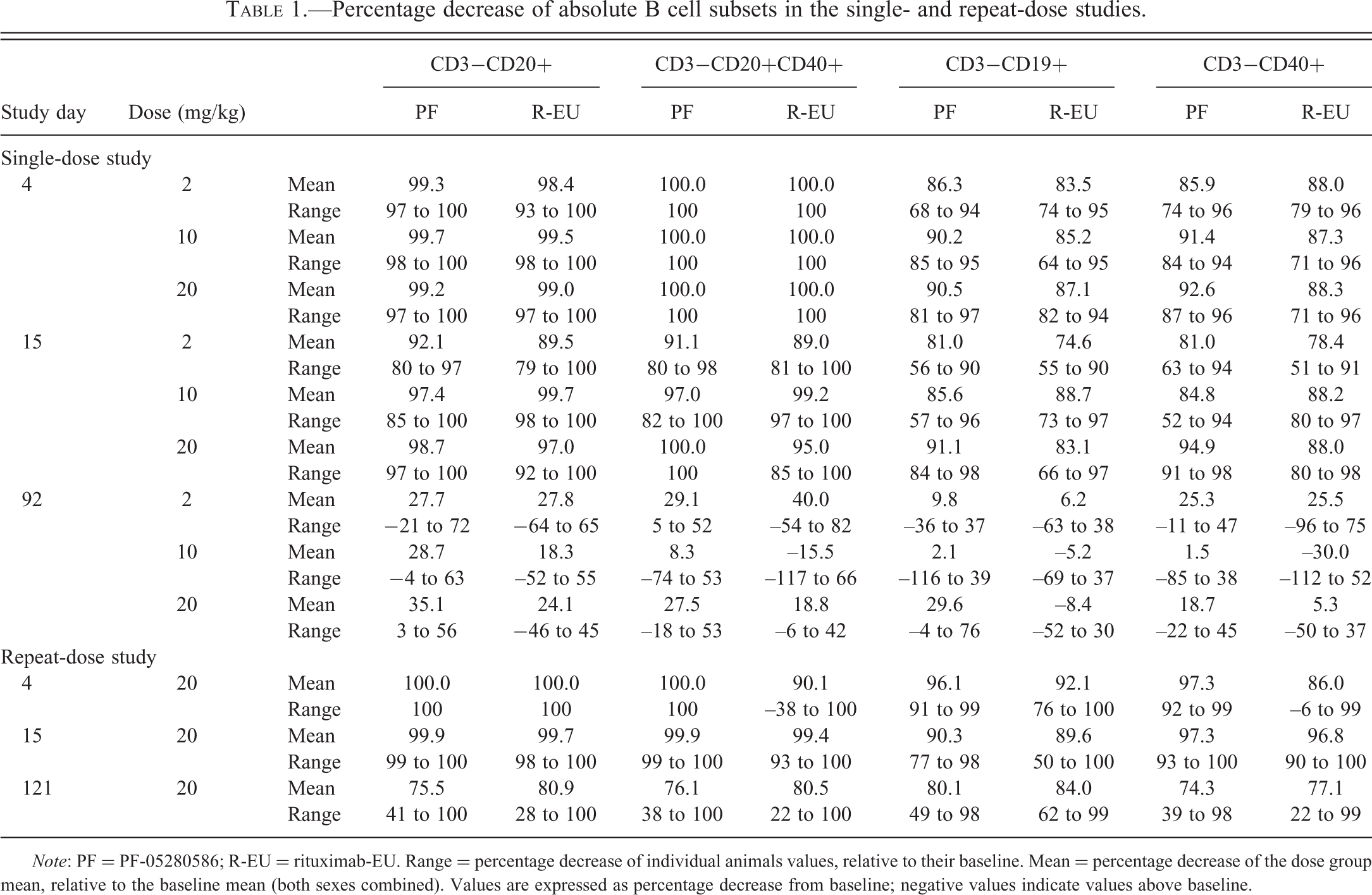
Note: PF = PF-05280586; R-EU = rituximab-EU. Range = percentage decrease of individual animals values, relative to their baseline. Mean = percentage decrease of the dose group mean, relative to the baseline mean (both sexes combined). Values are expressed as percentage decrease from baseline; negative values indicate values above baseline.
Since PF-05280586 and rituximab-EU are anti-CD20 mAbs that could potentially interfere with the detection and quantification of CD3−CD20+ B cells by flow cytometry, additional B cell markers (CD19 and CD40) were included in the immunophenotyping panel. Administration of PF-05280586 or rituximab-EU also resulted in almost complete depletion of CD3−CD20+CD40+ B cells on day 4 (Table 1) in both the single-dose (Figure 5A) and the repeat-dose (Figure 5B) studies. B cell depletion persisted through the dosing phase (day 30) in the repeat-dose study. Repletion was observed in some animals by day 92 in the single-dose study but, as expected with repeated administration, only partial repletion of CD3-CD20+CD40+ B cells was observed by day 121 in the repeat-dose study.
