
Abstract
The intestinal tract is inhabited by a large diverse community of bacteria collectively referred to as the gut microbiota. Alterations in gut microbiota composition are associated with a variety of disease states including obesity, diabetes, and inflammatory bowel disease (IBD). Transplant of microbiota from diseased persons (or mice) to germfree mice transfers some aspects of disease phenotype, indicating that altered microbiota plays a role in disease establishment and manifestation. There are myriad potential mechanisms by which alterations in gut microbiota might promote disease, including increasing energy harvest, production of toxic metabolites, and molecular mimicry of host proteins. However, our research indicates that an overarching mechanism by which an aberrant microbiota negatively impacts health is by driving chronic inflammation. More specifically, we hypothesize that the histopathologically evident gut inflammation that defines IBD is a severe but relatively rare outcome of an altered host–microbiota relationship, while a much more common consequence of such disturbances is “low-grade” inflammation characterized by elevated proinflammatory gene expression that associates with, and may promote, metabolic syndrome. In this context, a variety of chronic inflammatory diseases may stem from inability of the mucosal immune system to properly manage a stable healthy relationship with the gut microbiota. While one’s ability to manage their gut microbiota is dictated in part by genetics, it can be markedly influenced by the composition of the microbiota one inherits from their early environment. Moreover, the host–microbiota relationship can be perturbed by instigator bacteria or dietary components, which may prove to play a role in promoting chronic inflammatory disease states.
Metabolic Syndrome and Low-grade Inflammation
Humanity is facing an epidemic of metabolic syndrome, a group of interrelated metabolic disorders including obesity, insulin resistance, hyperglycemia, hyperlipidemia, and hepatic steatosis (Nathan 2008). Metabolic syndrome greatly increases the risk of diabetes, cardiovascular disease, and liver dysfunction. The increasing incidence of metabolic syndrome and its highly morbid, chronic, and exceptionally costly downstream diseases threatens to overwhelm the world’s health care systems and economies, thus making it a top public health priority in dire need of investigation. While obesity is the most recognizable aspect of metabolic syndrome, the central feature of this disorder is insulin resistance (Despres and Lemieux 2006). One potential explanation seeking to explain how obesity might lead to insulin resistance (outlined in Figure 1) is that nutrient excess promotes, sequentially, endoplasmic reticulum (ER) stress, chronic activation of proinflammatory-signaling pathways, and, ultimately, cross desensitization of insulin receptor signaling (Hotamisligil 2006). Such obesity-associated signaling is associated with broad, but relatively mild, systemic elevations in numerous cytokines and other inflammatory markers. While the precise cellular source of these cytokines (i.e., macrophages vs. adipocytes) remains under debate, that their elevated expression is systemically detectable and not associated with aggregates of immune cell infiltrates that, classically, histopathologically define inflammation, has led to this state being called “low-grade” and/or “systemic” inflammation. Such inflammation can be self-promoting in that its desensitization of insulin signaling (and likely that of other metabolic regulators, leptin would be one example), often results in hyperphagia that can further distort caloric intake thus inducing additional ER stress and exacerbating inflammatory processes (Tilg and Moschen 2006). However, importantly, the cyclical nature of this proposed mechanism holds that it need not necessarily commence with caloric excess but rather can also be initiated and/or promoted by directly inducing low-grade inflammation driven by other sources including, as discussed herein, the gut microbiota.
