
Abstract
The nonneoplastic diseases of the human pancreas generally comprise the inflammatory and degenerative conditions that include acute and chronic pancreatitis, with cystic fibrosis being arguably one of the most important diseases that induce the condition. Both acute and chronic conditions vary in severity, but both can be life threatening; and because of this fact, the study of their progression, and their responsiveness to therapy, is largely conducted by indirect means using serum markers of damage and repair such as amylase and lipase activities that normally occur at very low levels in the circulation but can be significantly increased during inflammatory episodes. Progress in the understanding the pathogenesis of both conditions has therefore been largely due to time course studies in animal models of pancreatitis, and it is in this context that animal model development has been so significant. In general terms, the animal models can be divided into the invasive, surgical procedures, and those induced by the administration of chemical secretagogues that induce hypersecretion of the pancreatic enzymes. The former include ligation and/or cannulation of the biliopancreatic ducts with infusion of solutions of various kinds, or the formation of closed duodenal loops. Secretagogue administration includes administration of caerulein or
Keywords
Introduction
The pancreas is a mixed exocrine and endocrine gland lying behind the stomach and stretching between the spleen on the left-hand side of the body and the duodenum on the right-hand side (Figure 1).
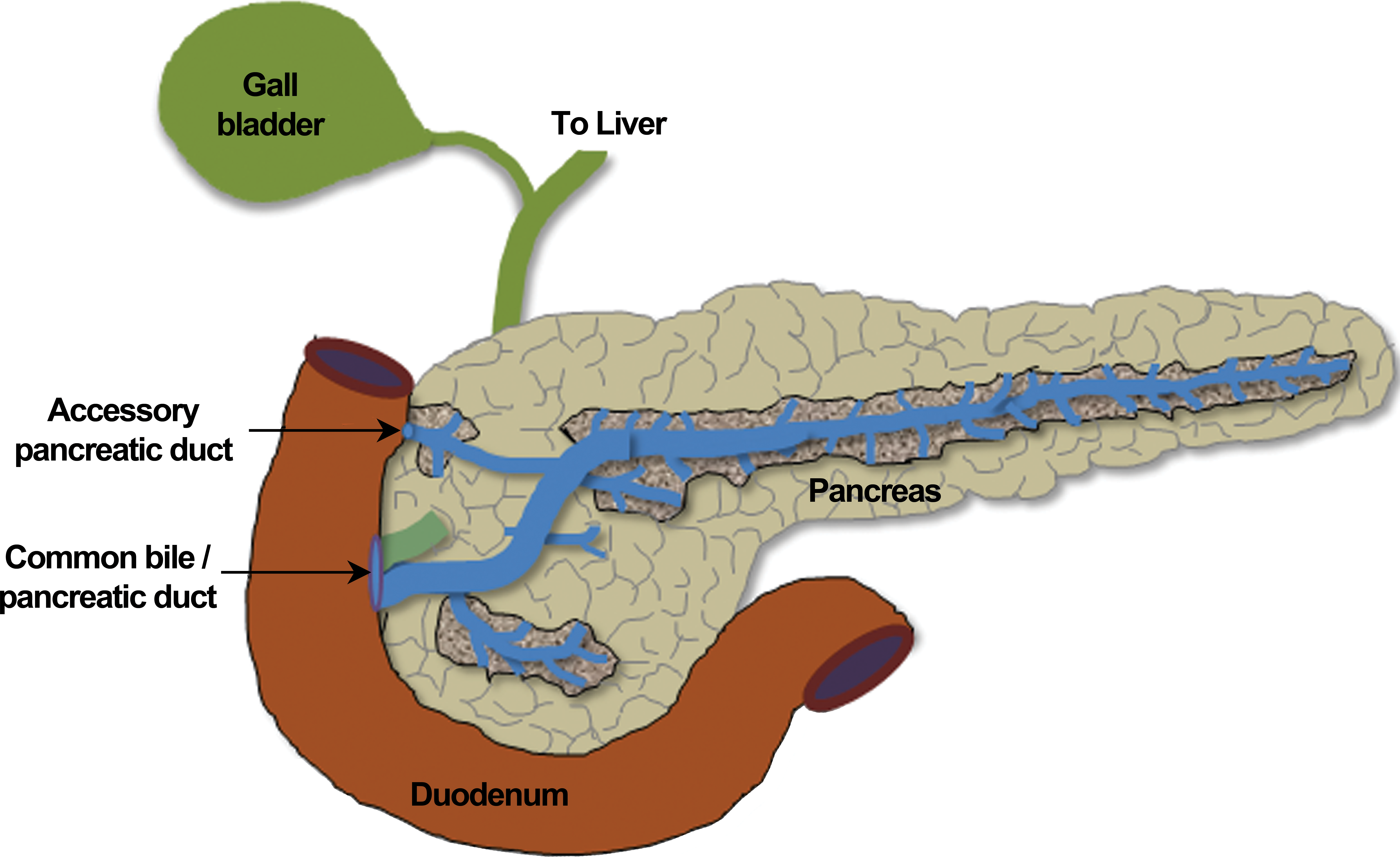
Anatomical localization of the pancreas.
The pancreas develops embryologically from 1 ventral and 2 dorsal buds that emerge from the endoderm of the developing gastrointestinal (GI) tract (Slack 1995; Tadokoro et al. 2003; Pieler and Chen 2006) at, or around, day 11 of gestation in the rat and at day 9.5 in the mouse (Pictet et al. 1972). Each bud has a pancreatic duct attached and the 2 buds fuse to form a single organ, with the ventral bud forming the inferior part of the head of the pancreas and the dorsal bud forming the superior part from which develops the head, body, and tail of the organ (Figure 2). The ducts from each part anastomose with the ventral and distal dorsal duct forming the main pancreatic duct (the duct of Wirsung) which, in man, enters the duodenum at the papilla of Vater. The proximal part of the dorsal duct either atrophies or persists as the accessory duct of Santorini which empties into the duodenum at a point closer to the stomach than the main duct. Failure of fusion of the 2 pancreatic ducts gives rise to a condition in humans called pancreas divisum where the duct of Wirsung drains only a small part of the pancreas, while the duct of Santorini drains the major part of the organ. This condition is associated with recurrent pancreatitis due to partial obstruction of the pancreatic flow and the tendency for bile stones to lodge at the opening of the duct of Wirsung into the common bile duct (Toskes 1985).
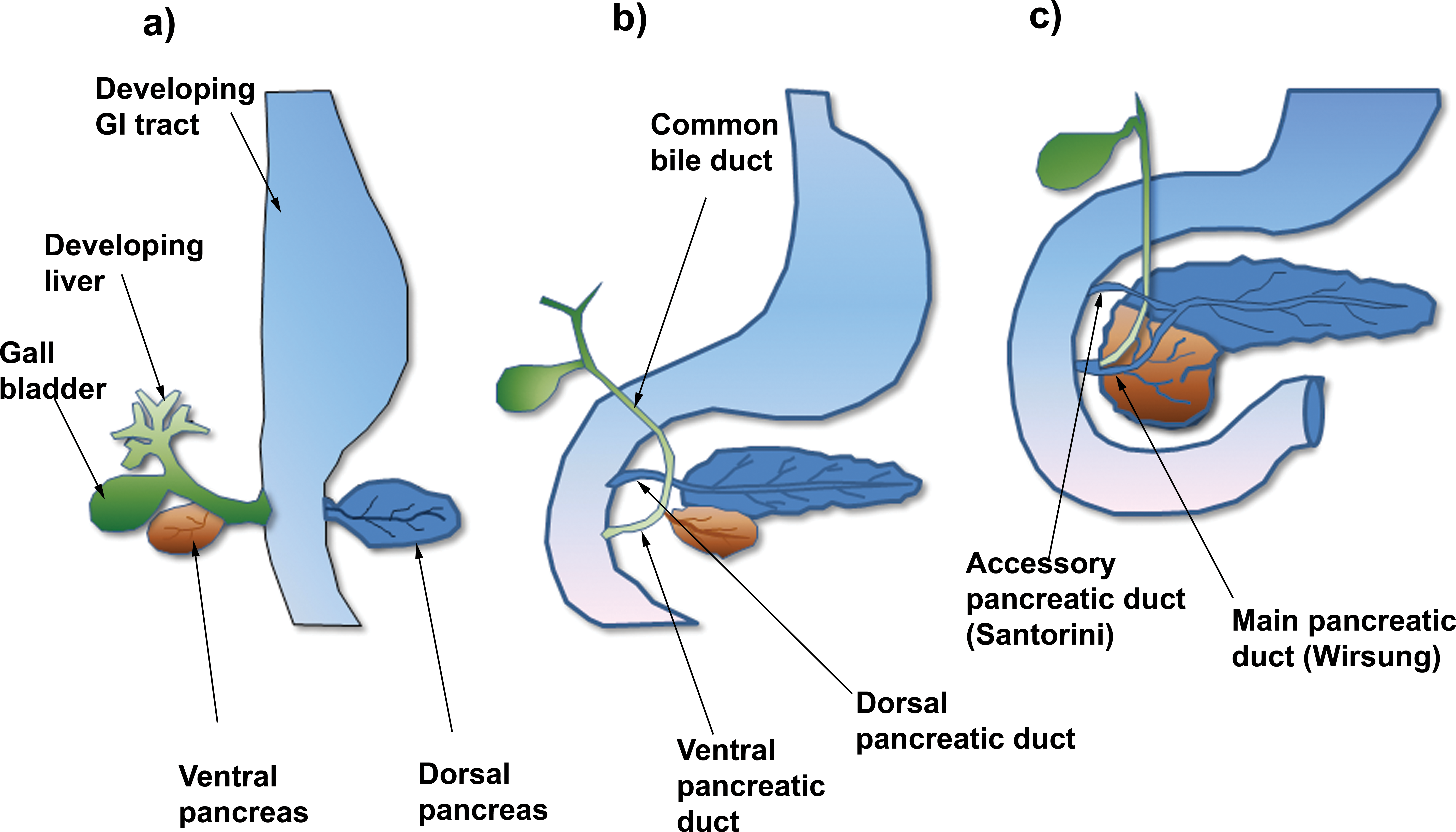
Organogenesis of the pancreas. Redrawn from Klimstra, Hruban, and Pitman (2007).
The endoderm of the pancreatic buds forms a network of primitive pancreatic ducts from which the exocrine pancreatic acini and the endocrine islets of Langerhans develop. The exocrine acini are organized into lobules, separated by thin fibrous septa, and groups of acini empty into intralobular pancreatic ducts (Figure 3). The intralobular ducts anastomose to form the larger interlobular ducts which, via an extralobular duct, eventually empty into the main pancreatic duct that combines with the common bile duct before entering the duodenum via an ampulla, which in man is termed the Sphincter of Oddi (Gavaghan 2002).