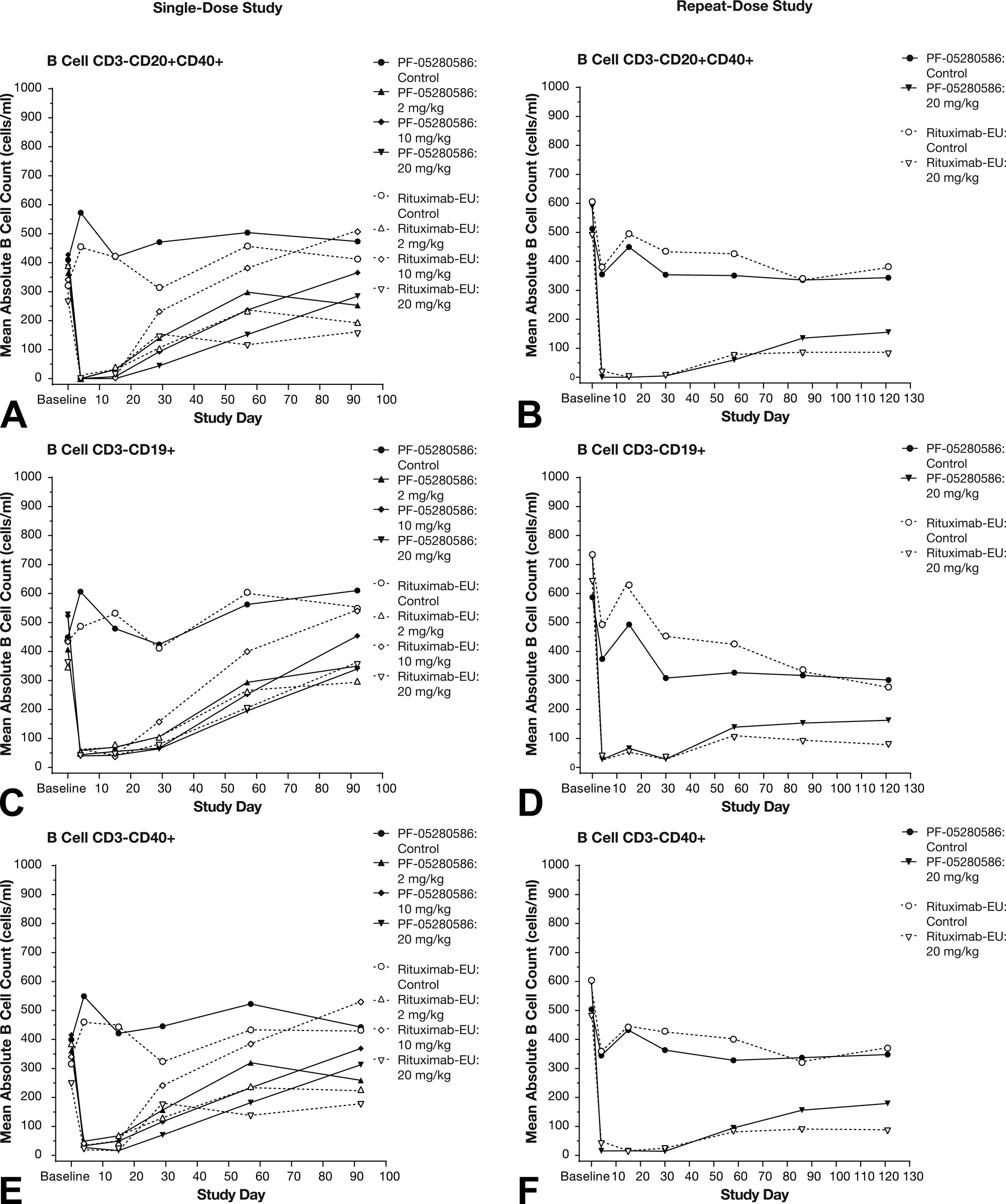
Effect of administration of PF-05280586 or rituximab-EU on additional B cell subsets: CD3−CD20+CD40+ (A and B), CD3−CD19+ (C and D), and CD3−CD40+ (E and F) mean counts in the single- (A, C, and E) and repeat-dose (B, D, and F) studies. The administration of the anti-CD20 mAbs PF-05280586 or rituximab-EU did not interfere with the ability to detect B cells changes in this study. Almost complete depletion of CD3−CD20+CD40+, CD3−CD19+ and CD3−CD40+ B cells was observed on day 4 in the single dose and repeat dose studies. Depletion persisted throughout the dosing phase (day 30) in the repeat-dose study. Repletion was observed in some animals by day 92 in the single-dose study, but only partial repletion was observed by day 121 in the repeat-dose study.