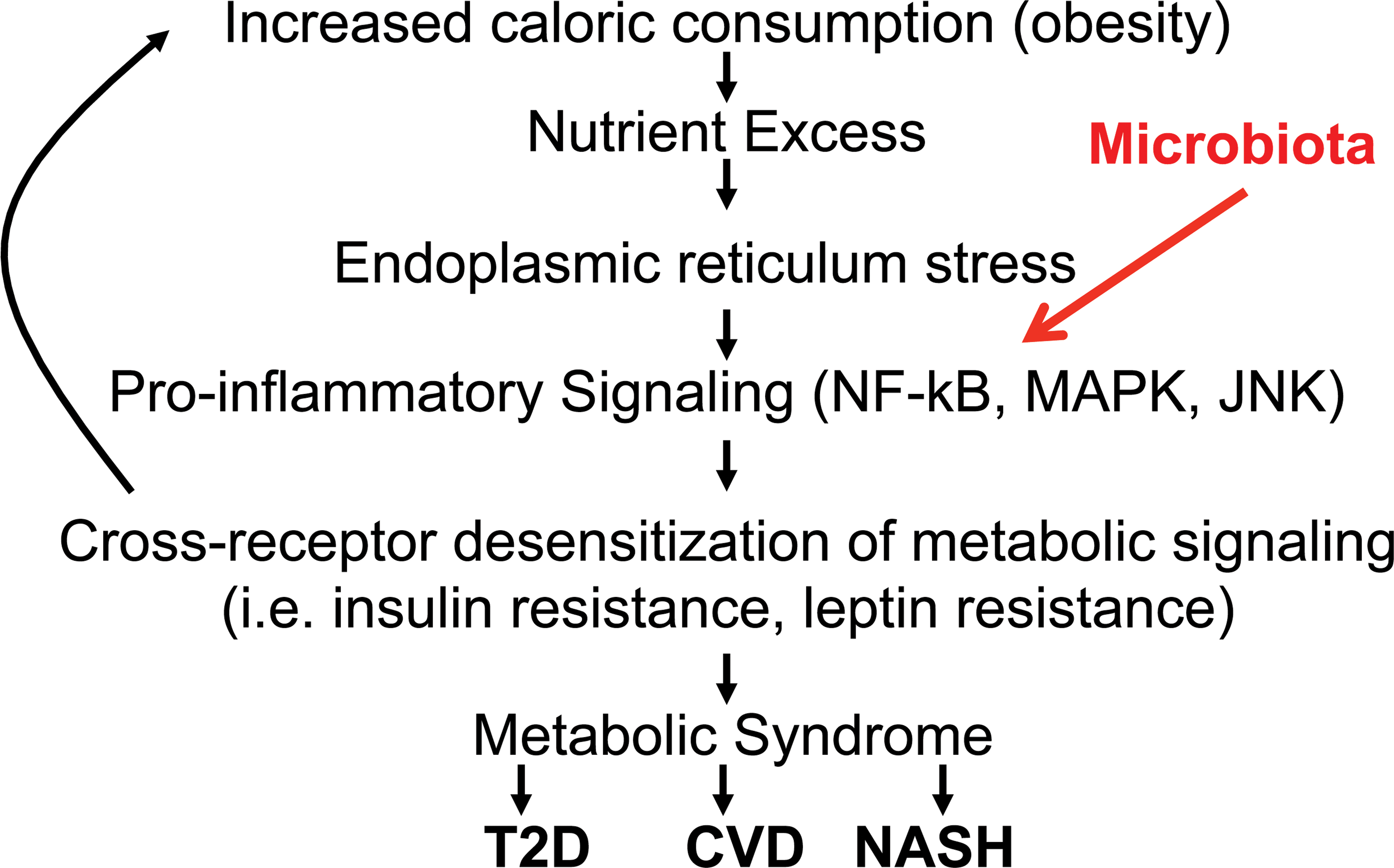
Potential mechanism by which altered gut microbiota can result in metabolic syndrome. Flowchart of widely proposed mechanism by which excess caloric consumption promotes metabolic syndrome and its downstream diseases such as type 2 diabetes (T2D), cardiovascular disease (CVD), and nonalcoholic steatohepatitis (NASH). We propose that the altered microbiota can initiate this cascade via directly promoting inflammation as described in the text.
Gut Microbiota: Central Player in Inflammation
The mammalian intestine is inhabited by a large diverse community of microbes collectively referred to as the gut microbiota. The human microbiota consists of approximately 100 trillion (1014) bacteria, weighing 1 to 2 kg, which are comprised of 6 to 10 major phyla and about 3,000 distinct species, sometimes referred to as operational taxonomic units (OTUs) to reflect that they are defined by the sequence of their 16s RNA-encoding gene (Hooper, Midtvedt, and Gordon 2002). When viewed holistically, the gut microbiota clearly has an overall benefit to the host in that “germfree” (also referred as gnotobiotic) mice, which lack a microbiota, have considerable immune and metabolic defects with the latter resulting in a requirement for greater caloric consumption relative to body mass (Wostmann et al. 1983). However, that multiple mouse models of inflammation require a gut microbiota and that the composition of the microbiota is a determinant of disease indicate that the microbiota can also constitute a major threat to its host. The central role of the microbiota in inflammation has been best defined in mouse models of colitis, wherein disease can be manipulated by altering the composition of the microbiota.