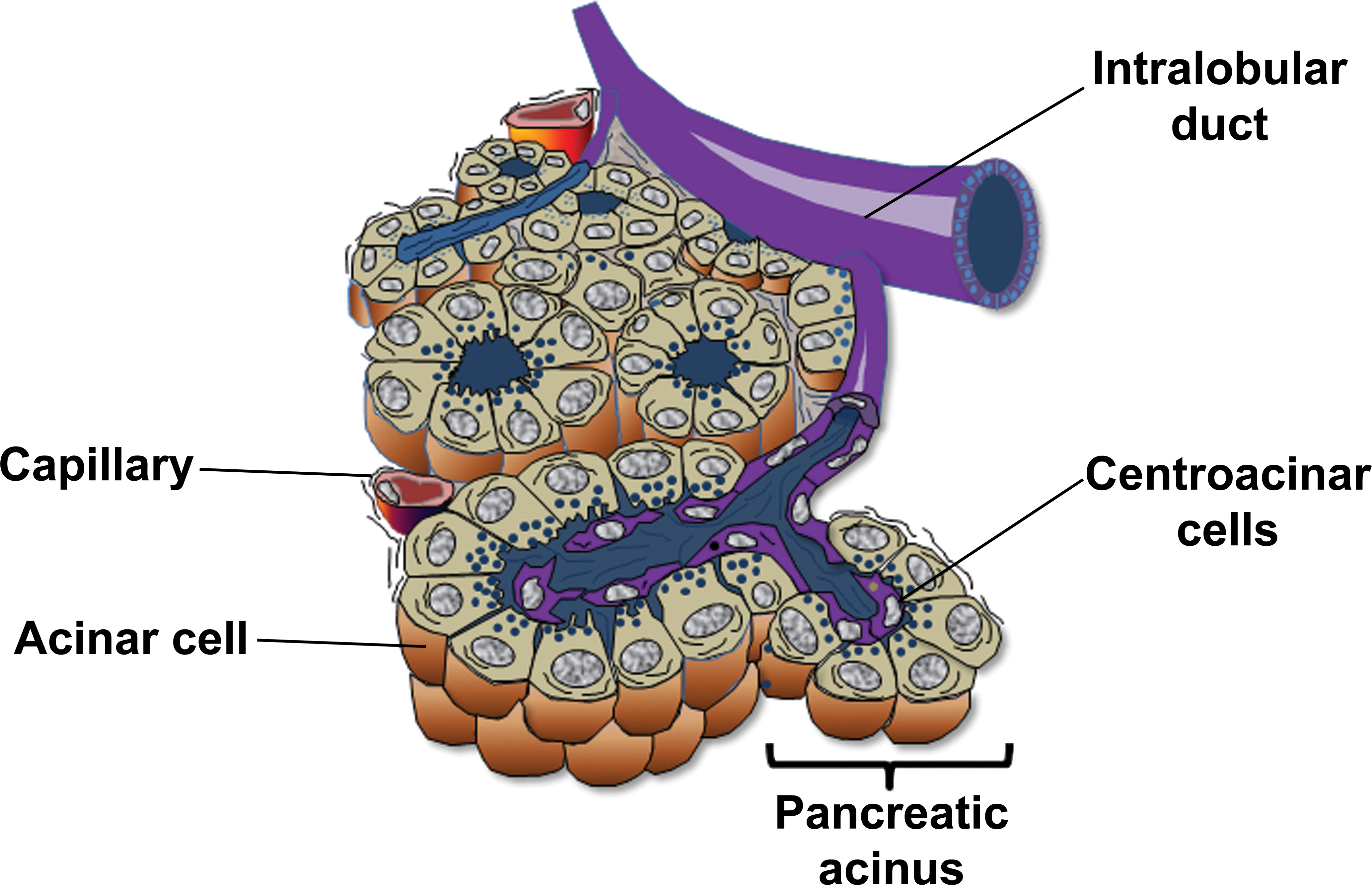
Diagram of relationship between the pancreatic acinus and the draining pancreatic duct. Redrawn from a figure in http://medcell.med.yale.edu/systems_cell_biology/liver_and_pancreas.php.
Nonneoplastic Pancreatic Disease in Man
There are several nonneoplastic pancreatic diseases of man including cystic fibrosis and pancreatitis. The pancreatic disease that develops in individuals carrying cystic fibrosis relates to the defect in chloride transport that occurs in affected individuals, and results in increased sodium and water reabsorption throughout the body and increased viscosity of the pancreatic secretions, among other effects. This leads to the pancreatic disease of mucoviscidosis, or fibrocystic disease, which develops in infants, adolescents, and young adults. The increased viscosity of the pancreatic secretions is thought to result in a blockage of the ducts with subsequent pancreatitis following damming of the flow of pancreatic enzymes, back diffusion into the pancreatic tissues, and degeneration and necrosis of the tissues (Park and Grand 1981).
Pancreatitis is an acute or chronic degeneration/inflammation characterized by varying degrees of edema, hemorrhage, and necrosis of the acini and associated pancreatic tissue and tends to be progressive with gradual replacement of the gland with fibrosis, glandular and ductular atrophy, and the development of partial or complete blockages due to the formation of proteinaceous calcifications that occur both in the interstitial tissue and within the glands and ducts (Figure 4). The incidence in the United States has increased from 2.94/100,000 during 1977 to 1986 to 4.35/100,000 person years during 1997 to 2006, with the increase being attributed to the increasing incidence of alcoholic chronic pancreatitis (Yadav et al. 2011). Acute abdominal pain is a cardinal sign of the disease, although it can subside within days of the onset of the condition, despite a continuation of the inflammation. Attacks can be very dangerous and in severe cases multiple organ failure, with significant morbidity and mortality, can result. The initiating event appears to be mediated via an inhibition of secretion from the exocrine cells (Braganza et al. 2011) and autodigestion by premature activation of the pancreatic enzymes within the organ, rather than following entry into the duodenum (Figure 5). Diagnosis can be problematic but is mainly based upon the finding of elevated serum amylase and lipase activities of 3 times the upper limit of normal, together with imaging of the pancreatic damage by computed tomography, ultrasound, or magnetic resonance imaging (Carroll et al. 2007). Histopathology is not the clinical diagnostic method of choice due to the risks involved in carrying out pancreatic biopsy.
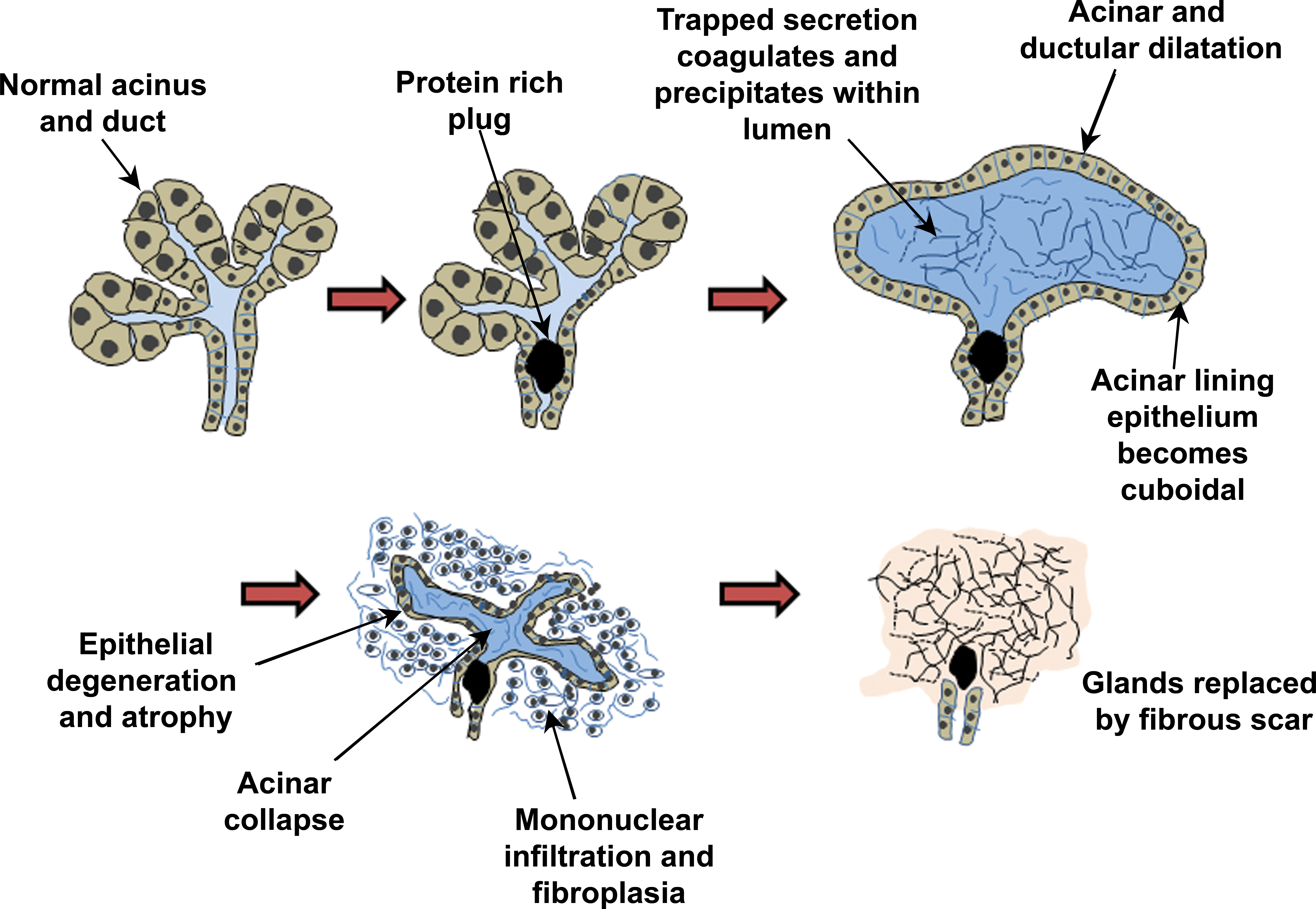
Diagram of the development of acute and chronic pancreatitis following partial or complete blockage of the draining pancreatic duct. Redrawn from http://www.4medstudents.com/Students/pancreatitisChronic.htm.