Decreased CD3−CD19+ and CD3-CD40+ B cells (Figures 5C and E, respectively) were also observed at all doses of PF-05280586 or rituximab-EU on day 4, with repletion in some animals by day 92 in the single-dose study (Table 1). In the repeat-dose study, marked decreases in peripheral blood CD3−CD19+ B cells (Table 1 and Figure 5d) and CD3−CD40+ B cells (Table 1 and Figure 5F) persisted throughout the dosing phase (day 30). Partial repletion of CD3−CD19+ and CD3−CD40+ B cells were observed by day 121, consistent with the repeated administration of PF-05280586 or rituximab-EU in these animals.
Anatomic Pathology (Repeat-dose Study Only)
There were no macroscopic findings at necropsy on day 30 or day 121 related to the administration of PF-05280586 or rituximab-EU. Microscopic findings were restricted to lymphoid organs (spleen, mesenteric, and axillary lymph nodes).
Relative to their respective control group, splenic weights (both sexes) were 12% to 42% and 15% to 44% lower in animals administered PF-05280586 or rituximab-EU, respectively. The lower splenic weights correlated with microscopic findings of decreased cellularity of lymphoid follicles and reduced numbers of CD20+ cells in the spleen. Microscopic findings with both molecules were restricted to the lymphoid system and consisted of reduced lymphoid follicles and CD20+ cells in the spleen, mesenteric lymph node, and axillary lymph node (Table 2 and Figure 6), consistent with the expected pharmacologic effects of anti-CD20 antibodies. The incidence and severity of these changes were similar between PF-05280586 and rituximab-EU (Table 2). In those animals necropsied on day 121, reduced lymphoid follicles and CD20+ cells in the spleen and mesenteric and axillary lymph nodes were noted in 9 of the 12 animals with an incidence and severity that was decreased relative to that observed in animals necropsied on day 30 (Table 2). Furthermore, these changes were not observed in the remaining 3 animals; together, these observations demonstrate partial recovery of B cells in lymphoid tissue.
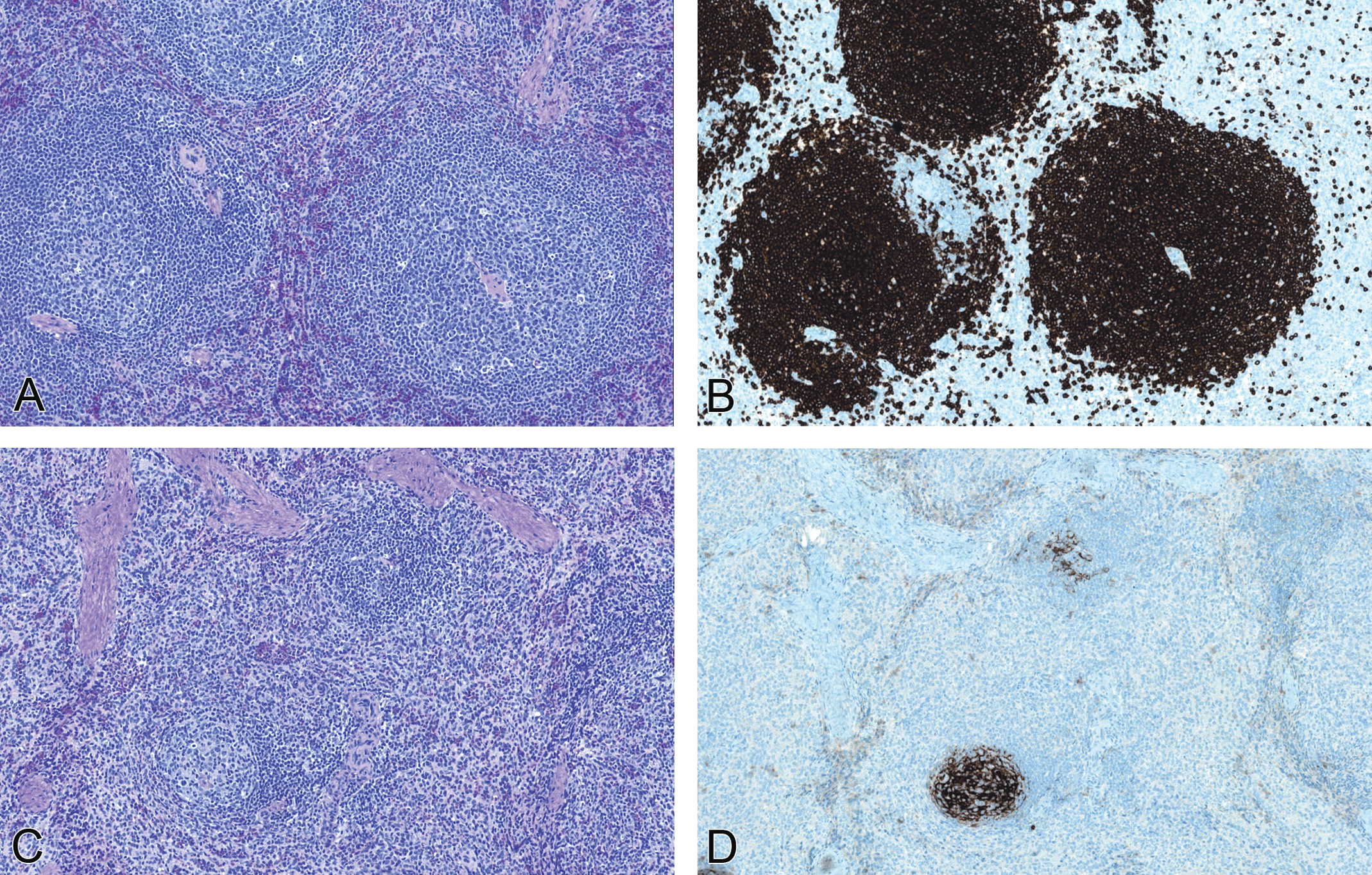
Representative photomicrographs of the spleen from vehicle control (A and B) and PF-05280586 (C and D) monkeys showing reduction in the size and cellularity of lymphoid follicles (C) and decreased CD20+ B cells (D) at the end of the dosing phase (day 30). Original magnification: 100×. These changes were of similar incidence and magnitude between PF-05280586 and rituximab-EU and were consistent with the expected pharmacology of anti-CD20 antibodies. These changes were partially recovered in those animals necropsied on day 121: 3 of the 12 animals had no microscopic findings of B cell depletion while the remaining 9 animals had changes that were less severe than observed at the day 30 necropsy.