Much of the impact of the gut microbiota on the host proinflammatory signaling is thought to be mediated by pattern recognition receptors (PRRs) of the innate immune system, particularly the toll-like receptors (TLRs) and Nod-like receptors (NLRs). Both TLRs and NLRs recognize a variety of broadly conserved microbial components and allow the innate immune system to sense a wide variety of bacteria, viruses, fungi, and parasites. In light of the benefits, the microbiota confers on key areas of host biology and its potential to harm its host if not managed properly, and considering that its composition is long lasting, we refer to the gut microbiota as the host’s best frenemy forever. While the role of the microbiota in driving gut inflammation has long been appreciated, more recent studies highlight that bacterial products might also drive the low-grade inflammation associated with metabolic syndrome. This concept was first suggested by Cani and colleagues (2007) who have shown that obesity can result in the loss of epithelial barrier function, leading to receptor TLR4 activation by the bacterial product lipopolysaccharide (LPS), which can result in the release of cytokines that promotes insulin resistance. As outlined below, our results indicate that alterations in the gut microbiota can, in fact, be a primary cause of obesity and other aspects of the metabolic syndrome.
TLR5: Keeping Gut Bacteria in Check
Our appreciation that altered microbiota can promote low-grade inflammation and metabolic syndrome originated in observations made while studying colitis. Briefly, we observed that mice with a genetic deficiency in the gene encoding TLR5, which detects bacterial flagellin—a protein made by motile bacteria—develop spontaneous colitis (Vijay-Kumar et al. 2007). Such colitis associates with alterations in gut bacteria and could be alleviated by antibiotic treatment or by rederiving the mice in a germfree state. However, interestingly, the colitic phenotype had only moderate penetrance. Specifically, while 15 to 30% of TLR5-deficient (T5KO) mice showed classic histopathologic evidence of colitis, including edema and presence of inflammatory cells, the majority of T5KO mice could not be distinguished from wild-type (WT) mice by histopathologic analysis. Yet, careful study of gene expression in such mice revealed that they harbor a mild elevation in proinflammatory gene expression, which is now referred to as low-grade inflammation. We have recently developed an important tool to help assess low-grade inflammation, specifically by measuring fecal levels of lipocalin-2 (Lcn2), whose levels closely correlate with extent of low-grade and robust (i.e., classic) gut inflammation (Chassaing et al. 2012). Use of this assay to track T5KO mice over time revealed that they uniformly exhibit low-grade inflammation upon weaning but that most mice control the inflammation without exhibiting clinical-type symptoms of colitis while a subset fail to control inflammation and develop chronic colitis (Carvalho, Koren, et al. 2012). Such colitis is driven by activation of other aspects of the innate immune system including TLR4 and inflammasome-mediated production of interleukin-1beta (IL-1β) (Carvalho, Nalbantoglu, Ortega-Fernandez, et al. 2012).
T5KO Mice with Low-grade Inflammation Develop Metabolic Syndrome
T5KO mice that did not develop colitis were prone to developing obesity. To eliminate potential opportunistic pathogens that may have been present in T5KO and WT littermates, and to make their gut microbiota similar to that of mice from the Jackson Laboratory (the world’s largest supplier of research mice), we “rederived” T5KO mice by transplanting embryos into mice purchased from this supplier. Such standardization of the microbiota in the T5KO mice eliminated histopathologically evident colitis and resulted in a more uniform degree of low-grade inflammation associated with obesity (Vijay-Kumar et al. 2010). Metabolic characterization of noncolitic T5KO mice revealed a phenotype very reminiscent of human metabolic syndrome. Briefly, both female and male mice exhibited modest (10–20%) increases in body mass by 20 weeks of age. Much of the increased weight gain occurred in fat mass with the epididymal fat pads increasing in mass by 50 to 120%. The increase in fat mass was accompanied by the increased presence of macrophage infiltration into the adipose tissue, which was associated with increased expression of proinflammatory cytokines. Like humans with metabolic syndrome, T5KO mice displayed moderate hypertension and a glucose regulatory phenotype characterized by insulin resistance, which was compensated for by increased production of insulin, which together results in mild hyperglycemia and mild loss of glucose tolerance associated with increased numbers and sizes of pancreatic islets. When placed on a “high-fat diet,” commonly purported to mimic a western diet, T5KO mice developed more severe dysglycemia and, moreover, developed hepatic steatosis.