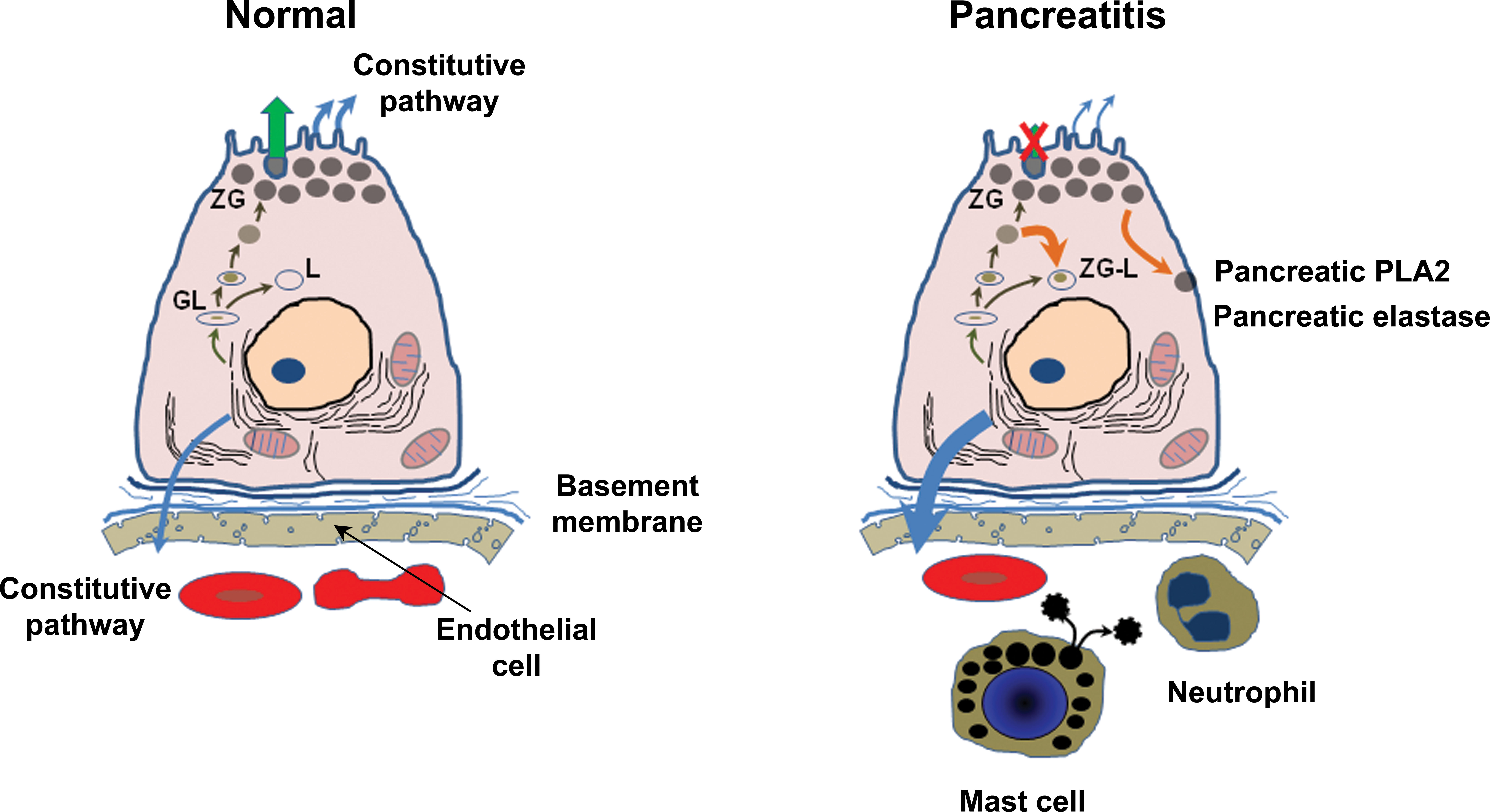
Diagram of disruption of acinar cell production and release of zymogen during development of acute pancreatitis. The constitutive pathway, at the basal and apical membrane, normally transports a fraction of the newly synthesized enzyme. RER = rough endoplasmic reticulum; GC = golgi complex; ZG = zymogen; L = lysosomes; ZG-L = activated zymogen within lysosomes; PLA2 = phospholipase A2. Modified from Braganza et al. (2011).
The genetic and environmental risk factors for the development of acute pancreatitis in man are shown in Figure 6, with alcohol being the most important of the preventable factors with an incidence of 90 in 100,000 in heavy drinkers (Van Woerkom and Adler 2009). The alcohol-induced disease also has the highest odds ratio for overall mortality when compared to biliary pancreatitis, and this is thought to be related to the relatively poor nutrition associated with alcoholics (Frey et al. 2006).
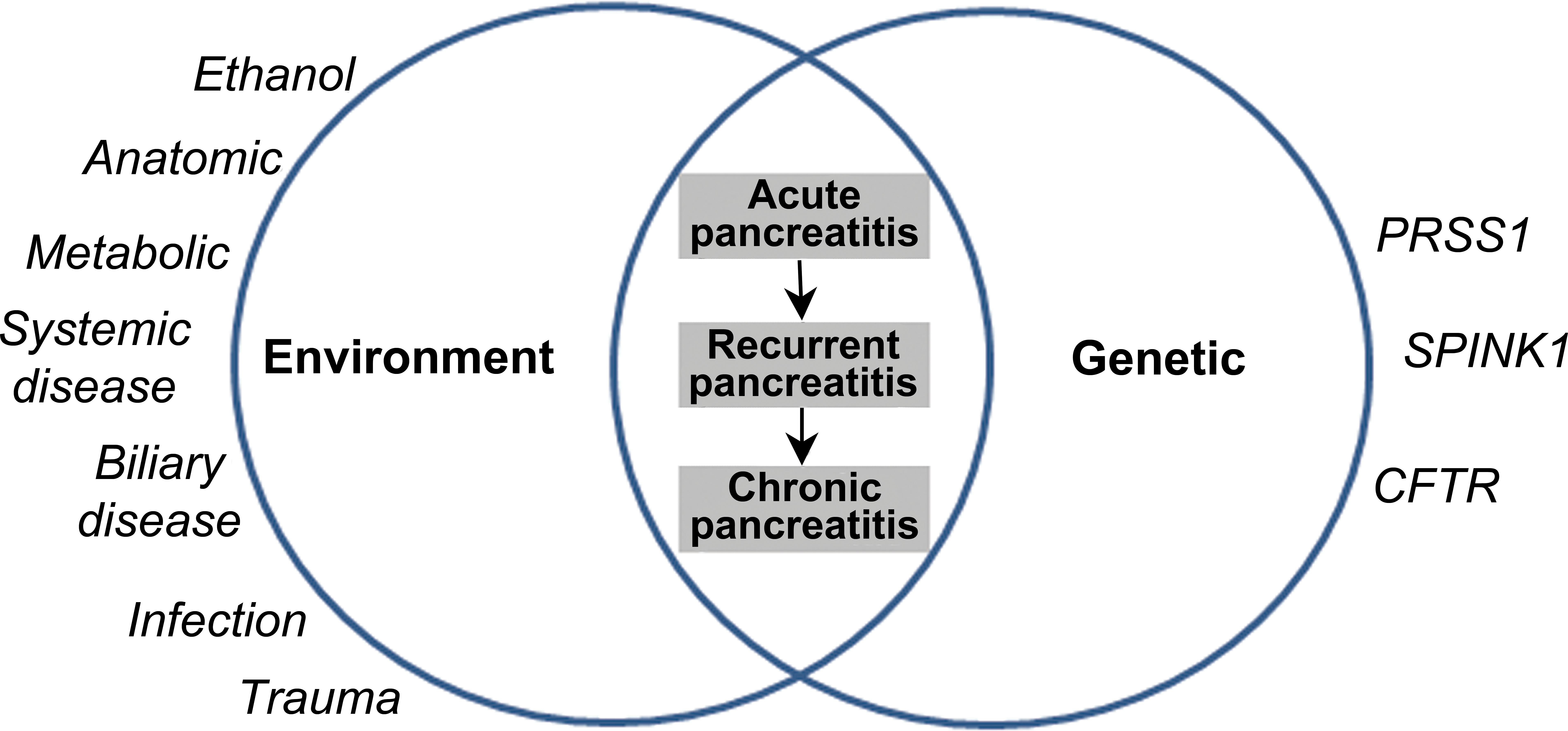
Interaction of genetic and environmental influences on the development of pancreatitis. Redrawn from Larrosa-Haro, Sánchez-Ramírez, and Gómez-Nájera (2012).
As with all of the inducers of the disease, the pathogenesis of alcohol-induced pancreatitis is thought to be multifactorial, and the combination of cigarette smoking and alcohol significantly increases the risk of developing both the acute and the chronic forms of the disease (Figure 7). One hypothesis for the role of environmental factors involves the generation of increased amounts of free radicals within the pancreas, and the production of cytotoxic metabolites of ethanol and other organic chemicals within the pancreas, leading to premature activation of the pancreatic enzymes, is again thought to be responsible for the initiating acinar cell damage (Gorelick 2003; Braganza 1998). Support for the metabolic theory of pancreatic damage was given by the demonstration of higher pancreatic levels of certain cytochromes P450 in patients with chronic pancreatitis (Foster et al. 1993) over that present in healthy subjects.
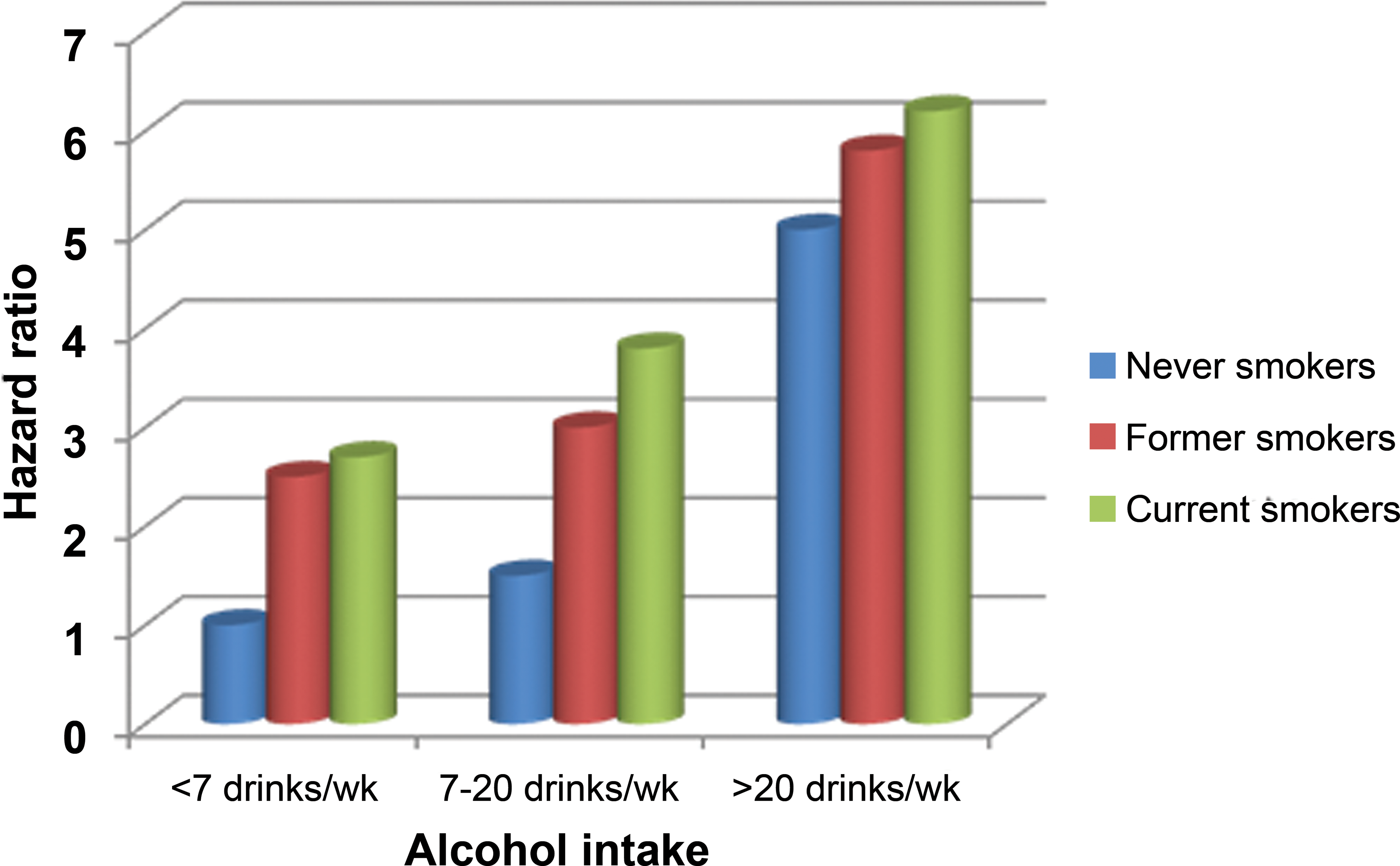
Hazard ratios of pancreatitis by smoking status and weekly alcohol intake in men and women combined. One drink corresponds to 12 g of pure alcohol. Adapted from Tolstrup et al. (2009).