Repeat-dose study: Incidence and severitya of microscopic and immunohistochemistry findings.
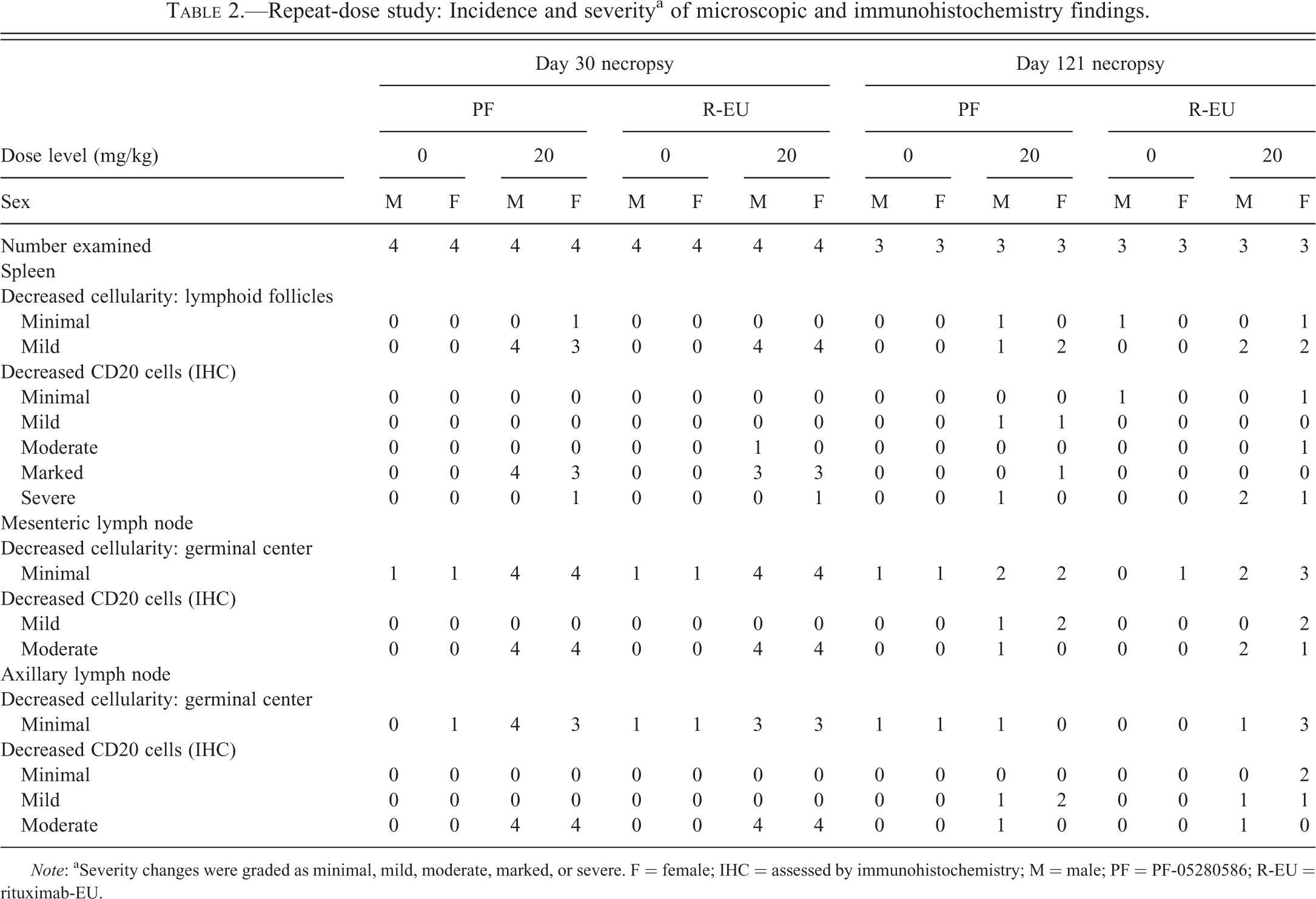
Note: aSeverity changes were graded as minimal, mild, moderate, marked, or severe. F = female; IHC = assessed by immunohistochemistry; M = male; PF = PF-05280586; R-EU = rituximab-EU.