T5KO Metabolic Syndrome Is Driven by Low-grade Inflammation
In light of our earlier work that demonstrated that TLR5 helped maintain the intestinal–gut microbiota relationship, and work from Jeff Gordon and colleagues demonstrating that altered microbiota composition could contribute to obesity (Ley et al. 2006), we hypothesized that T5KO metabolic syndrome was driven in this manner. In accordance, the metabolic syndrome phenotype was eliminated by either maintaining these mice on broad-spectrum antibiotics or rederiving them into a germfree state. Moreover, metabolic syndrome could be transferred via microbiota transplant to germfree mice, further substantiating that microbiota composition can be a determinant of metabolic syndrome. Thus, alterations in gut microbiota that can result from loss of TLR5 are necessary and sufficient to drive metabolic syndrome. The connection between microbiota and obesity, described by Gordon and colleagues, was mediated by microbiota influencing the efficiency of energy harvest. In contrast, loss of TLR5 did not affect energy harvest but rather resulted in increased food consumption. Restricting food consumption ameliorated most aspects of T5KO metabolic syndrome but did not completely rescue metabolic syndrome. As outlined below, and illustrated in Figure 1, we hypothesize that altered microbiota drives low-grade inflammation, particularly moderate elevations in levels of proinflammatory cytokines. The receptors by which such cytokines signal share some of the same signaling pathways as metabolic receptors such as those that respond to insulin and leptin. Consequently, chronically elevated levels of such cytokines can result in insulin resistance and likely resistance to leptin and numerous other metabolic regulators. Insulin and leptin are both involved in mediating satiety and, hence, not only are insulin resistance and leptin resistance a consequence of increased adiposity but may also promote adiposity in accordance with the notion that microbiota promoting inflammation can be an initiating event in development of obesity and metabolic syndrome.
Mechanisms by Which Altered Microbiota Promotes Inflammation
While observations from various investigators that microbiota transplants confer susceptibility to obesity, metabolic syndrome, and colitis all support the notion that altered microbiota plays a central role in these phenotypes, deciphering underlying mechanisms remains a major challenge in this field. We have been using the T5KO mouse model to attempt to unravel the interrelationship between altered microbiota and intestinal inflammation. In a recent “prospective study,” we collected fecal samples weekly from a large number of WT and T5KO mice from weaning to adulthood (14 weeks of age) at which point mice were assayed for development of colitis or metabolic syndrome. We then retrospectively analyzed fecal inflammatory markers and gut microbiota composition throughout the course of the study. We observed that a uniform consequence of innate immune deficiency was that loss of TLR5 resulted in the microbiota exhibiting greater volatility, that is, showed greater week-to-week changes (Carvalho, Nalbantoglu, Aitken, et al. 2012). Such increased microbiota volatility correlated with elevated intestinal inflammation, as revealed by levels of fecal Lcn-2. Interestingly, during this volatile period, the elevations in fecal Lcn2 bifurcated primarily in the postweaning period in that levels of this marker soared in colitic mice and stabilized at modest elevations in T5KO mice that exhibited metabolic syndrome.
Loss of TLR5 also eventuated in a change in composition of the microbiota, which was evident at the family level in colitic mice and at the species level in T5KO with metabolic syndrome. Together, these results are in accord with a model wherein the altered microbiota that results from loss of TLR5 causes compensatory activation of other aspects of the immune system that often contains/stabilizes a modestly altered microbiota associating with low-grade inflammation. This type of compensation and “new equilibrium” can be viewed as successful in that the mice breed normally and appear generally healthy—metabolic syndrome notwithstanding. When T5KO mice do not manage to effectively contain their altered microbiotas, classic chronic inflammation ensues. The factors that may tip this balance toward such robust colitis, which in mice typically results in lack of breeding and is hence a clearly more severe phenotype than metabolic syndrome, remain to be defined, but include the presence of select bacteria, termed pathobionts, in that they promote disease in select circumstances. In any case, we speculate that the immune-mediated compensation that eventuates in insulin resistance may reflect that the insulin resistance state is associated with modestly elevated glucose levels may help immune cells contain the perturbed microbiota and hence be important for the new equilibrium—outlined in Figure 2.