The disease of chronic pancreatitis is considered to result from multiple episodes of acute acinar damage leading to pancreatic dysfunction and the production of increased amounts of digestive enzymes in the absence of increased fluid or bicarbonate from the duct cells. The increased protein, in combination with calcium salts, is thought to form plugs within the inter- and intralobular ducts, leading to an inhibition of flow, and resulting in blockage and reflux of the secreted pancreatic enzymes back into the gland. This results in a progressive degeneration of the acinar cells and a chronic inflammatory/fibrotic destruction of the functional compartments of the pancreatic acini (Figure 4). Grossly, the pancreas becomes enlarged and frequently demonstrates the presence of intraductal calcifications at intervals throughout the organ (Bastid et al. 1990), which can be visualized by radiography and which is considered to be pathognomic of the disease.
Pancreatitis is frequently accompanied by abdominal pain, malabsorption as a result of progressive loss of digestive enzyme output, malnutrition, weight loss, and in the late stages, following destruction of the islets of Langerhans, diabetes mellitus (Steer, Waxman, and Freedman 1995). Chronic pancreatitis has an incidence ranging from 0.04 to 5% (Owyang and Levitt 1991); and in developed countries, around 70% of patients have a history (6–12 years) of heavy alcohol consumption (Sarles et al. 1979). The mortality rate is reported to be up to 50% within 20 years of its onset (Ammann et al. 1984), and pancreatitis predisposes to pancreatic cancer in roughly 4% of patients within 20 years of an initial diagnosis of the disease (Lowenfels et al. 1993).
Histopathology of Pancreatitis in Man
There are relatively few reports of the histopathology of human acute pancreatitis except in very severe cases that have resulted in the death of the patient. Biopsy is to be generally avoided in acute pancreatitis due to the risks of exacerbation of an already serious condition, and histopathology is not a diagnostic tool for this condition. In severe cases, the histopathology is characterized by interstitial tissue necrosis, involving both acinar and ductal cells, ductal necrosis, and acinar necrosis. Interstitial tissue necrosis appears to be autodigestive in nature and is typical of the more common forms of acute pancreatitis associated with alcohol, bile duct disease, metabolic conditions, and other rare factors. The milder form has also been called edematous pancreatitis and shows edema of the pancreas combined with foci of fat necrosis (Klöppel 2004). Severe or necrotizing pancreatitis shows large areas of hemorrhagic necrosis of the pancreatic and peripancreatic tissue. The ductal type of necrosis is rare and may be seen in pancreatitis associated with prolonged circulatory failure. The acinar type of necrosis is associated with sepsis and is caused by infectious agents (Schmid et al. 1999). Complications of acute pancreatitis, such as pseudocyst, bleeding, and infection, determine the course or resolution of the disease.
The histopathology of patients with chronic pancreatitis demonstrates large, and variable, areas of coagulative necrosis of the acini, with hemorrhage, fat necrosis, and foreign body granuloma, with varying degrees of fibrosis (Vuong et al. 1984). Figure 8 shows a dog pancreas from an animal that was diagnosed with chronic fibrosing pancreatitis and illustrates the concentric nature of the fibrotic reaction centered on an interlobular pancreatic duct with surrounding necrosis and apoptosis of the acini. Ductal distension and vascular damage, through the presence of thrombi, were reported in the 20 subjects studied in the review alongside the acinar necrosis, and mineralized concretions were present in the larger pancreatic ducts in the more chronically affected organs. In areas of the organ less affected, a generalized decrease in zymogen was observed in many of the samples. Varying degrees of inflammatory cell infiltrate composed of mononuclear cells and neutrophils with edema were also observed generally throughout the samples. Blood vessels included in the affected areas frequently showed fibrinoid necrosis of the smooth muscle lining. In a study of 26 patients with alcohol-associated chronic pancreatitis (De Angelis et al. 1992), mineralized ductal plugs were present, which the authors believed could have led to the progressive fibrosis and acinar atrophy on the obstructed pancreatic lobule. A constant finding in this series of alcoholic chronic pancreatitis was the presence of inflammatory foci of lymphocytes associated with nerve bundles, this being postulated as the cause of the pain commonly associated with the disease.
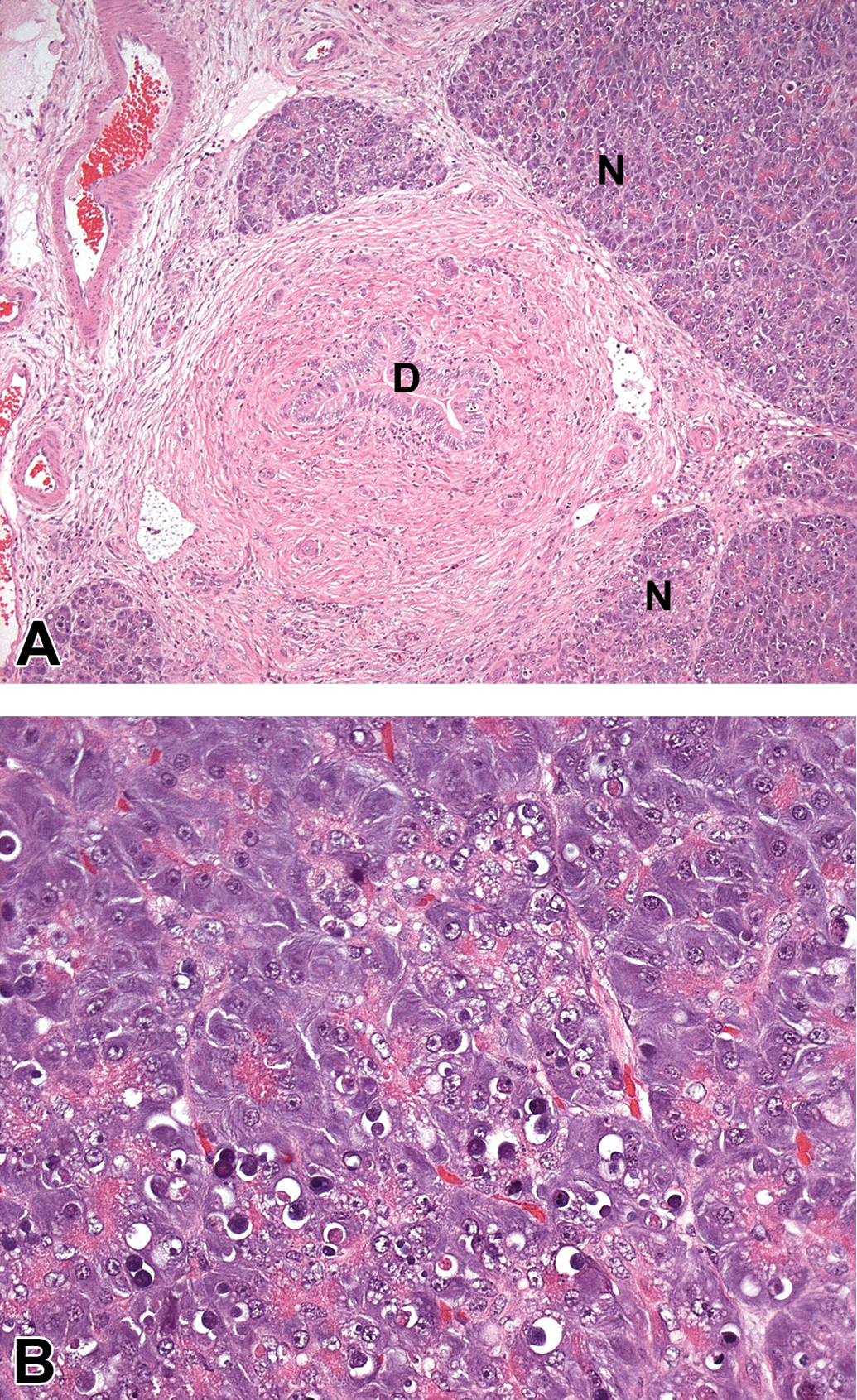
Chronic pancreatitis in dog given a serine/threonine kinase inhibitor. (A) Concentric rings of fibrosis centered upon an interlobular pancreatic duct (D) and surrounded by necrotic acini, a 5× magnification. (B) Necrosis and apoptosis of exocrine cells of the acini 25× magnification.
In nonalcohol-induced, or idiopathic, pancreatitis cases, while fibrosis and acinar atrophy were always present, neutrophils were never observed (De Angelis et al. 1992), suggesting either significantly different etiologies of the two diseases or different time delays in the onset of inflammation. In contrast, in obstructive pancreatitis, the histopathological picture was similar to that seen with the alcoholic cases.
Animal Models of Acute Pancreatitis
Claude Bernard is widely credited for inducing the first case of acute pancreatitis in dogs by injecting bile and olive oil retrograde into the pancreatic duct to produce an acute necrotizing pancreatitis (Bernard 1856). Since this time, a number of animal models have been developed, which show varying degrees of faithfulness to the developing lesions in man and which have been used to address the possibility of therapeutic intervention to modify the course of the disease (Büchler et al. 1992; Al-Mufti and Williamson 1999; Lerch and Adler 1994). The ability to investigate the development of the disease, following a known insult, has significantly contributed to our understanding of the underlying pathogenesis of the human disease and have guided some of the available therapies in use today (Foster 2009). The animal models that are readily available tend to be in rodents, although useful models have been developed in the dog, the possum, and the cat (Lerch et al. 1992; Su, Cuthbertson, and Christophi 2006; Wedgwood, Farmer, and Reber 1986).
Invasive Models of Pancreatitis
These model systems were developed to test the reflux hypothesis of pancreatitis whereby pancreatic fluid/bile is forced, in a retrograde or anterograde direction, from the main pancreatic ducts back into the body of the pancreas. This was the first model employed by Bernard (1856) in his experiments in the dog and can be achieved in a number of ways.
Pancreatic Duct Injection
By this method, the pancreatic duct is cannulated and solutions are perfused along the pancreatic ducts (Figure 9). The original model was carried out in a cat (Reber, Robert, and Way 1979), whereby the cannula was implanted into the pancreatic duct at the tail of the pancreas with solutions being perfused in an anterograde direction to exit into the duodenum. When activated pancreatic enzymes were perfused in this model, an edematous pancreatitis was produced (Farmer, Maslin, and Reber 1983). Because of the considerable surgical skill required in cannulating the pancreatic ducts at the tail end of the pancreas, a more commonly used variant of this model involves cannulating the ducts at the head end, either directly into the pancreatic duct or via cannulation of the common bile duct. In this model, solutions, such as activated digestive enzymes (trypsin, elastase, lipase, or phospholipase A), bile, with or without trypsin, can be introduced in a retrograde direction from the head of the pancreas toward the tail (Aho, Koskensalo, and Nevalainen 1980), and Figure 10 illustrates the effect of infusing a solution of enterokinase and bile into the pancreatic duct of a guinea pig. The pressure and volume of solution used in injecting these agents into the pancreatic duct is critical in determining the severity of the induced pancreatitis, and in severe cases can lead to the death of the animal within 2 to 3 days (Hansson 1967). To prevent flow of solutions into the liver, the bile duct is clamped and bile salts are infused via a perfusion pump where they flow into the pancreatic duct and into the pancreas. Unconjugated bile salts have been shown to induce a greater degree of pancreatitis than do conjugated bile salts (Adler, Kern, and Scheele 1986). Infusion of sodium taurocholate induced edema and a hemorrhagic necrosis immediately following the infusion, which progressed to an acute inflammatory response with neutrophils (Wanke 1970). A number of potential therapeutic agents have been applied to this model of bile salt–induced pancreatitis, including glucagon (Lankisch et al 1974), somatostatin (Lankisch, Koop, Winckler, Folsch, et al. 1977), cimetidine (Evander and Ihse 1980), cholecystokinin (CCK) receptor antagonists (Wisner and Renner 1988), and other fibrinolytic agents (Lankisch, Koop, Winckler, Kostering, et al. 1977).