Comparative PK
PF-05280586 or rituximab-EU was not detected in any samples from vehicle control animals in the single- or repeat-dose studies. All remaining study animals had documented exposure to the mAbs. In both studies, the PK profiles for PF-05280586 and rituximab-EU were similar (Table 3 and Figures 7 and 8). In the single-dose study, dose-dependent increases in C max (Table 3) and AUC (Figure 7) were similar following administration of PF-05280586 or rituximab-EU. In the repeat-dose study, C max (Table 3) and AUC to 168 h (AUC0-168h) values were similar between PF-05280586 and rituximab-EU on day 1 and on day 22 (Figure 8). Sex differences in exposure were not noted in these studies, based on high interanimal variability and less than 2.0× differences in serum mAb concentrations between dose-matched males and females.
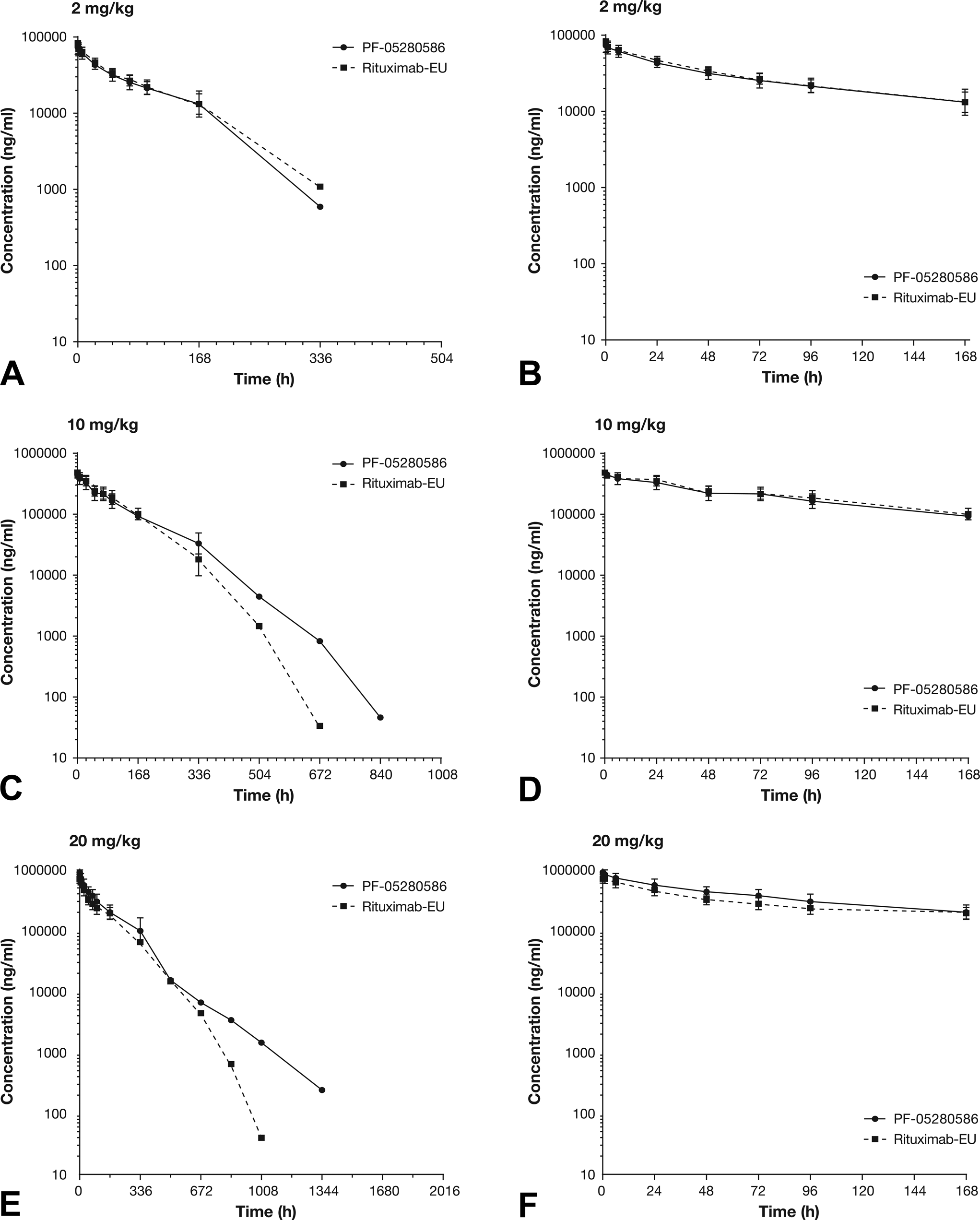
Effect of adminstration of PF-05280586 or rituximab-EU (2, 10, and 20 mg/kg) on serum concentration–time profiles in the single-dose study: full profiles (A, C, and E) and AUC0-168h profiles (B, D, and F). The impact of ADA induction (first detected on day 29, the first time point examined for ADA) on exposure can be seen in the full study profiles (A, C and E; X and Y axes optimized to fully display the concentration time curves). The limit of quantification for the assay was 15.0 ng/mL.