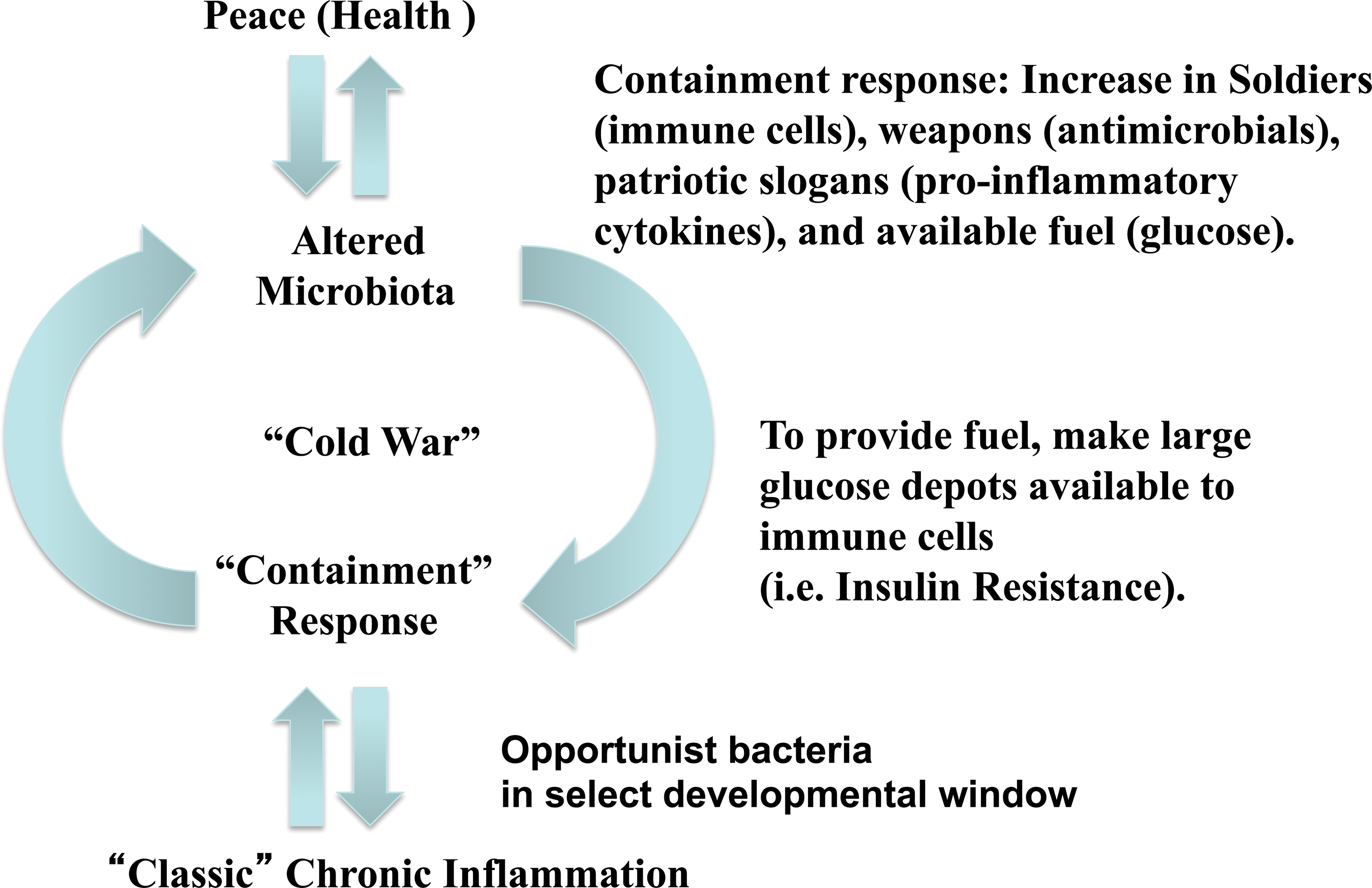
Development of low-grade inflammation analogous to a Cold War. This illustration points out that most perturbations in the microbiome or the host result in a new equilibrium with changes in the host and microbiota that suggest preparation to defend against each other. We speculate that such changes might invoke insulin resistance as a means to give more fuel to immune cells.