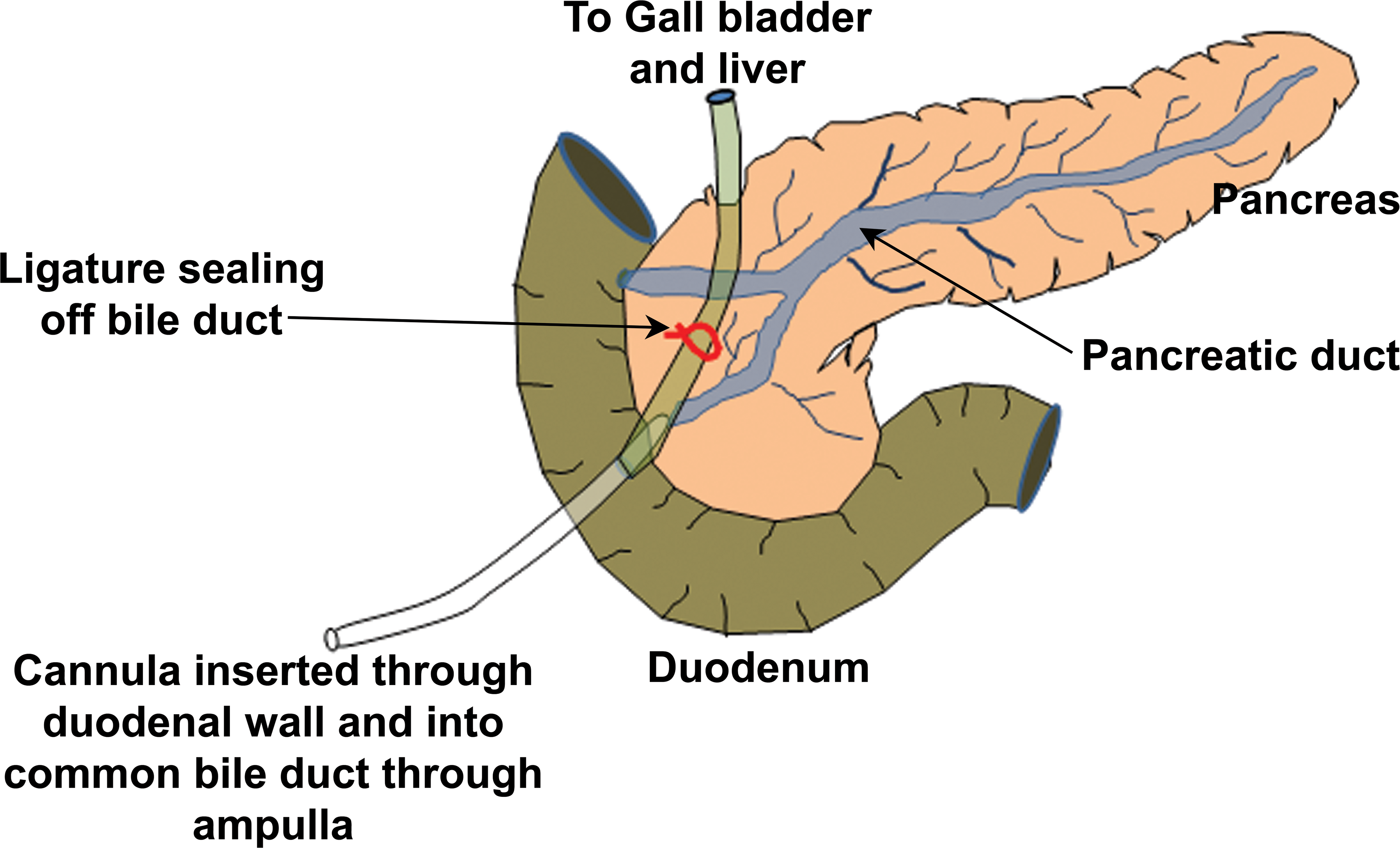
Surgical cannulation of the common bile/pancreatic duct and ligation of the bile duct to prevent retrograde infusion of solutions into the liver. Adapted from http://www.physio-pedia.com/Pancreatitis.
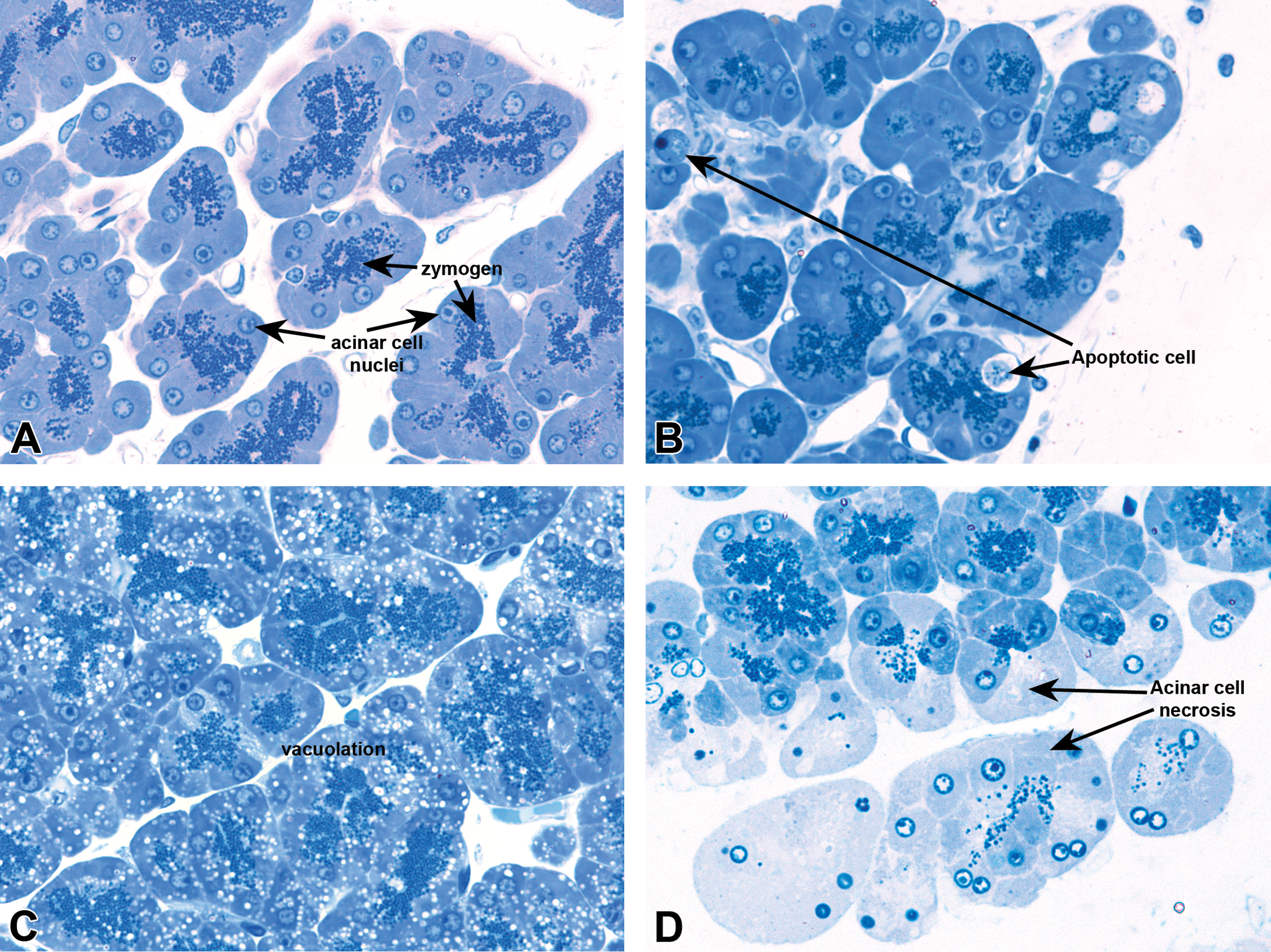
Exocrine pancreas following retrograde injection of a solution of enterokinase and bile in a retrograde direction into the pancreatic ducts of male guinea pigs with animals being killed 36 hr later. (A) Control. (B) Apoptotic bodies within the acini (arrows). (C) Diffuse vacuolation in the acinar cells. (D) Acinar coagulative necrosis. Note the relative lack of inflammation in this model. Toluidine blue staining 40× magnification.
Closed Duodenal Loop
This model was first described in dogs and is accomplished by ligating the duodenum either side of the entry of the common bile duct (Seidel 1910; Pfeffer, Stasior, and Hinton 1957). In a refinement of the model, the bile duct is also cannulated to divert the bile away from the duodenum and avoid involvement of bile in the ensuing pancreatitis (Figure 11). The cannulated bile is normally diverted into the jejunum in this version of the model (Adler, Kern, and Scheele 1986). The combination of duodenal and pancreatic secretions accumulates in the closed duodenal loop and ease reflux of the combination of fluids back into the pancreatic duct.