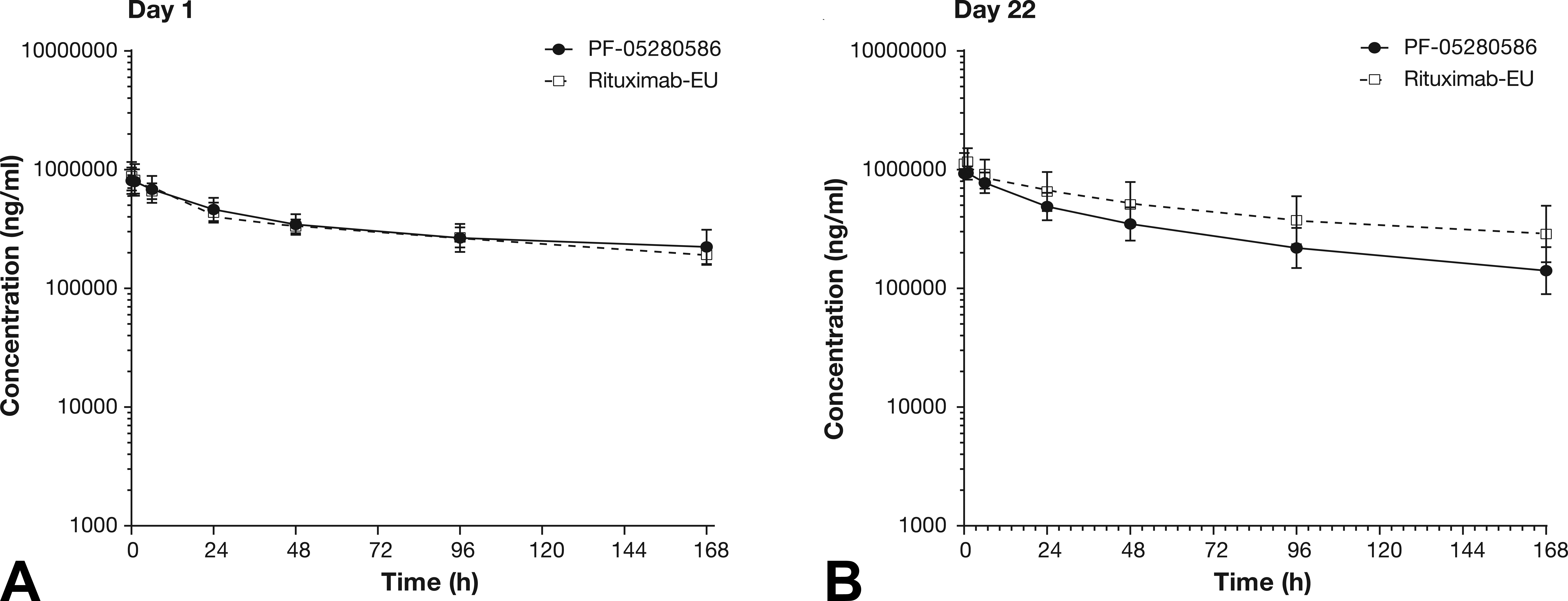
Effect of administration of PF-05280586 and rituximab-EU (20 mg/kg) on serum concentration–time profiles from the repeat-dose study after dosing on day 1 (A) and day 22 (B). The AUC0-168h profiles were similar between PF-05280586 and rituximab-EU on day 1 and day 22, with only minimal increase in AUC0-168h between the first and last dose. In addition, the day 1 AUC0-168h data were also similar to that of the 20 mg/kg group in the single-dose study (Figure 7F). ADA were detected on day 22 (the first time point examined for ADA) and was present in 3 of the 6 (50%) of the animals in each 20 mg/kg group on day 114 (the last time point examined for ADA).
Summary comparison of pharmacokinetic (PK) parameters of PF-05280586 and rituximab-EU (combined sexes).