Our ongoing studies are seeking to understand how alterations in microbiota composition promote inflammation. We recently considered the possibility that the altered bacteria might simply have an inherently greater potential to activate the innate immune system. Indeed, it has long been appreciated that LPS, flagellin, and likely other components of bacteria exhibit great variance in their potency as TLR/NLR ligands depending upon the specific bacterial species from which they were isolated. This is indeed the case in T5KO mice. Specifically, the altered microbiota in TLR5 mice, especially when colitic, has greater levels of proinflammatory LPS and flagellin. Such inherently increased potential to activate innate immunity might promote both classic and low-grade inflammation (Chassaing et al. 2013). It will be important to determine if similar changes are seen in mice, and humans, that develop intestinal inflammation due to other underlying causes.
Causes of Altered Microbiota–Host Relationship in Humans
That many of our studies of gut inflammation have focused on a single molecule, flagellin, reflects the great importance of motility to many bacteria. Yet, the general paradigm our studies reflect is not specific to flagellin and/or its innate immune recognition. Rather, we hypothesize that a wide array of genetic alterations that make a host unable to manage gut bacteria would predispose to a variety of chronic inflammatory diseases including colitis and metabolic syndrome. In accordance, many mouse strains and humans with mutations in genes that mediate innate immunity are prone to developing colitis. Moreover, although less studied than T5KO mice, mice lacking TLR2 and NLRP6 are predisposed to developing metabolic syndrome (Caricilli et al. 2011; Henao-Mejia et al. 2012). Recent studies have shown that humans with reduced levels of TLR5 function are prone to develop type 2 diabetes. Thus, any alterations in factors that mediate host–bacterial interactions can predispose to chronic inflammatory diseases. However, the dramatic increase in inflammatory diseases during the past 75 years despite relatively constant human genetics suggests that either the human microbiota has changed and/or the presence of another environmental factor has altered the way in which the microbiota and host interact. A number of studies have indeed suggested that the predominant microbiota of persons in developed countries has changed although the evidence is largely indirect (Cho and Blaser 2012). Specifically, it has been observed that the microbiome in developing countries is more diverse than that of developed countries, suggesting a change in the latter. The best defined example of a societal change in the microbiota is the ongoing disappearance of Helicobacter, which colonized about 80% of the U.S. population 80 years ago but has become almost eliminated in young persons who will likely never be colonized (Chen and Blaser 2012). The cause of this disappearance of Helicobacter is not clear, but it seems unlikely to be specific for this microbe and more likely reflects a broader change in gut microbiota composition.
A number of environmental (i.e., nonhost genetic) factors that might have altered the gut microbiota composition, and/or its interaction with the host, can be envisaged that can broadly be placed in two categories, namely microbial or dietary factors. Microbial factors include increased use of antibiotics, general changes in hygiene resulting in less exposure to microbes, loss of parasites, which were once ubiquitous in many populations and/or presence or absence of specific bacteria or viruses that might broadly influence behavior of other microbes. Dietary factors that might have influenced microbiota include changes in macronutrient content, which is known to alter microbiota composition (Wu et al. 2011). Moreover, regimens of modern food production include numerous small molecules that might alter the microbiota and/or its interaction with the host. One example of this notion is emulsifying agents that are added to many processed foods. These detergent-like molecules have been shown to promote bacterial translocation and have been hypothesized to play a role in the increased rates of inflammatory bowel disease (IBD) that have afflicted developed societies.
Conclusion
The gut microbiota plays an important role in maintaining the health of the host. Yet, this complex microbial community must be well managed to protect the host from this large microbial biomass. Failure to properly manage the gut microbiota can result in numerous flavors of inflammation, including IBD, metabolic syndrome, and perhaps cancer. Use of genetically modified mice has proven invaluable in defining microbial and host determinants that help to maintain a stable microbiota–host relationship. While there remains to be many aspects of such mechanisms to be elucidated, another arising challenge is to define environmental factors that may underlie increased incidence of chronic inflammatory diseases. Moreover, it will be important to define modalities to therapeutically restore order to gut microbiota–host interactions to treat such diseases.
Footnotes
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The author(s) received no financial support for the research, authorship, and/or publication of this article.