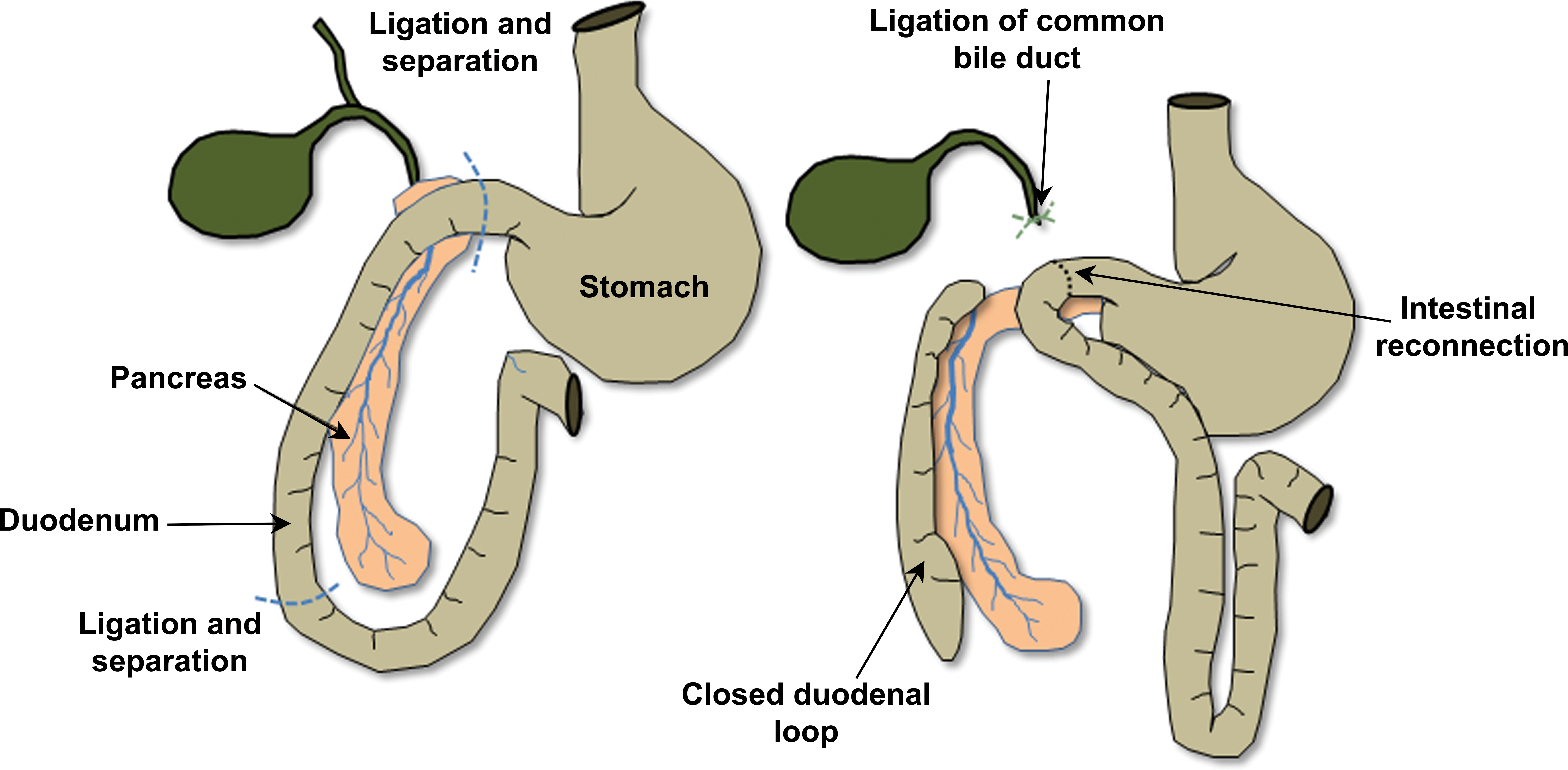
Diagram of the surgical production of a closed duodenal loop. Redrawn from Weber et al. (1992).
Intralobular edema, with a low-grade acinar necrosis, is seen in this model, and the changes become progressively more severe until the pancreatic ductules separate and hemorrhagic pancreatic necrosis develops usually within the first 12 hr after ligation. Although fat necrosis does not appear to occur in this model and inflammation is not a major feature, bacteria are present within the pancreas and this model has been considered potential for investigating the role of bacterial infection in the development and resolution of certain forms of human acute pancreatitis (Byrne and Joison 1964; Schmid et al. 1999). The model in primates appears to require additional stimulation of secretion of the pancreas to develop pancreatitis (Johnson and Doppman 1967). Findings from this model support the hypothesis that reflux of duodenal contents into the pancreatic duct is a possible etiologic factor in the development of acute pancreatitis (Su, Cuthbertson, and Christophi 2006). While ligation of the duodenum is ultimately lethal, the model has efficacy with a number of treatments that affect both the course and severity of the ensuing pancreatitis including the coadministration of protease inhibitors (Murakami et al. 1990; Ono et al. 1990), somatostatin (De Rai et al. 1988), prostaglandin E1 (Crocket et al. 1984), and vasopressin (Schapiro et al. 1976).
Duct Ligation Model
Pancreatitis can be induced by directly ligating the common bile duct, as it enters the duodenum (Baxter et al. 1985). In the rat, this procedure induces an acute pancreatitis and an obstructive hepatic cholestasis and cholangitis in the liver. This clearly has similarities to gallstone-induced pancreatitis in humans and, to some degree, models certain types of the abnormalities of the sphincter of Oddi and strictures at the papillae that rarely occur in man. Surgical ligation of the pancreatic duct alone has not been found to be sufficient to produce pancreatitis even though they will develop atrophy and acinar apoptosis without significant necrosis or inflammation (Walker 1987). Complete ligation of the common biliopancreatic duct in the rat has been described as resembling the multiple organ effects seen in man including pancreatic necrosis, hemorrhages, inflammatory cell infiltration, and the development of multiple thrombi in the lung, stomach, and kidney (Vasilescu and Tasca 1991).
The possum is an animal species that has been used to produce a model of acute hemorrhagic pancreatitis and studies where ligation of the pancreatic duct alone, or bile and pancreatic duct separately, leads to a severe pancreatitis that kills the majority of animal after approximately 2 weeks postoperation (Senninger et al. 1986). While in other species duct ligation is followed by acinar cell apoptosis in the opossum, acinar cell necrosis is the initiating feature (Gukovaskaya et al. 1996).
Noninvasive Models of Pancreatitis
Secretagogue Model—Acetylcholine Stimulation
Cholinergic hyperstimulation alone is able to induce pancreatic pathology in the dog through excessive pancreatic enzyme release and was shown in the early studies by Mouret (1895) and Babkin (1906). Pancreatic pathology was subsequently shown to occur following excessive injections of acetylcholine (Villaret and Justin-Besançon 1929). Cholinergic stimulation of acinar secretion via the vagus nerves is one of the controlling mechanisms for the pancreas (Figure 12), and they work through activating muscarinic M1 receptors located on the acinar cell membranes (Gautam et al. 2005) to stimulate secretion of the pancreatic digestive enzymes. Of equal if not more importance in the regulation of pancreatic enzyme secretion is via the release of peptide hormones, such as CCK, gastrin, and secretin (Niebergall-Roth and Singer 2001), secreted from the GI tract under the ingestion of fats or proteins (Liddle 1997). As its name suggests, cholecystokinin, a peptide synthesized by I-cells in the mucosa of the small intestine, causes the release of not only the digestive enzymes from the pancreas but also bile from the gallbladder.
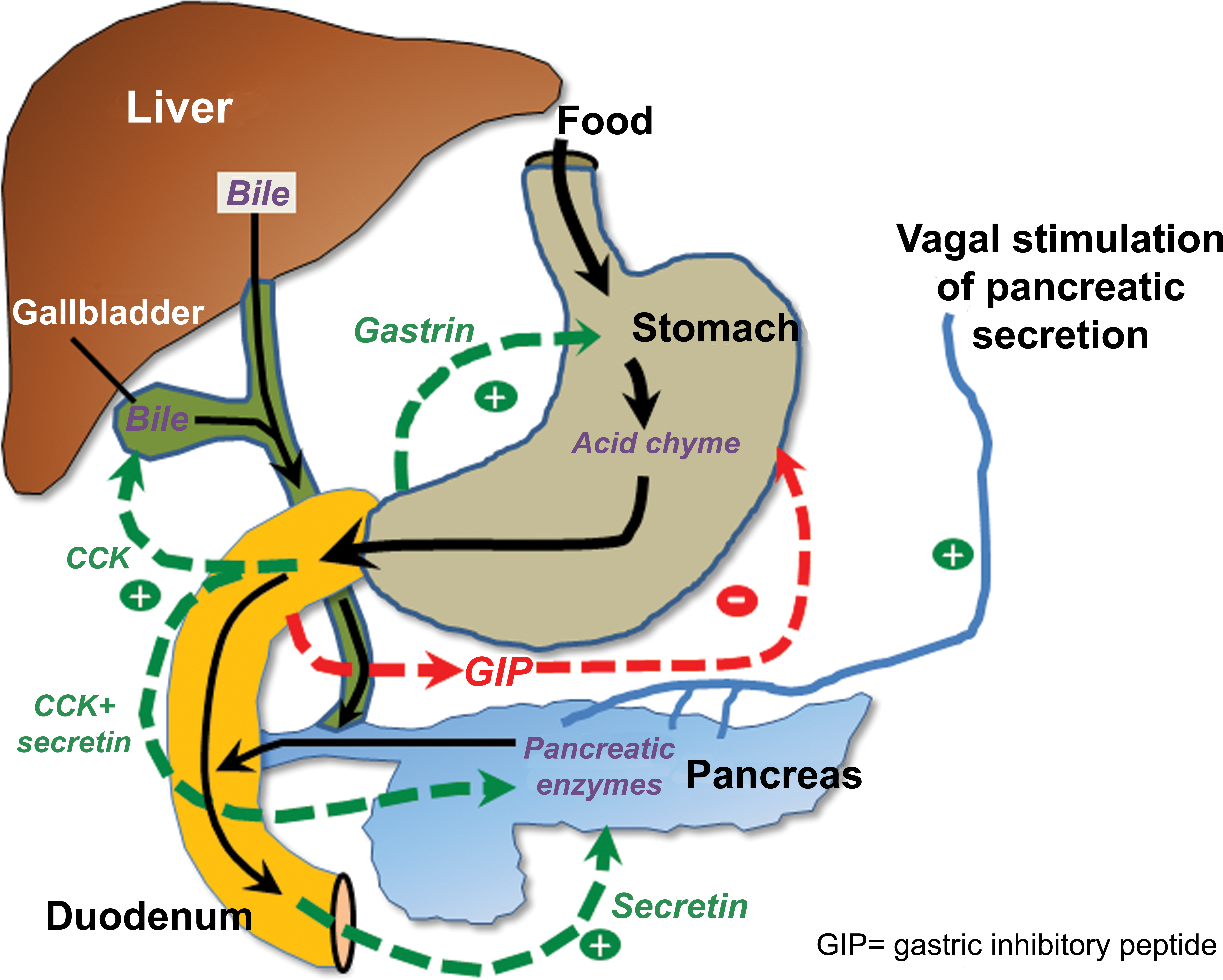
Diagram illustrating the control of exocrine pancreatic secretions by both neural and hormonal factors. Redrawn from http://bioserv.fiu.edu/∼walterm/Fund_Sp2004/digestion/present.htm.
Caerulein Localization Model
Caerulein is a CCK analog and has been used to induce acute pancreatitis in a number of species including the rat and the mouse (Niederau, Ferrell, and Grendell 1985; Watanabe et al. 1984; Lampel and Kern 1977). Acute pancreatitis can be induced by a single intravenous infusion (5 µg/kg body weight), a single intraperitoneal injection (50 µg/kg body weight), or multiple subcutaneous injections of caerulein (24 μg/kg, every 8 hr for 2 days), which stimulate proteolytic enzyme secretion by the acinar cells, and causes acinar cell necrosis preceded by progressive interstitial edema (Konturek et al 1992; Lankisch, Koop, Winckler, Folsch, et al. 1977; Adler, Hupp, and Kern 1979; Adler et al. 1983). Multiple injections increase the severity of the response and decrease the interanimal variability (Bhatia et al. 1998, 2001; Song et al. 2002).
The exact mechanism by which caerulein induces pancreatitis is unclear, but studies suggest that administration induces an abnormal localization of the zymogen and lysosomal hydrolases that are then activated intracellularly within the acinar cells rather than secreted in the protected form into the acinar lumen (Steer and Mendolesi 1987). This model has also been used to assess the potential for moderating the induced pancreatitis with therapeutic intervention, and the cholecystokinin antagonists, proglumide and benzotript, have been shown to reduce the biochemical, and pathological, abnormalities induced by caerulein in the mouse (Niederau, Ferrell, and Grendell 1985). The positive responses with these two antagonists suggest that the effects of caerulein are at the level of the CCK receptor, rather than by any nonspecific toxicity resulting from accumulation/activation of the analog. Other CCK antagonists have also been shown to modify the pancreatitis induced by caerulein (Bilchik et al 1990).