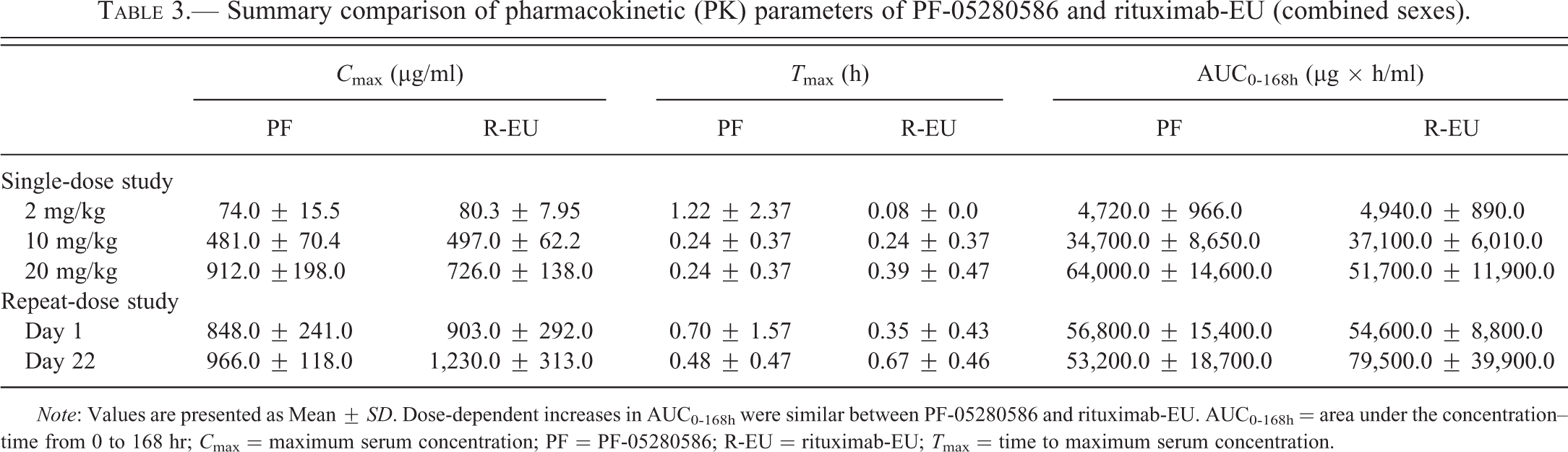
Note: Values are presented as Mean ± SD. Dose-dependent increases in AUC0-168h were similar between PF-05280586 and rituximab-EU. AUC0-168h = area under the concentration–time from 0 to 168 hr; C max = maximum serum concentration; PF = PF-05280586; R-EU = rituximab-EU; T max = time to maximum serum concentration.
Anti-drug Antibody
In the single-dose study, all animals administered PF-05280586 or rituximab-EU were positive for ADA by day 29 (the earliest time point examined) and remained positive until day 85 (the last time point examined). ADA was detected in 1 of 6 (16.7%) animals in both vehicle control groups on a single occasion (day 43 for rituximab-EU vehicle control; day 85 for the PF-05280586 vehicle control); neither of these animals had PF-05280586 or rituximab-EU detected in any of their serum samples.
In the repeat-dose study, 1 of 14 (7%) of PF-05280586 vehicle control animals was positive for ADA pre-study and on day 22 but PF-05280586 was not detected in serum samples from any of the control animals. ADA was detected on day 22 (the first time point examined) in 10 of the 14 (71%) and 5 of the 14 (36%) animals administered PF-05280586 or rituximab-EU, respectively. On day 30 (24 hr after the last dose), the number of animals with ADA decreased to 1 of 14 (7%) and 2 of 14 (14%) animals administered PF-05280586 and rituximab-EU, respectively. (Note that circulating PF-05280586 or rituximab-EU may have interfered with the ability to detect ADA at this time point.) On day 114 (the last time point sampled), ADA was detected in 3 of 6 (50%) animals in each 20 mg/kg dose group (log10 titers of 1.87–6.13 and 2.01–5.70 for animals administered PF-05280586 and rituximab-EU, respectively). Most, but not all, of the ADA-positive animals had lower exposure compared with animals in the same group that were negative for ADA.
Discussion
Biotherapeutics are by nature different from small molecule drugs. Since their production depends on a biologic process that determines the final product and even small changes can occur in the product under controlled manufacturing processes (Ündey et al. 2010), it is virtually impossible to develop an exact replica of the licensed product (Kozlowski et al. 2011). Biosimilars are designed to be similar to an approved marketed biologic product with respect to key parameters including the amino acid sequence, physiochemical attributes, in vitro functional activity, pharmacologic activity, efficacy, and safety. Thus, various regulatory agencies require substantial evidence demonstrating a high degree of similarity, based on physicochemical and functional analysis, quality attributes, safety, and efficacy endpoints, between the proposed biosimilar and the marketed product (CHMP 2005; FDA 2012; [CHMP] 2012). In these studies, we demonstrated the nonclinical similarity of the proposed biosimilar PF-05280586 to the licensed product rituximab-EU (MabThera®).
PF-05280586 and rituximab-EU are genetically engineered chimeric mouse/human IgG1 mAbs directed against the CD20 antigen expressed on the surface of normal and malignant B cells. Comparison of the tryptic peptide maps of PF-05280586 and rituximab-EU demonstrated that the chromatographic profiles were essentially superimposable and 98.6% of the peptides were identical between the mAbs. Trace differences that were observed were typical of profiles resulting from trypsin digestion or represent expected modifications in the NH2-terminal (Q to PyroQ) and truncation of the COOH-terminal lysine residue expected in the production of biologics (Chirino and Mire-Sluis 2004; Schellekens 2004). No difference in in vitro CDC function was detected between PF-05280586 and rituximab-EU using an optimized CDC assay employing Ramos cells, a B lymphoblast cell line expressing CD20. In this biological activity assay, the dose–response profiles were superimposable, indicating similar in vitro functional properties across the range of concentrations examined.
In both in vivo studies, the B cell depletion-repletion profile of PF-05280586 was similar to rituximab-EU. Multiple B cell markers (CD19, CD20, and CD40) demonstrated that administration of the anti-CD20 mAbs PF-05280586 or rituximab-EU did not interfere with the detection of cynomolgus B cells in these studies. CD3−CD20+, CD3−CD20+CD40+, CD3−CD19+, and CD3−CD40+B cell depletion was evident by day 4, the first time point examined. In the single-dose study, B cell repletion was observed by day 92, consistent with the maturation kinetics of B cells. Following repeated administration, B cell repletion was incomplete on day 121, consistent with the effects of repeated PF-05280586 and rituximab-EU administration over the 30-day dosing period. There were no effects on total T cells, helper T cells, cytotoxic T cells, or natural killer cells observed in these studies.
Administration of PF-05280586 or rituximab-EU resulted in decreased lymphoid cellularity and CD20+ B cells in the spleen and axillary and mesenteric lymph nodes (repeat-dose study day 30 necropsy). These changes were of similar incidence and magnitude between PF-05280586 and rituximab-EU and were consistent with the expected pharmacologic activity of anti-CD20 antibodies. These changes were partially recovered in animals necropsied on day 121: 3 of the 12 animals had no microscopic findings of B cell depletion, while the remaining 9 animals had changes generally less severe than those observed in animals necropsied on day 30. Administration of PF-05280586 or rituximab-EU also resulted in decreased splenic weights, consistent with the microscopic observations of decreased lymphoid cellularity in the spleen.
Both studies demonstrated similar PK profiles for PF-05280586 and rituximab-EU. In the single-dose study, both molecules had similar dose-dependent increases in C max and AUC. Furthermore, the AUC0-168h data for the 20 mg/kg dose groups were similar between the single- and repeat-dose studies. In the repeat-dose study, there was only a minimal increase in C max and AUC0-168h between the first and the last dose, consistent with data reported for rituximab (Vaidyanathan et al. 2011).
PF-05280586 and rituximab-EU were both immunogenic in cynomolgus monkeys, with similar incidence and titers. This was not unexpected, since both molecules were heterologous (chimeric mouse/human) proteins. Most, but not all, of the ADA-positive animals had lower exposure compared with animals in the same group that were negative for ADA. Effects of ADA on the rate or extent of B cell repletion were not discernible because of intra- and interanimal variability in lymphocyte counts and the small number of animals in these studies. Because immunogenicity in animals has limited predictive value for immunogenicity in humans (Brinks, Jiskoot, and Schellekens 2011; Bugelski and Treacy 2004), potential differences in the immunogenicity profiles of PF-05280586 and rituximab-EU will be assessed in clinical studies.
Nonclinical studies in cynomolgus monkeys administered rituximab-EU reported B cell depletion/repletion in peripheral blood and lymphoid organs, emesis, testicular hypospermatogenesis, and sex-related difference in PK (European Medicines Agency 2005). The investigators ascribed the testicular finding to immaturity. However, the PK differences between sexes were unexplained. In our studies, we specifically used sexually-mature animals, and we did not observe hypospermatogenesis or sex-related PK differences with PF-05280586 or rituximab-EU. Similar to what was reported for rituximab-EU, our studies replicated the findings of B cell depletion/repletion and emesis with PF-05280586 and rituximab-EU.
Overall, responses to PF-05280586 and rituximab-EU appeared similar in a number of in vitro and in vivo evaluations, including tryptic peptide mapping, CDC activity, B cell depletion/repletion, PK profiles, and ADA response. These data support development of PF-05280586 as a potential biosimilar to the licensed rituximab product.
Footnotes
Acknowledgments
The authors thank QPS (Newark, DE) for its analytical work and pharmacokinetic and immunogenicity assays, and Sharon Ripp and Vanessa D’Souza for their assistance with the pharmacokinetic analyses. Editorial support for this article was provided by Mukund Nori, PhD, MBA, CMPP, of UBC Scientific Solutions and was funded by Pfizer Inc, New York, NY.
Authors’ Note
Authors Ryan, Sokolowski, Ng, Shirai, Collinge, Shen, Radi, Cummings, Hurst, Finch, and Leach are employees of Pfizer Inc. Authors Arrington, Ploch, Stephenson, and Tripathi are employees of Covance Laboratories, Inc., who conducted the in vivo studies for Pfizer, Inc.
The authors are employees of Pfizer Inc. or Covance Laboratories Inc.