Other Secretagogues
Other pancreatic secretagogues that have been used to produce acute pancreatitis include the muscarinic receptor agonist, carbachol (Chaudhuri, Kolodecik, and Gorelick 2005; Sandstrom et al. 2005), and anticholinesterases such as the organophosphate pesticides, fenthion (Ikizceli et al. 2005; Costa et al. 1984) and diazonin (Dressel et al. 1979). Irreversible acetylcholinesterase inhibition, as is seen with some organophosphate pesticides, leads to an accumulation of acetylcholine at the postsynaptic receptors to initiate the sustained stimulation of the nerves controlling the enzymic secretion from the acinar cells. Both this, and parasympathetic stimulation with acetylcholine and pilocarpine, augment the normal pancreatic flow and increase the intrapancreatic ductal pressure, but while the edema can be similar, the degree of inflammation present with these chemicals has been shown to be less than that seen with caerulein, the CCK agonist (Adler, Rohr, and Kern 1982; Adler et al. 1983).
Alcohol-induced Pancreatitis
An association between alcohol and chronic pancreatitis has been known since the early 19th century (Friedreich 1878; Siech, Heinrich, and Letko 1991; Andrzejewska, Dlugosz, and Jurkowska 1998), and indeed currently it has been estimated that over 70% of all cases of chronic pancreatitis are due to alcohol abuse even though less than 10% of chronic alcoholics develop chronic pancreatitis (Shimizu 2008; Li et al. 2008). The increased incidence of pancreatitis that has occurred between 1997 and 2006 is thought to be a direct consequence of the increased alcohol consumption (Yadav et al. 2011; Irving et al. 2009; Tolstrup et al. 2009) and hence the search for an animal model of alcohol-induced pancreatitis was seen as a priority in order to investigate the pathogenesis of the disease.
The administration of alcohol alone, either in the diet or in drinking water, to laboratory animals was not successful in inducing pancreatitis (Siech, Heinrich, and Letko 1991; Andrzejewska, Dlugosz, and Jurkowska 1998). Several years of effort were required before a successful model was developed, during which time it emerged that ethanol-induced pancreatitis in man, as with ethanol-induced hepatitis, was a multifactorial problem of over consumption and malnutrition (Yadav and Whitcomb 2010). Long-term administration of ethanol to healthy animals was found to cause minimal pancreatic damage in laboratory animals (Gukovsky et al. 2008; Singh, LaSure, and Bockman 1982; Schneider, Whitcomb, and Singer 2002) and generally required combination treatments with secretagogues, lipopolysaccharide, or surgical intervention, to allow the required pancreatic damage to develop (Vonlaufen 2011; Foitzik et al. 1994; Quon et al. 1992). However, when the alcohol administration was combined with diets rich in unsaturated fat, in a gradually ascending ethanol administration regime, a reproducible, chronic, alcohol-induced pancreatitis was produced with the key determinants being high ethanol and high doses of dietary fat administered (Kono et al. 2001). This model showed elevated serum amylase and lipase activities and acinar cell atrophy, fatty vacuolation of the acinar and islet cells, a mild inflammatory cell infiltration and focal necrosis in the pancreas. With more chronic administration, interstitial fibrosis was also a prominent feature. The development of pathological changes was associated with the measurement of increased free radicals within the pancreas and supported the hypothesis that ethanol increased gut permeability, leading to increased plasma endotoxin levels, which sequentially promoted inflammatory cell infiltration into the pancreas, which consequently increased acinar cells damage through the generation of free radicals within the organ (Figure 13).
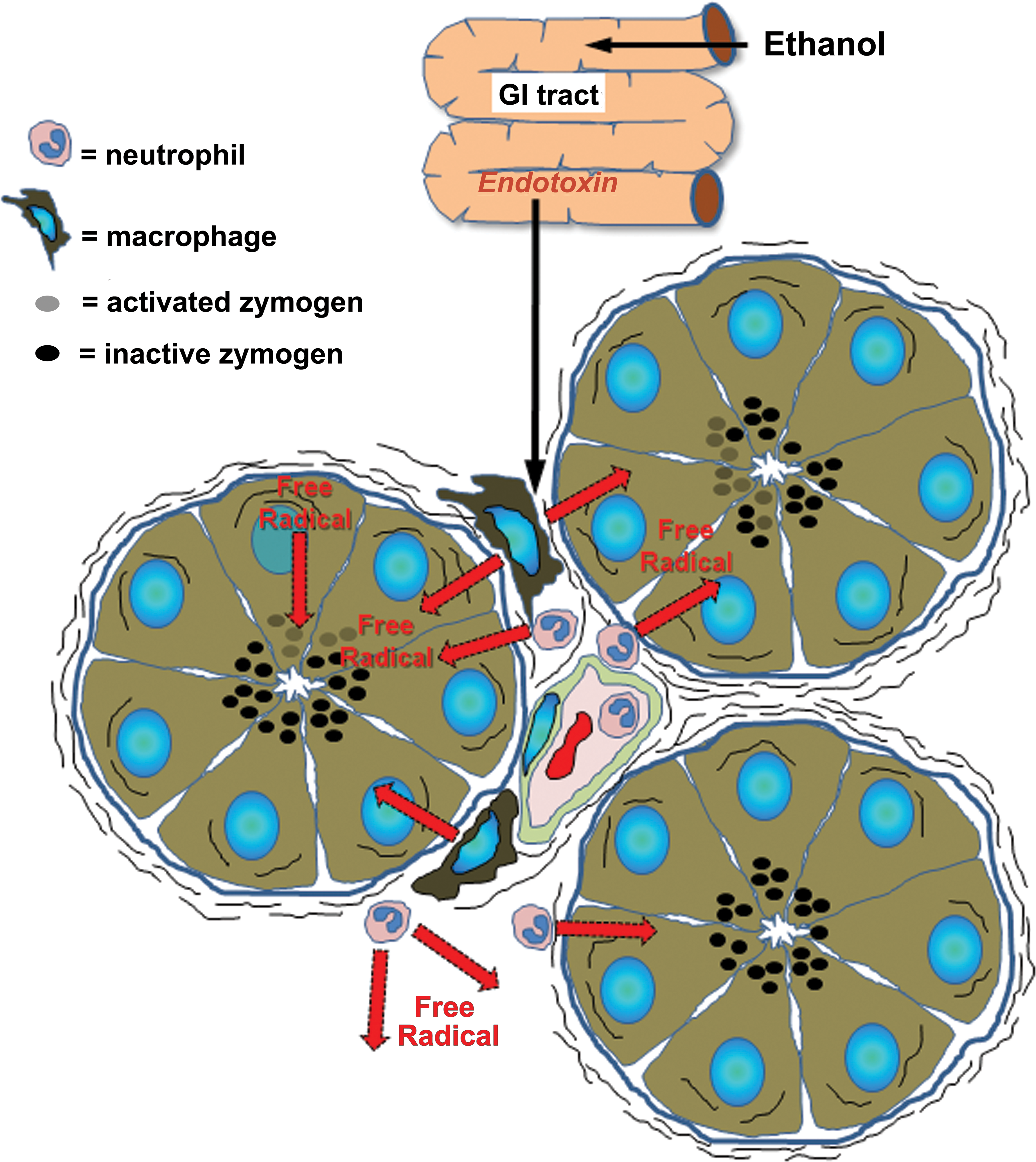
Diagram for proposed effect of ethanol in inducing acute pancreatitis mediated through increased permeability of gut mucosa and introduction of endotoxin and production of free radicals through release of inflammatory cytokines. Redrawn from Kono et al. (2001).
Dibutyltin Dichloride (DBTC)–induced Pancreatitis
A single intravenous administration of 6 to 8 mg/kg DBTC is an effective model for inducing a progressive interstitial pancreatitis that develops chronic features of the human disease with time (Merkord et al. 1997). The pathogenesis appears to be via an initial necrosis of the epithelium lining the common bile duct which, when lost from the mucosa, are passed down the duct with the bile and form plugs at the distal common bile duct. Because of the significant extrahepatic cholestasis that results from the damage to the common bile duct, peribiliary hepatic inflammation and elevations in plasma alkaline phosphatase and bilirubin are seen with this model as early as 1 day following administration (Merkord et al. 2001). The earliest indications of pancreatitis were seen during the first 4 days, and this was accompanied by increased plasma levels of amylase and lipase activities. Seven days after administration, extensive infiltration of the pancreatic interstitium with mononuclear cell infiltration which progressed to frank periductal and interstitial fibrosis, and a chronic active inflammation in animals killed after 28 days (Merkord et al. 1997, 1999). While this model appears to mimic the duct obstruction hypothesis of human pancreatitis, it was subsequently adapted by adjusting various other parameters, such as pancreatic duct ligation, and by the addition of ethanol (Merkord et al. 1998), and has several of the histopathology features seen in both the acute and the chronic forms of the human diseases. The model has been criticized in its use as an experimental tool for studying chronic pancreatitis because of its lack of reproducibility and because of the concomitant involvement of hepatic damage that alternative models lack (Otsuki, Yamamoto, and Yamaguchi 2010).
Ethionine-induced Pancreatitis
The methionine analog, ethionine, when given in the diet, or injected intraperitoneally, to a range of laboratory animals can produce a pancreatitis which can mimic the human disease especially in those models where the corresponding methionine content of the diet had been reduced (De Almeida and Grossman 1952). The onset of pancreatitis can be seen within 1 to 4 days and, following an initial inflammatory cell reaction, fibrosis and acinar regeneration occurred (Sato et al. 2003). The model has been used to study the pancreatic regeneration that occurs following damage to the exocrine pancreas. Feeding of a choline-deficient diet supplemented with ethionine to young female mice over a 5-day period was shown to be especially effective in inducing a necrotizing, hemorrhagic, pancreatitis leading to the death of the animals (Lombardi, Estes, and Longnecker 1975). Male mice, or female mice larger than 20 g, did not develop the pancreatitis; and administration of estradiol to male mice made them susceptible to developing the same pancreatic disease as the young female mice (Rao, Eagon, and Okamura 1982). In this model, biochemical defects develop within the acinar cells leading to an inhibition of secretion with massive accumulation of zymogen being observed within the acinar cells (Koike, Steer, and Meldolesi 1982). Elastase, trypsin, and chymotrypsin have been found in the interstitial tissue during the early phases of the disease, while the pancreatic content of lysosomal enzymes increases, suggesting that the premature activation and resulting autodigestion of the organ were critical events in triggering pancreatitis (Rao, Tuma, and Lombardi 1976; Rao et al. 1980). The model shows acinar cell necrosis and extensive inflammatory cell infiltration and has been used to test the efficacy of a number of therapeutics including anti-inflammatory approaches (Imamura et al. 2003), cortisone (Daugharty, Sullivan, and Gantner 1959), and green tea catechins (Takabayashi, Harada, and Hara 1995).
l -arginine-induced Pancreatitis
Genetic Animal Models of Pancreatitis
Genetic analysis of the human pancreatic diseases has shown several predisposing genetic abnormalities that affect the development of both acute and chronic pancreatitis (Braganza et al. 2011). Since premature activation of digestive enzymes within the acinar cells is one hypothesis for its onset, genetic abnormalities in the genes associated with the production of the proenzymes and their inhibitors, or in the genes controlling the secretory processes involved in pancreatic enzyme function, have been key targets for investigation. There are three genes in human chronic pancreatitis that have been associated with the disease—serine protease inhibitor Kazal-type 1 (SPINK1), cationic trypsinogen (PRSS1), and the cystic fibrosis transmembrane conductance regulator (CFTR). Considerable interest was focused on cystic fibrosis and the CFTR where abnormalities of the chloride ion channels have been shown to be responsible for the pancreatic fibrosis and functional defects in the lungs, and small intestine, that typify the disease (Strausbaugh and Davis 2007).
The advent of transgenic technology has enabled the development of mice with the 3 dominant genetic defects in human pancreatitis which, together with some mouse and rat strains that spontaneously develop pancreatic dysfunction, have provided yet another angle to study several features of the human disease.
The CFTR Deficient Tg Mouse
The first mouse model of cystic fibrosis was established in 1992 (Snouwaert et al. 1992), and since that time more specific models that more closely mimic the mutations found most commonly in the human disease have been developed (Delaney et al. 1996; Colledge et al. 1995; Zeiher et al. 1995). While the majority of these mutations alone showed little, or very mild, pancreatic changes (Rozmahel et al. 1996), the severity of the intestinal disease can be high and, for some, the intestinal pathologic changes are lethal at or around day 40 (Snouwaert et al. 1992). Further breeding of these models with other mouse strains have selectively developed models that show varying degrees of pancreatic disease comparable with that seen in the human situation (Durie et al. 2004). These animals show significant atrophy of the pancreatic acinar cells, while the intercalated, interlobular, and intralobular pancreatic ducts were dilated and filled with inspissated granular material with a mild degree of periductular inflammatory cells. The animals additionally showed a progressive liver disease with steatohepatitis, cholangitis, and biliary fibrosis by the time they were 1 year of age (Durie et al. 2004). While potentially these gene knockout animal models held great promise for their relevance to the comparable specific human genetic changes, their current utility to study the pathogenesis of the human diseases has been limited due to the apparent intractability of the murine homologue to show the disease states commonly seen in man.
The SPINK3 KO Mouse
Trypsin is stored in the zymogen granules, alongside a secretory trypsin inhibitor that prevents the premature intracellular activation of trypsinogen (Hirota, Ohmuraya, and Baba 2006). The rat homologue of this protein is called pancreatic secretory trypsin 1 or PST-1, while that in the human pancreas has been termed the serine protease inhibitor Kazal-type or SPINK (Graf and Bimmler 2006). It is highly expressed both in the pancreatic exocrine cells and in the pancreatic secretions to prevent activation before the proenzyme reaches the duodenum. Along with PRSS1 (cationic trypsinogen) and CFTR, SPINK1 is the third member of a gene group known to increase the susceptibility to developing pancreatitis in man (Chen et al. 2000; Liddle 2006; Whitcomb et al. 1996; Keim 2008; Cohn et al. 1998), but not in themselves sufficient to cause pancreatitis.
As a consequence of the perceived importance of the SPINK protein in pancreatitis in man, a homozygous mouse deficient in SPINK3, the mouse homologue of human SPINK 1, was generated (Ohmuraya et al. 2006). In these mice, the pancreas developed normally up to day 15.5 postconception, but from this time on, the pancreas atrophies and the mice never survive past 15 days postpartum (Ohmuraya et al. 2005). Active trypsin was detected within the pancreas in the 1.5-day-old SPINK3 −/− pups. Because of the lethality associated with the homozygous knockout mice, the role of this protein in increasing the susceptibility to pancreatitis was studied in a heterozygous SPINK3 +/− mouse, which demonstrated the expected decrease in the amount of SPINK3 protein in the exocrine cells (Romac et al. 2010). Surprisingly, using caerulein as an initiator of acute pancreatitis, the SPINK3 +/− mice failed to show any enhanced susceptibility to developing acute pancreatitis over that seen in the wild-type mouse. Once again this model has underlined the importance of environmental conditions, in addition to genetic susceptibility, in determining the onset and progression of pancreatitis in the mouse, and by inference, its development in man.
Transgenic Mouse Model Carrying Overexpression of PRSS1
One of the hypotheses for the trigger event for pancreatitis invokes the premature activation of trypsinogen within the exocrine cells (Braganza et al. 2011). This can then activate a number of other digestive enzymes within the pancreatic acinar cells and precipitate the necrosis and inflammation seen in pancreatitis. An arginine–histidine mutation (classified as R122H) on the human cationic trypsinogen protein (PRSS1) is associated with a rare type of hereditary human pancreatitis characterized by necrosis and chronic inflammation (Whitcomb et al. 1996). An untreated transgenic mouse, carrying an overexpressing mis-sense mutation, R122H, in trypsin 4, shows pancreatic fibrosis (Archer et al. 2006), while a second transgenic mouse strain carrying the R122H mutated human trypsinogen showed increased serum amylase activity in the absence of morphological pancreatitis (Selig et al. 2006). Both transgenes showed increased susceptibility to caerulein-induced acute pancreatitis in comparison to their wild-type littermates (Gaiser et al. 2011) emphasizing the role of the PRSS1 mutations as triggering events in pancreatitis.
The WBN/Kob Rat Model
This rat strain was developed at the University of Bonn for the study of cancer of the glandular stomach (Kobori, Gedigk, and Totovic 1977; Tsuchitani et al. 1985). Male rats of the WBN/Kob rat strain spontaneously develop a form of chronic pancreatitis which begins around 3 months of age with a periductular fibrosis leading to progressive loss of exocrine tissue which results in diabetes at 60 to 90 weeks of age. Despite their use as a model of diabetes, the accompanying morphological and biochemical changes also make them a useful model for studying chronic pancreatitis (Aghdassi et al. 2011). In this rat strain, the males only begin to develop pancreatitis when they become sexually mature and plasma androgen levels increase. Estradiol treatment in males has also been shown to significantly reduce the development of fibrosis, while ovariectomy of females has been shown to cause pancreatic fibrosis, pointing to the critical role of hormonal status in the development of the disease in this species. The precise relevance of this model to the human disease is debatable considering the absence of a sex preference in man, and a significant disadvantage of the model is the prolonged, and variable, onset and progression of the developing pancreatitis.
Drug-induced Pancreatitis
While not a common adverse reaction, from reports issued between 1968 and 1993, drug-induced pancreatitis in patients had been reported for over 500 separate drugs (Lancashire, Cheng, and Langman 2003). While the diagnosis of drug-induced pancreatitis requires that the affected individual is actually on medication, it is generally arrived at by exclusion of all other known causes and by a resolution of symptoms once the drug has been withdrawn (Nitsche et al. 2010). A classification system devised by Karch and Lasagna (1975) divided drugs associated with pancreatitis into “Definite,” “Probable,” and “Possible,” and using these criteria mesalazine, azathioprine, and simvastatin were assigned as being definite pancreatitis-inducing drugs in a review submitted to the Danish Committee on Adverse Drug Reactions between 1968 and 1999 (Andersen, Sonne, and Andersen 2001).
Of particular interest, because of their fairly recent introduction into therapeutic indications, and because of the fairly large number of different drugs that have been registered, are the class of tyrosine kinase inhibitors used against a range of different cancers (Pezzilli, Corinaldesi, and Morselli-Labate 2010). Imatinib is one such drug that has been shown to be remarkably effective against myeloid leukemia but, due to the development of resistance, a number of similar targeting agents, including dasatinib, nilotinib, and bosutinib have been introduced, and all of these have been associated with elevations in serum amylase activity and/or lipase activity with incidences of up to 70% (Kantarjian et al. 2006; Britten et al. 2008; Akaza et al. 2007). Despite these high levels of pancreatic enzymes, frank pancreatitis has not been reported for this class of drugs (National Cancer Institute 2006; Pezzilli, Corinaldesi, and Morselli-Labate 2010). The mechanism behind this extremely common pancreatic effect of these drugs remains unknown although for one, Sorafenib, a definite association between administration and acute pancreatitis has been made (Amar, Wu, and Tan 2007). Since most if not all of the patients receiving these drugs have serious, life-threatening cancers, the significant contribution of the patient’s ongoing disease in inducing the observed pancreatic effects cannot be excluded (Saadati and Saif 2010; Li and Srinivas 2007).
The question of how drugs can provoke episodes of acute pancreatitis is an interesting one. With regard to Sorafenib, altered GI motility has been proposed to change the functioning of the sphincter of Oddi, possibly leading to bile/pancreatic secretion reflux into the pancreatic ducts (Li and Srinivas 2007). Other drugs are thought to selectively accumulate within the acinar cells and either cause toxicity directly or as a consequence of metabolic activation (Foster et al. 1993; Braganza 1998). Yet others, such as the angiotensin converting enzyme inhibitors, are thought to induce cellular lesions through their interference with the pancreatic microcirculation by affecting the renin–angiotensin, or the kalikrein–kinin, system (Balani and Grendell 2008).
Clinical Relevance of the Models
The clinical relevance of the various animal models of both acute and chronic pancreatitis has received considerable criticism from practicing clinicians, and an equally forcible support from their advocates in experimental pathology (Bilchik et al. 1990; Aghdassi et al. 2011; Su, Cuthbertson, and Christophi 2006; Büchler et al. 1992; Schmidt et al. 1992; Al-Mufti and Williamson 1999).
The spectrum of the acute form of the human disease is broad ranging from a mild edematous lesion, which recovers without therapy, to severe hemorrhagic necrosis resulting in death. Modern clinical management of the shock reaction, which occurs with severe disease, has prevented early deaths but can be followed by sepsis and organ failure which are by far the most common causes of death (De Bolla and Obeid 1984). The complexity of the human diseases are such that any animal model developed will have a considerable hill to climb if they are to mimic all the varied aspects of the human disease, and in essence the available models show some, but generally not all, of the features of the spectrum of the human disease. Nevertheless they have and will continue to provide invaluable tools with which to study the components of the disease from the very early onset of pancreatic change that precedes necrosis and inflammation, through to the molecular events that govern the development of fibrosis, which could not ethically be carried out in human cases. They also allow the study of the early therapeutic intervention in disease models known to progress to greater severity in the absence of therapy, a situation that would equally be unethical in human patients. Perhaps more importantly selective animal models show varying degrees of reproducibility, in terms of the onset and progression of the disease, and which provide groups of subjects with which to rigorously test the effectiveness of drug, supplement, and surgical interventions at specific times postinitiation of pancreatitis (Büchler et al. 1992). Several of the models, including caeurulein hyperstimulation and infusion of bile salts into the pancreatic duct, either produce too mild a disease, too severe an outcome, or are simply too variable to permit the acquisition of useful information (Lampel and Kern 1977). The ethionine-supplemented, choline-deficient diet has been reported to mimic the human disease in its pathology but because it has to be used with young female mice has been criticized as having little relevance to the pathogenesis of the human disease (Lombardi, Estes, and Longnecker 1975).
Reflections
The method by which pancreatitis is initiated in animals may be irrelevant if the subsequent sequence of events mirrors the progression seen in some variants of the human disease since even in man the onset, development, and outcome of the disease may vary considerably (Braganza et al. 2011). Probably, because of the inability to study the early disease in man, much of the hypothesis-driven, therapeutic, and dietary intervention studies have their origins in work in animal models (Amano et al. 2010; McClave 2012). While it is unlikely that wholly novel animal models would significantly improve our understanding and treatment of the human disease, the potential of combining the existing models in the genetically modified mice and rats promises to significantly advance our knowledge of the underlying molecular events that predict the outcome of acute episodes of the disease and to provide intervention strategies that could significantly improve the outcomes.
Footnotes
Abbreviations
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The author(s) were employed by AstraZeneca Pharmaceuticals during the preparation and publication of this article.